
VYDURA - Tabletas orodispersables
Sustancia(s):
- Rimegepant
Presentaciones:
- 1 Caja, 2 Tabletas, 75 mg
- 1 Caja, 8 Tabletas, 75 mg
FORMA FARMACÉUTICA Y FORMULACIÓN:
Cada TABLETA contiene:
Rimegepant 75 mg
Excipiente cbp 1 tableta
INDICACIONES TERAPÉUTICAS:
• Tratamiento agudo de la migraña con o sin aura en adultos.
• Tratamiento preventivo de la migraña episódica en adultos que tienen al menos 4 ataques de migraña por mes.
FARMACOCINÉTICA Y FARMACODINAMIA:
Propiedades farmacocinéticas:
Absorción:
Después de la administración oral de VYDURA®, el rimegepant se absorbe con la concentración máxima a las 1.5 horas. La biodisponibilidad oral absoluta del rimegepant es de aproximadamente un 64%.
Después de la administración de VYDURA® en condiciones de alimentación con una comida alta o baja en grasas, el Tmáx se retrasó de 1 a 1.5 horas. Una comida rica en grasas redujo la Cmáx de un 42% a un 53% y el ABC de un 32% a un 38%. Una comida baja en grasas redujo la Cmáx en un 36% y el ABC en un 28%. Se administró VYDURA® sin tener en cuenta los alimentos en los estudios clínicos de seguridad y eficacia.
Distribución:
El volumen de distribución en estado de equilibrio del rimegepant es de 120 L. La unión a proteínas plasmáticas del rimegepant es de aproximadamente un 96%.
Eliminación:
Metabolismo:
El rimegepant se metaboliza principalmente mediante el CYP3A4 y, en menor medida, mediante el CYP2C9. El rimegepant se elimina principalmente en forma inalterada (~77% de la dosis) sin metabolitos importantes (es decir, que representaban > 10% del material relacionado con el medicamento) detectados en el plasma humano.
Excreción:
La vida media de eliminación del rimegepant es de aproximadamente 11 horas en sujetos sanos. Después de la administración oral del [14C]-rimegepant a sujetos masculinos sanos, el 78% de la radioactividad total se recuperó en las heces y el 24% en la orina. El rimegepant sin cambios es el componente único más importante en las heces excretadas (42%) y en la orina (51%).
Linealidad/no linealidad:
El rimegepant muestra incrementos en exposición mayores a la dosis proporcional después de la administración oral de una dosis única, lo que parece estar relacionado con un aumento dependiente de la dosis en la biodisponibilidad.
Interacción medicamentosa:
Estudios in vitro:
• Enzimas
El rimegepant es un sustrato del CYP3A4 y CYP2C9 (consulte los Estudios in vivo). El rimegepant no es un inhibidor del CYP1A2, 2B6, 2C9, 2C19, 2D6 o UGT1A1 en concentraciones clínicamente relevantes. Sin embargo, el rimegepant es un inhibidor débil de CYP3A4 con inhibición dependiente del tiempo. El rimegepant no es un inductor de CYP1A2, CYP2B6 o CYP3A4 en concentraciones clínicamente relevantes.
• Transportadores
In vitro, el rimegepant es un sustrato de los transportadores de eflujo de la proteína de resistencia al cáncer de mama (BCRP por sus siglas en inglés) y P-gp. Los inhibidores de los transportadores de eflujo de P-gp y BCRP pueden aumentar las concentraciones plasmáticas del rimegepant. El rimegepant se puede administrar de forma concomitante con inhibidores del transportador BCRP y con inhibidores débiles a moderados solamente de la P-gp. Los inhibidores fuertes de la P-gp pueden coadministrarse con una frecuencia no mayor que una vez cada 48 horas, según un estudio de interacción clínica con un inhibidor dual fuerte de la P-gp y de la BCRP (ciclosporina) y con un inhibidor selectivo de la P-gp (quinidina) que dio lugar a aumentos significativos de magnitud similar en la exposición al rimegepant (ABC y Cmáx en > 50%, pero menos de dos veces).
El rimegepant no es un sustrato de OATP1B1 o de OATP1B3. Teniendo en cuenta su baja depuración renal, no se evaluó el rimegepant como sustrato de OAT1, OAT3, OCT2, MATE1 o MATE2-K.
El rimegepant no es un inhibidor de P-gp, BCRP, OAT1 o MATE2-K en concentraciones clínicamente relevantes. Es un inhibidor débil de OATP1B1 y OAT3.
El rimegepant es un inhibidor de OATP1B3, OCT2 y MATE1. No se esperan interacciones medicamentosas clínicas para VYDURA® con estos transportadores en concentraciones clínicamente relevantes.
Estudios in vivo:
Inhibidores de la CYP3A4:
En un estudio dedicado a interacciones medicamentosas, la administración concomitante de 75 mg del rimegepant (dosis única) con itraconazol, un inhibidor fuerte del CYP3A4 en estado de equilibrio resultó en una mayor exposición de rimegepant (ABC en 4 veces y Cmáx en ~1.5 veces). No se llevó a cabo ningún estudio dedicado a las interacciones medicamentosas para evaluar el efecto de la administración concomitante de un inhibidor débil del CYP3A4 sobre la farmacocinética del rimegepant. La administración concomitante del rimegepant con un inhibidor moderado del CYP3A4 puede aumentar las exposiciones del rimegepant (ABC) en menos de 2 veces. No se espera que la administración concomitante del rimegepant con un inhibidor débil del CYP3A4 tenga un efecto clínicamente significativo sobre las exposiciones al rimegepant (ver sección Interacciones medicamentosas y de otro género).
Inductores del CYP3A4:
En un estudio dedicado a las interacciones medicamentosas, la administración concomitante de 75 mg del rimegepant (dosis única) con la rifampicina, un inductor fuerte del CYP3A4 en estado de equilibrio provocó una disminución de las exposiciones del rimegepant (ABC en un 80% y Cmáx en un 64%), lo que puede provocar la pérdida de eficacia. No se llevó a cabo ningún estudio dedicado a las interacciones medicamentosas para evaluar el efecto de la administración concomitante de un inductor débil o moderado del CYP3A4 sobre la farmacocinética del rimegepant. Debido a que el rimegepant es un sustrato moderadamente sensible del CYP3A4, los medicamentos que son inductores moderados del CYP3A4 también pueden causar una reducción significativa en la exposición al rimegepant, lo que da como resultado la pérdida de eficacia. No se espera una interacción clínicamente significativa con la administración concomitante de inductores débiles del CYP3A4 y el rimegepant (ver sección Interacciones medicamentosas y de otro género).
Inhibidores del CYP2C9:
En un estudio dedicado a las interacciones medicamentosas, la administración concomitante de 75 mg del rimegepant (dosis única) con fluconazol, un inhibidor moderado combinado de CYP3A4 y CYP2C9, dio como resultado mayores exposiciones al rimegepant (ABC en 1.8 veces) sin efecto relevante sobre la Cmáx. El rimegepant se metaboliza principalmente mediante el CYP3A4 y, en menor medida, mediante CYP2C9. El aumento en la exposición del rimegepant se puede atribuir a la inhibición combinada de CYP2C9 y CYP3A4 con la administración del fluconazol, lo que sugiere una contribución menor del CYP2C9. Por lo tanto, no se espera que la inhibición de CYP2C9 sola afecte significativamente las exposiciones al rimegepant (ver sección Interacciones medicamentosas y de otro género).
Inhibidores de BCRP y/o P-gp solamente:
En un estudio dedicado a las interacciones medicamentosas, la administración concomitante de 75 mg del rimegepant (dosis única) con ciclosporina, un inhibidor fuerte de los transportadores P-gp y BCRP, así como también por separado con quinidina, un inhibidor fuerte de P-gp solamente, produjo un aumento similar de las exposiciones de rimegepant de > 50%, pero menos de dos veces a través de la inhibición de P-gp. Basado en la totalidad de los resultados, no se espera que la inhibición de la BCRP afecte significativamente las exposiciones al rimegepant.
Sustratos del MATE1:
En un estudio dedicado a las interacciones medicamentosas, la administración concomitante de VYDURA® con metformina, un sustrato del transportador MATE1, no produjo un efecto clínicamente significativo en la farmacocinética de la metformina ni en la utilización de glucosa.
Otros medicamentos:
En estudios dedicados de interacción medicamentosa, no se observaron interacciones farmacocinéticas significativas cuando se administró de manera concomitante el rimegepant con anticonceptivos orales (norelgestromina, etinilestradiol), midazolam (un sustrato sensible de CYP3A4) o sumatriptán.
Poblaciones específicas:
Insuficiencia renal:
No existe una diferencia clínicamente significativa en la exposición al rimegepant en sujetos con insuficiencia renal en comparación con los sujetos con función renal normal. En un estudio clínico dedicado a comparar la farmacocinética del rimegepant en sujetos con insuficiencia renal leve (depuración de creatinina estimada [CLCR] 60-89 mL/min), moderada (CLCR 30-59 mL/min), y grave (CLCR 15-29 mL/min) con el de los sujetos normales (control emparejado sano), se observó un aumento de menos del 50% en la exposición total a rimegepant después de una dosis única de 75 mg. VYDURA® no se ha estudiado en pacientes con enfermedad renal en estado terminal (CLCR < 15 mL/min) (ver sección Dosis y vía de administración).
Insuficiencia hepática:
No hubo diferencias clínicamente significativas en la exposición al rimegepant en sujetos con insuficiencia hepática leve (Child-Pugh clase A) y moderada (Child-Pugh clase B) en comparación con los sujetos con función hepática normal (ver sección Dosis y vía de administración). En un estudio clínico dedicado a comparar la farmacocinética del rimegepant en sujetos con insuficiencia hepática leve, moderada y grave con la de sujetos normales (control sano emparejado), la exposición del rimegepant (Cmáx y ABC) tras una dosis única de 75 mg fue aproximadamente 2 veces mayor en sujetos con insuficiencia grave (clase C de Child-Pugh).
Otras poblaciones específicas:
No se observaron diferencias clínicamente significativas en la farmacocinética del rimegepant en función de la edad (ver sección Dosis y vía de administración), el sexo, la raza, la etnia, el peso corporal o el genotipo CYP2C9.
Farmacogenómica:
La Cmáx y ABC0-inf del rimegepant fueron similares en los metabolizadores intermedios de CYP2C9 (es decir, *1/*2, *2/*2, *1/*3, n = 43) en comparación con los metabolizadores normales (es decir, *1/*1, n = 72). No se dispone de datos farmacocinéticos adecuados de los metabolizadores pobres de CYP2C9 (es decir, *2/*3). Dado que la contribución de CYP2C9 al metabolismo de rimegepant se considera menor, no se espera que el polimorfismo de CYP2C9 afecte significativamente su exposición.
Propiedades farmacodinámicas:
El rimegepant se une selectivamente con alta afinidad al receptor del péptido relacionado con el gen de la calcitonina humana (CGRP por sus siglas en inglés) y antagoniza la función del receptor de CGRP.
No se observaron diferencias clínicamente relevantes en la presión arterial en reposo cuando se administró de manera concomitante el rimegepant con el sumatriptán (12 mg por vía subcutánea, administrados en dos dosis de 6 mg separadas por una hora) en comparación con el sumatriptán sólo en voluntarios sanos.
Electrofisiología cardiaca:
Con una dosis única 4 veces superior a la dosis recomendada, el rimegepant no prolonga el intervalo QT en una medida clínicamente relevante.
Eficacia clínica: tratamiento agudo:
La eficacia de VYDURA® para el tratamiento agudo de la migraña con y sin aura en adultos se demostró en un ensayo aleatorizado, doble ciego, controlado con placebo: Estudio 1. El estudio aleatorizó a los pacientes para que recibieran 75 mg de VYDURA® (N = 732) o placebo (N = 734). Los pacientes fueron instruidos para tratar una migraña de intensidad de dolor de cabeza de moderada a grave. Se permitió el uso de medicamento de rescate (es decir, AINE, acetaminofén y/o un antiemético) 2 horas después del tratamiento inicial. No se permitieron otras formas de medicamentos de rescate, como triptanos, dentro de las siguientes 48 horas del tratamiento inicial. Aproximadamente el 14% de los pacientes tomaba medicamentos preventivos para la migraña en el periodo inicial. Ninguno de los pacientes en el Estudio 1 se encontraba en tratamiento con medicamentos preventivos concomitantes que actúan sobre la vía peptídica relacionada con el gen de la calcitonina.
Los análisis de eficacia primaria se realizaron en pacientes que trataron una migraña con dolor moderado a grave. VYDURA® demostró una reducción estadísticamente significativa de la ausencia de dolor y de la ausencia del síntoma más molesto (SMM) a las dos horas de la dosis, en comparación con el placebo. La ausencia de dolor se definió como la reducción del dolor de cabeza moderado o grave a la ausencia de dolor de cabeza, y la ausencia de SMM se definió como la ausencia del SMM autoidentificado (es decir, fotofobia, fonofobia o náuseas). Entre los pacientes que seleccionaron un SMM, el síntoma seleccionado con mayor frecuencia fue la fotofobia (54%), seguida de náuseas (28%) y fonofobia (15%).
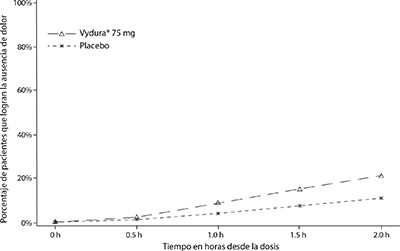
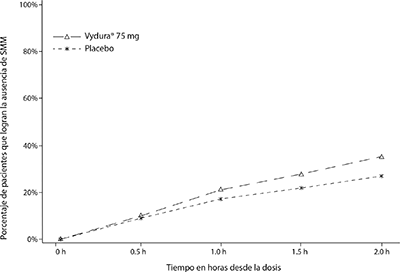
En el Estudio 1, el porcentaje de pacientes que lograron la ausencia de dolor de cabeza y la ausencia de SMM dos horas después de una sola dosis fue estadística y significativamente mayor en los pacientes que recibieron VYDURA® en comparación con los que recibieron placebo (tabla 1). La eficacia fue similar en dos ensayos pivotales adicionales, doble ciego y controlados con placebo en pacientes con migraña que recibieron una forma de dosificación oral bioequivalente de 75 mg de rimegepant.
Tabla 1. Criterios de valoración de la eficacia de la migraña para el Estudio 1
|
Estudio 1 |
||
|
VYDURA® 75 mg |
Placebo |
|
|
Ausencia de dolor luego de 2 horas |
||
|
n/N* |
142/669 |
74/682 |
|
% de sujetos que respondieron al tratamiento |
21.2 |
10.9 |
|
Diferencia en comparación con el placebo (%) |
10.3 |
|
|
Valor p |
< 0.001 |
|
|
Ausencia de SMM luego de 2 horas |
||
|
n/N* |
235/669 |
183/682 |
|
% de sujetos que respondieron al tratamiento |
35.1 |
26.8 |
|
Diferencia en comparación con el placebo (%) |
8.3 |
|
|
Valor p |
0.001 |
|
* n = cantidad de pacientes que respondieron al tratamiento; N = cantidad de pacientes en ese grupo de tratamiento; SMM = síntoma más molesto.
En la Figura 1, se presenta el porcentaje de pacientes que lograron la ausencia de dolor por migraña en el plazo de 2 horas después del tratamiento en el Estudio 1.
Figura 1. Porcentaje de pacientes que lograron la ausencia de dolor en el plazo de 2 horas en el Estudio 1
En la Figura 2, se presenta el porcentaje de pacientes que lograron la ausencia de SMM en el plazo de 2 horas en el Estudio 1.
Figura 2. Porcentaje de pacientes que lograron la ausencia de SMM en el plazo de 2 horas en el Estudio 1
En el Estudio 1, se demostraron los efectos estadísticamente significativos de VYDURA® en comparación con el placebo para los criterios de valoración de la eficacia adicionales del alivio del dolor a las 2 horas, la ausencia sostenida del dolor de 2 a 48 horas, el uso de medicamentos de rescate dentro de las siguientes 24 horas, y el porcentaje de pacientes que informaron una función normal dos horas después de la dosificación (tabla 2). El alivio del dolor se definió como una reducción del dolor por migraña de severidad moderada o grave a leve o ninguno. La determinación del porcentaje de pacientes que informaron una función normal dos horas después de la dosificación se derivó de un cuestionario de un solo dato, donde se les pidió a los pacientes que seleccionaran una respuesta en una escala de 4 puntos; función normal, deterioro leve, deterioro grave o reposo en cama requerido. Se observaron resultados similares en dos estudios pivotales adicionales con una forma de dosificación oral bioequivalente de 75 mg de rimegepant.
Tabla 2. Criterios de valoración adicionales de la eficacia sobre la migraña en el Estudio 1
|
Estudio 1 |
||
|---|---|---|
|
VYDURA® 75 mg |
Placebo |
|
|
Alivio del dolor a las 2 horas |
||
|
n/N* |
397/669 |
295/682 |
|
% de sujetos que respondieron al tratamiento |
59.3 |
43.3 |
|
Diferencia en comparación con el placebo |
16.1 |
|
|
Valor p |
< 0.001 |
|
|
Ausencia sostenida de dolor de 2 a 48 horas |
||
|
n/N* |
90/669 |
37/682 |
|
% de sujetos que respondieron al tratamiento |
13.5 |
5.4 |
|
Diferencia en comparación con el placebo (%) |
8.0 |
|
|
Valor p |
< 0.001 |
|
|
Administración de medicamento de rescate en el plazo de 24 horas** |
||
|
n/N* |
95/669 |
199/682 |
|
% de sujetos que respondieron al tratamiento |
14.2 |
29.2 |
|
Diferencia en comparación con el placebo (%) |
-15.0 |
|
|
Valor p |
< 0.001 |
|
|
Porcentaje de pacientes que informaron función normal a las 2 horas |
||
|
n/N* |
255/669 |
176/682 |
|
% de sujetos que respondieron al tratamiento |
38.1 |
25.8 |
|
Diferencia en comparación con el placebo (%) |
12.3 |
|
|
Valor p |
< 0.001 |
|
* n = cantidad de sujetos que respondieron al tratamiento; N = cantidad de pacientes en ese grupo de tratamiento.
** En este análisis se incluye solamente la administración de AINE, acetaminofén o antieméticos en el plazo de 24 horas después de la dosis. No se permitió la administración de triptanos u otros medicamentos para la migraña aguda.
La incidencia de fotofobia y fonofobia se redujo a las 2 horas después de la administración de 75 mg de VYDURA® en comparación con el placebo. También se demostró un beneficio significativo de VYDURA® en comparación con el placebo (valor p < 0.05) para el alivio del dolor y la ausencia de discapacidad funcional a los 60 minutos; el alivio del dolor, la ausencia de dolor, la ausencia de discapacidad funcional y la ausencia de SMM a los 90 minutos; ausencia de discapacidad funcional a las 2 horas; ausencia sostenida de SMM, alivio sostenido del dolor, ausencia sostenida del dolor y ausencia sostenida de la discapacidad funcional de 2 a 24 horas y de 2 a 48 horas.
Eficacia clínica: profilaxis:
La eficacia del rimegepant se evaluó como un tratamiento preventivo de la migraña en un estudio aleatorizado, doble ciego, controlado con placebo (Estudio 2).
El Estudio 2 incluyó a adultos masculinos y femeninos con al menos 1 año de antecedentes de migraña (con o sin aura). Los pacientes tenían antecedentes de 4 a 18 ataques de migraña de intensidad del dolor de moderada a grave cada 4 semanas en el periodo de 12 semanas previas a la visita de selección. El estudio aleatorizó a los pacientes para que recibieran 75 mg de rimegepant (N = 373) o placebo (N = 374) durante un máximo de 12 semanas. Se indicó a los pacientes que tomaran el tratamiento aleatorizado cada dos días (CDD) para el periodo de tratamiento de 12 semanas. Se permitió que los pacientes utilizaran otros tratamientos agudos para la migraña (p. ej., triptanos, AINE, acetaminofén, antieméticos) según lo necesitaran. Aproximadamente el 22% de los pacientes tomaban medicamentos preventivos para la migraña en el periodo inicial. Se permitió a los pacientes continuar en un estudio de extensión abierto durante 12 meses adicionales.
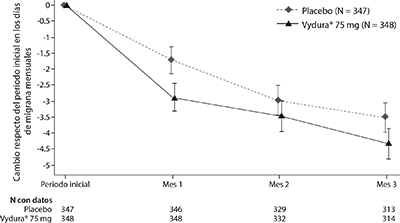
El criterio primario de valoración de la eficacia para el Estudio 2 fue el cambio desde el periodo inicial en la cantidad media de días de migraña mensuales (DMM) durante las semanas 9 a 12 de la fase de tratamiento doble ciego. Los criterios secundarios de valoración incluyeron el logro de una reducción ≥ 50% desde el periodo inicial en los días de migraña mensual moderada o grave, la frecuencia del consumo de medicamentos de rescate, el cambio desde el periodo inicial en la función de rol del Cuestionario de Calidad de Vida específico de la migraña (MSQ, por sus siglas en inglés) durante las semanas 9 a 12, y también el cambio desde el periodo inicial en los DMM durante las semanas 1 a 12. El rimegepant de 75 mg dosificado cada dos días demostró mejoras estadísticamente significativas para los criterios clave de valoración de la eficacia en comparación con el placebo, como se resume en la tabla 3 y se muestra gráficamente en la Figura 3.
Tabla 3. Criterios clave de valoración de la eficacia para el Estudio 2
|
Rimegepant 75 mg CDD |
Placebo CDD |
|
|
Días mensuales de migraña (DMM), semanas 9 a 12 |
N = 348 |
N = 347 |
|
Cambio desde el periodo inicial |
-4.3 |
-3.5 |
|
Cambio en comparación con el placebo |
-0.8 |
|
|
Valor p |
0.010a |
|
|
Reducción ≥ 50% en los DMM moderados o graves de las semanas 9 a 12 |
N = 348 |
N = 347 |
|
% de sujetos que respondieron al tratamiento |
49.1 |
41.5 |
|
Diferencia en comparación con el placebo |
7.6 |
|
|
Valor p |
0.044a |
|
|
Días de migraña mensual (DMM), semanas 1 a 12 |
N = 348 |
N = 347 |
|
Cambio desde el periodo inicial |
-3.6 |
-2.7 |
|
Diferencia en comparación con el placebo |
-0.8 |
|
|
Valor p |
0.0017a |
|
|
MSQ Función de rol: puntaje de dominio restrictivo en la semana 12 |
N = 269 |
N = 266 |
|
Cambio medio respecto del periodo inicial |
18.0 |
14.6 |
|
Diferencia en comparación con el placebo |
3.5 |
|
|
Valor p |
0.036b |
a Valor p significativo en pruebas jerárquicas.
b Valor p nominal en pruebas jerárquicas.
Figura 3. Cambio respecto del periodo inicial en los días de migraña mensuales en el Estudio 2
Eficacia a largo plazo:
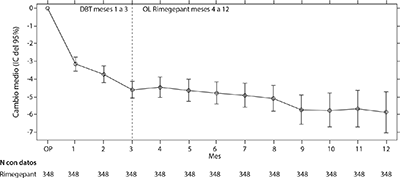
Se permitió a los pacientes que participaron en el Estudio 2 continuar en un estudio de extensión abierto durante 12 meses adicionales. La eficacia se mantuvo por hasta 1 año en un estudio de extensión abierto en la que los pacientes con migraña episódica o crónica (con un valor inicial promedio de 10 DMM mensuales) recibieron 75 mg de rimegepant cada dos días más rimegepant según se necesitara, en días de dosificación no programados (Figura 4). Una parte compuesta por 203 pacientes asignados a rimegepant completaron el periodo de tratamiento general de 16 meses. En estos pacientes, la reducción media general desde el periodo inicial en el número de DMM promediada durante el periodo de tratamiento de 16 meses fue de 6.2 días.
Figura 4. Gráfico longitudinal del cambio en la cantidad media de días de migraña mensuales (DMM) desde el periodo de observación a lo largo del tiempo durante el tratamiento doble ciego (TDC) (meses 1 a 3) y durante el tratamiento abierto (TA) con rimegepant (meses 4 a 16)
CONTRAINDICACIONES:
VYDURA® está contraindicado en pacientes con antecedentes de reacciones de hipersensibilidad a rimegepant o cualquiera de sus componentes.
Este medicamento no se recomienda en pacientes con insuficiencia hepática grave.
RESTRICCIONES DE USO DURANTE EL EMBARAZO Y LA LACTANCIA:
Embarazo:
No existen datos adecuados en humanos sobre el riesgo de desarrollo asociado con la administración del rimegepant en el embarazo. Los estudios en animales demuestran que, en exposiciones clínicamente relevantes, rimegepant no produce muerte embriofetal ni malformaciones fetales. No hubo efectos sobre el desarrollo en ratas con dosis de hasta 60 mg/kg/día (exposiciones 46 veces el ABC humana a la dosis humana máxima recomendada [MRHD, por sus siglas en inglés] de 75 mg/día) o en conejos con hasta la dosis más alta analizada de 50 mg/kg/día (exposiciones 10 veces la MRHD de 75 mg/día).
Lactancia:
Se realizó un estudio de lactancia en 12 mujeres adultas sanas en periodo de lactancia que tenían entre 2 semanas y 6 meses después del parto y se les administró una dosis oral única de rimegepant 75 mg. Los resultados han establecido una proporción promedio de leche a plasma de 0.20 y una dosis infantil relativa de menos del 1% de la dosis materna ajustada en función del peso. Estos datos respaldan que la transferencia de rimegepant a la leche materna es baja. No existen datos sobre los efectos de rimegepant en el lactante o en la producción de leche.
Se deben considerar los beneficios de la lactancia para la salud y el desarrollo, junto con la necesidad clínica de la administración del rimegepant a la madre y cualquier posible efecto adverso en el lactante amamantado debido a la administración del rimegepant o de la condición materna subyacente.
REACCIONES SECUNDARIAS Y ADVERSAS:
Más de 2100 pacientes con migraña han sido tratados con VYDURA® o con una forma de dosificación oral bioequivalente en estudios para registro controlados con placebo. En el programa de desarrollo general, más de 670 pacientes estuvieron expuestos durante al menos 12 meses.
La reacción adversa más frecuente fue la náusea para el tratamiento agudo (1.2% frente al 0.8% en pacientes que recibieron placebo) y para la prevención de la migraña (1.4% frente al 0.8%). La mayoría de las reacciones fueron de severidad leve o moderada. Hipersensibilidad, incluida la disnea y la erupción grave, ocurrió en menos del 1% de los pacientes tratados.
Lista tabulada de reacciones adversas:
Las reacciones adversas se enumeran por clasificación por órganos y sistemas por MedDRA en la tabla 4. La categoría de frecuencia correspondiente para cada reacción adversa al medicamento se basa en la siguiente convención (CIOMS III): muy frecuente (≥ 1/10), frecuente (≥ 1/100 a < 1/10), poco frecuente (≥ 1/1000 a < 1/100), rara (≥ 1/10,000 a < 1/1000), muy rara (< 1/10,000).
Tabla 4. Lista de reacciones adversas
|
Clasificación por |
Reacción adversa |
Frecuencia |
|
Tratamiento agudo |
||
|
Trastornos gastrointestinales |
Náusea |
Frecuente |
|
Trastornos del sistema inmunológico |
Hipersensibilidad, incluida disnea y erupción grave |
Poco frecuente |
|
Prevención |
||
|
Trastornos gastrointestinales |
Náusea |
Frecuente |
Se realizaron extensiones a los estudios abiertos para el tratamiento agudo y preventivo con dosificación durante 1 año y no se identificaron nuevas señales de seguridad.
Descripción de las reacciones adversas seleccionadas:
Reacciones de hipersensibilidad:
La hipersensibilidad, incluida la disnea y la erupción grave, ocurrieron en menos del 1% de los pacientes tratados en los estudios clínicos. Las reacciones de hipersensibilidad pueden ocurrir días después de la administración y se ha producido una hipersensibilidad seria retardada.
PRECAUCIONES EN RELACIÓN CON EFECTOS DE CARCINOGÉNESIS, MUTAGÉNESIS, TERATOGÉNESIS Y SOBRE LA FERTILIDAD:
Carcinogénesis:
La administración oral de rimegepant a ratones Tg.rasH2 sensibles a carcinógenos humanos genotóxicos y no genotóxicos durante 26 semanas y a ratas durante 91 a 100 semanas, no produjo evidencia de tumores inducidos por medicamentos en ninguna de las especies. En estos estudios en animales, la exposición plasmática (ABC) en la dosis más alta analizada fue aproximadamente 350 veces (ratones) y 30 veces (ratas) en la MRHD de 75 mg/día.
Mutagénesis:
El rimegepant fue negativo en los ensayos in vitro e in vivo.
Deterioro de la fertilidad:
La administración oral del rimegepant a ratas macho y hembra antes y durante el apareamiento, y la continuación en hembras hasta el día 7 de gestación, dio como resultado una disminución de la fertilidad con la dosis más alta (150 mg/kg/día) analizada.
La dosis sin efecto para el deterioro de la fertilidad y el desarrollo embrionario temprano en ratas con 60 mg/kg/día se asoció con exposiciones plasmáticas al medicamento (ABC) aproximadamente 30 veces la MRHD de 75 mg/día.
Fertilidad:
Los estudios en animales no mostraron ningún impacto clínicamente relevante en la fertilidad femenina y masculina.
INTERACCIONES MEDICAMENTOSAS Y DE OTRO GÉNERO:
Se debe consultar al médico cuando se utilicen otros medicamentos además del rimegepant.
Administración concomitante con inhibidores fuertes o moderados del CYP3A4:
Se debe evitar la administración concomitante de VYDURA® con inhibidores fuertes del CYP3A4, ya que la administración concomitante da como resultado un aumento significativo en la exposición al rimegepant (ver sección Farmacocinética y farmacodinamia).
Se debe evitar otra dosis del VYDURA® dentro de las siguientes 48 horas cuando se administra concomitantemente con inhibidores moderados del CYP3A4, ya que la administración concomitante puede dar como resultado un aumento de la exposición de VYDURA® (ver sección Farmacocinética y farmacodinamia).
Administración concomitante con inductores fuertes o moderados del CYP3A4:
Se debe evitar la administración concomitante de VYDURA® con inductores fuertes o moderados del CYP3A4. La administración concomitante puede dar como resultado una reducción significativa en la exposición al rimegepant, lo que puede llevar a la pérdida de eficacia de VYDURA® (ver sección Farmacocinética y farmacodinamia).
Administración concomitante con inhibidores fuertes del P-gp:
Se debe evitar otra dosis del VYDURA® dentro de las siguientes 48 horas cuando se coadministra con inhibidores fuertes del P-gp, ya que la coadministración puede provocar un aumento de la exposición del rimegepant (ver sección Farmacocinética y farmacodinamia).
Otros medicamentos:
No se observaron interacciones farmacocinéticas significativas cuando se coadministró VYDURA® con anticonceptivos orales (norelgestromina, etinilestradiol), midazolam (un sustrato sensible de CYP3A4), metformina (un sustrato de la MATE1) o sumatriptán (ver sección Farmacocinética y farmacodinamia).
ALTERACIONES EN LOS RESULTADOS DE PRUEBAS DE LABORATORIO:
No se dispone de datos hasta la fecha.
PRECAUCIONES GENERALES:
Se han producido reacciones de hipersensibilidad, incluida la disnea y la erupción en menos del 1% de los pacientes tratados con VYDURA® en estudios clínicos. Las reacciones de hipersensibilidad, incluida la hipersensibilidad seria, pueden ocurrir días después de la administración. Si se produce una reacción de hipersensibilidad, interrumpa la administración de VYDURA® de inmediato e inicie el tratamiento adecuado (ver sección Contraindicaciones).
Cefalea por uso excesivo de medicación:
El uso excesivo de cualquier tipo de medicamento para las cefaleas puede empeorarlas. Si se presenta o se sospecha esta situación, se debe acudir al médico e interrumpir el tratamiento. Se debe sospechar el diagnóstico de cefalea por uso excesivo de medicación en pacientes que presentan cefaleas frecuentes o diarias a pesar (o a causa) del uso habitual de medicamentos para la cefalea aguda.
Efectos en la capacidad para conducir y operar máquinas:
En función de los eventos adversos informados, VYDURA® no debe tener influencia identificada sobre la capacidad de conducir y utilizar máquinas.
DOSIS Y VÍA DE ADMINISTRACIÓN:
Vía de administración: Oral.
Instrucciones de uso:
• Despegue la cubierta de aluminio que cubre un blíster y retire con cuidado la tableta orodispersable.
• Tome la tableta inmediatamente después de sacarla del empaque.
Posología:
Prevención de la migraña: La dosis recomendada de VYDURA® es de 75 mg (base libre del rimegepant) tomado por vía oral cada dos días.
Tratamiento agudo de la migraña: La dosis recomendada de VYDURA® es de 75 mg (base libre del rimegepant) tomado por vía oral, según se necesite.
La dosis máxima en un periodo de 24 horas es una tableta de VYDURA®, igual a 75 mg de base libre del rimegepant.
Método de administración:
Colocar la tableta orodispersable (ODT) sobre la lengua o debajo de la lengua. La tableta se desintegrará en la boca y se puede tomar sin líquido.
Para obtener instrucciones sobre la manipulación del medicamento antes de su administración ver sección Leyendas de protección.
Poblaciones de pacientes especiales:
Población pediátrica:
No se ha establecido la seguridad ni la eficacia del VYDURA® en los pacientes pediátricos.
Uso geriátrico:
En los estudios farmacocinéticos, no se observaron diferencias farmacocinéticas clínicamente significativas entre los sujetos de edad avanzada y los sujetos más jóvenes. Los estudios clínicos de VYDURA® no incluyen cantidades suficientes de pacientes de 65 años y mayores como para determinar si responden de forma diferente que los pacientes más jóvenes.
En función del perfil de eventos adversos observados en adultos jóvenes y de mediana edad, la administración en pacientes de edad avanzada se considera aceptable.
Insuficiencia hepática:
No se requiere ajustar la dosis de VYDURA® en pacientes con insuficiencia hepática leve (Child- Pugh A) o moderada (Child-Pugh B). Las concentraciones plasmáticas del rimegepant fueron significativamente mayores en los sujetos con insuficiencia hepática grave (Child-Pugh C) (ver sección Farmacocinética y farmacodinamia). Se debe evitar el uso de VYDURA® en pacientes con insuficiencia hepática grave.
Insuficiencia renal:
No se requiere ajustar la dosis de VYDURA® en pacientes con insuficiencia renal leve, moderada o grave. de VYDURA® no ha sido estudiado en pacientes con enfermedad renal en estado terminal ni en pacientes en diálisis. Se debe evitar el uso de VYDURA® en pacientes con enfermedad renal en estado terminal (CLCR < 15 mL/min) (ver sección Farmacocinética y farmacodinamia).
MANIFESTACIONES Y MANEJO DE LA SOBREDOSIFICACIÓN O INGESTA ACCIDENTAL:
La experiencia clínica con sobredosis de VYDURA® es limitada. No se dispone de un antídoto específico para el tratamiento de la sobredosis del rimegepant. El tratamiento de la sobredosis con VYDURA® debe constar de medidas generales de apoyo, incluido el monitoreo de los signos vitales y la observación del estado clínico del paciente. Es poco probable que el rimegepant se elimine significativamente mediante diálisis debido a la alta unión a proteínas séricas (ver sección Farmacocinética y farmacodinamia).
PRESENTACIONES:
Caja con 2, 8 o 16 tabletas de 75 mg.
RECOMENDACIONES SOBRE ALMACENAMIENTO:
Consérvese a no más de 25 °C. Consérvese la caja bien cerrada. Mantenga la tableta en su empaque.
No guarde la tableta fuera del empaque para usarla en el futuro.
LEYENDAS DE PROTECCIÓN:
Su venta requiere receta médica. No se deje al alcance de los niños. No se use en el embarazo o lactancia. No se administre en menores de 18 años. Disuélvase debajo o sobre la lengua. No almacenar fuera del empaque. No manipule la tableta con las manos húmedas. No presione la tableta a través del blíster.
Reporte las sospechas de reacción adversa al correo: farmacovigilancia@cofepris.gob.mx y
MEX.AEReporting@pfizer.com
PFIZER, S.A. de C.V.
Km. 63 Carretera México-Toluca,
Zona Industrial, C.P. 50140, Toluca, México, México.
Reg. Núm. 147M2023 SSA