VENTANA PD-L1 (SP263) ASSAY
Para visualizar el contenido completo de la IPPA (información para prescribir amplia), deberá iniciar sesión.
INTERPRETACIÓN DE LAS TINCIONES Y RESULTADOS PREVISTOS:
El procedimiento de inmunotinción automatizada VENTANA hace que un producto de reacción de color marrón DAB se precipite en los sitios del antígeno localizados por el anticuerpo de VENTANA PD-L1 (SP263) ASSAY. Un anatomopatólogo cualificado interpreta el portaobjetos teñido usando la microscopía óptica. Un anatomopatólogo cualificado con experiencia en procedimientos de inmunohistoquímica (IHC) debe evaluar los controles tisulares y calificar el producto con tinción antes de interpretar los resultados.
El patrón de tinción celular de VENTANA PD-L1 (SP263) ASSAY es membranoso y/o citoplasmático en células tumorales. Las células inmunitarias manifiestan tinción de membrana lineal, citoplasmática difusa, y/o punteada.
Consulte la guía de interpretación VENTANA PD-L1 (SP263) ASSAY Interpretation Guide (1015317) para obtener información más específica y ver imágenes.
Control tisular placentario:
El control tisular placentario contiene elementos de tinción positivos y negativos para la proteína PD-L1 y, por tanto, es adecuado para su uso como control tisular. Deben examinarse los elementos de tinción positivos y negativos para comprobar que todos los reactivos han funcionado correctamente. Si estos elementos no muestran una tinción adecuada, los resultados de las muestras de la prueba se deben considerar no válidos.
El tejido de placenta teñido con VENTANA PD-L1 (SP263) ASSAY muestra una tinción uniforme de moderada a fuerte de la membrana y una tinción uniforme de débil a fuerte del citoplasma de las células de linaje trofoblástico. La vasculatura y el tejido estrómico placentario pueden utilizarse para evaluar cualquier tinción de fondo (Tabla 6).
Tabla 6. Criterios de evaluación del control tisular de placenta para VENTANA PD-L1 (SP263) ASSAY
|
Interpretación |
Descripción de la tinción |
|
Aceptable |
Tinción uniforme de moderada a fuerte de la membrana de células de linaje trofoblástico, estroma placentario y vasculatura sin tinción. |
|
Inaceptable |
La membrana de las células de linaje trofoblástico no presenta tinción o es débil y/o se observa una tinción específica en el tejido estrómico placentario y vascular. |
Control de reactivo negativo:
La tinción no específica, de estar presente, puede tener un aspecto difuso y se puede evaluar mediante el portaobjetos de control de reactivo negativo teñido con Rabbit Monoclonal Negative Control Ig. Se deben utilizar células intactas para la interpretación de los resultados de tinción, ya que la tinción de las células necróticas o degeneradas suele ser no específica. Si la tinción de fondo es excesiva, los resultados de las muestras de la prueba se deben considerar como no válidos. Consulte la Tabla 8 para obtener información sobre los criterios de aceptabilidad de la tinción no específica. Los ejemplos de tinción de fondo de este ensayo se pueden encontrar en la guía de interpretación (P/N 1015317).
Tejido del paciente:
El tejido del paciente debe evaluarse de acuerdo con el algoritmo de puntuación de VENTANA PD-L1 (SP263) ASSAY que se facilita en la Tabla 7 y la Tabla 8. Consulte la guía de interpretación (Interpretation Guide) de la tinción de NSCLC con VENTANA PD- L1 (SP263) ASSAY P/N 1015317 para ver imágenes representativas y obtener información sobre las instrucciones para la puntuación.
El patrón de tinción celular para VENTANA PD-L1 (SP263) ASSAY es membranoso y/o citoplasmático en células tumorales. Las células inmunitarias manifiestan tinción de membrana lineal, citoplasmática difusa, y/o punteada. La tinción citoplasmática de células tumorales, si está presente, no se considera positiva a efectos de puntuación.
Las células tumorales se puntúan como el porcentaje de células tumorales con tinción de membrana de PD-L1 de cualquier intensidad sobre la tinción de fondo, tal y como se indica en el correspondiente control negativo.
El tejido del paciente debe evaluarse según el algoritmo de puntuaciones de VENTANA PD-L1 (SP263) ASSAY.
Algoritmo de puntuación: NSCLC
Las muestras del NSCLC deben evaluarse de acuerdo con el algoritmo de puntuaciones de VENTANA PD-L1 (SP263) ASSAY para NSCLC (Tabla 7) y los criterios no específicos de puntuación de fondo (Tabla 8). Consulte la guía de interpretación para obtener instrucciones adicionales e imágenes representativas.
Tabla 7. Algoritmo de puntuación de VENTANA PD-L1 (SP263) ASSAY para NSCLC
|
Interpretación de PD-L1 |
Descripción de la tinción |
|---|---|
|
≥ 1% |
≥ 1% de células tumorales con positividad de membrana para PD-L1 a cualquier intensidad sobre la tinción de fondo, como se indica en el correspondiente control negativo. |
|
< 1% |
< 1% de células tumorales con positividad de membrana para PD-L1 a cualquier intensidad sobre la tinción de fondo, como se indica en el correspondiente control negativo. |
|
≥ 5% |
≥ 5% de células tumorales con positividad de membrana para PD-L1 a cualquier intensidad sobre la tinción de fondo, como se indica en el correspondiente control negativo. |
|
< 5% |
< 5% de células tumorales con positividad de membrana para PD-L1 a cualquier intensidad sobre la tinción de fondo, como se indica en el correspondiente control negativo. |
|
≥ 10% |
≥ 10% de células tumorales con positividad de membrana para PD-L1 a cualquier intensidad sobre la tinción de fondo, como se indica en el correspondiente control negativo. |
|
< 10% |
< 10% de células tumorales con positividad de membrana para PD-L1 a cualquier intensidad sobre la tinción de fondo, como se indica en el correspondiente control negativo. |
|
≥ 50% |
≥ 50% de células tumorales con positividad de membrana para PD-L1 de cualquier intensidad sobre la tinción de fondo, como se indica en el correspondiente control negativo. |
|
< 50% |
< 50% de células tumorales con positividad de membrana para PD-L1 de cualquier intensidad sobre la tinción de fondo, como se indica en el correspondiente control negativo. |
Tabla 8. Criterios de puntuación de la tinción de fondo no específica para VENTANA PD-L1 (SP263) ASSAY
|
Interpretación |
Descripción de la tinción |
|
Aceptable |
Tinción no específica que no interfiere con la interpretación de tinción específica. |
|
Inaceptable |
Tinción no específica que interfiere con la interpretación de tinción específica. |
RESOLUCIÓN DE PROBLEMAS:
Se facilita una guía de resolución de problemas en la Tabla 28. En caso de que algún problema no pueda atribuirse a ninguna de estas causas o la acción correctiva sugerida no consiga resolver el problema, diríjase a su representante local de asistencia técnica de Roche.
Tabla 28. Directrices para la resolución de problemas de VENTANA PD-L1 (SP263) ASSAY
|
Problema |
Causa probable |
Acción sugerida |
|---|---|---|
|
Tinción ligera o nula de los portaobjetos |
Protocolo de tinción seleccionado incorrecto |
Verificar que se usó el procedimiento de tinción recomendado. |
|
Verificar que se ha seleccionado VENTANA PD-L1 (SP263) como anticuerpo primario. |
||
|
Degradación del tejido |
Verificar que el tejido se tiñó dentro del marco temporal recomendado después del corte. |
|
|
Mal funcionamiento del dispensador |
Verificar que el tapón de la boquilla está quitado. |
|
|
Asegurarse de que el dispensador está cebado. |
||
|
Revisar la cámara de cebado para ver si hay partículas o cuerpos extraños, tales como fibras o precipitados. |
||
|
Consulte la hoja de datos del dispensador en línea asociado con P/N 741-4905 que se encuentra en navifyportal.roche.com. |
||
|
Asegurarse de que solo se usan los fijadores y tiempos de fijación recomendados. |
||
|
Reactivo de fluidos incorrecto o ausente |
Asegurarse de que los reactivos de fluidos estén correctamente llenos. |
|
|
Tinción de fondo excesiva de los portaobjetos |
Protocolo de tinción seleccionado incorrecto |
Verificar que se usó el procedimiento de tinción recomendado. |
|
Reactivo de fluidos incorrecto o ausente |
Asegurarse de que los reactivos de fluidos estén correctamente llenos. |
|
|
Método de fijación usado inapropiado |
Asegurarse de que solo se usan los fijadores y tiempos de fijación recomendados. |
|
|
Tejido desprendido del portaobjetos |
Uso de portaobjetos de microscopio incorrectos |
Asegurarse de que se utilizan portaobjetos para microscopio con carga positiva. |
ALMACENAMIENTO Y ESTABILIDAD:
Tras la recepción y cuando no se utilice, consérvese de 2-8°C. No lo congele.
Para garantizar una dispensación adecuada del reactivo y la estabilidad del anticuerpo, vuelva a poner el tapón del dispensador después de cada uso y almacene inmediatamente el dispensador en la nevera, en posición vertical.
Todos los dispensadores de anticuerpos tienen una fecha de caducidad. Si se almacena correctamente, el reactivo se mantendrá estable hasta la fecha indicada en la etiqueta. No usar el reactivo después de la fecha de caducidad.
ADVERTENCIAS Y PRECAUCIONES:
1. Para uso diagnóstico in vitro (IVD).
2. Solo para uso profesional.
3. No utilizar por encima del número especificado de ensayos.
4. La solución ProClin 300 se utiliza como conservante en este reactivo. Está clasificada como irritante y puede ocasionar sensibilización por contacto con la piel. Adopte precauciones razonables cuando la manipule. Evite el contacto de los reactivos con los ojos, la piel y las membranas mucosas. Utilice ropa protectora y guantes.
5. Los ensayos clínicos que se describen en esta hoja de datos no han validado el uso de bloques celulares de NSCLC procedentes de FNA en el marco de prescripción de un tratamiento específico.
6. Los portaobjetos con carga positiva pueden verse afectados por presiones ambientales, dando lugar a una tinción incorrecta. Póngase en contacto con su representante de servicio de Roche para obtener más información sobre el uso de este tipo de portaobjetos.
7. Los materiales de origen animal o humano deben manipularse como materiales biopeligrosos y eliminarse con las precauciones adecuadas. En caso de exposición, deberán seguirse las directivas sanitarias de las autoridades responsables.7,8
8. Evite el contacto de los reactivos con los ojos y las membranas mucosas. Si los reactivos entran en contacto con zonas sensibles, lávelas con agua abundante.
9. Evite la contaminación microbiana de los reactivos, dado que podría dar lugar a resultados incorrectos.
10. Para obtener más información sobre el uso de este producto, consulte el Manual del usuario del instrumento BenchMark IHC/ISH y las instrucciones de uso de todos los componentes necesarios que puede encontrar en navifyportal.roche.com.
11. Consultar a las autoridades locales y/o estatales sobre el método de eliminación recomendado.
12. El etiquetado de seguridad de los productos sigue principalmente las directrices del SGA de la UE. Está disponible bajo petición la hoja de datos de seguridad para los usuarios profesionales.
13. Para comunicar la sospecha de incidentes graves relacionados con este dispositivo, contacte con su representante local de servicio Roche y con las autoridades competentes del Estado o País Miembro de residencia del usuario.
14. Es posible que los tratamientos KEYTRUDA®, OPDIVO®, IMFINZI™, LIBTAYO® o TECENTRIQ® no se encuentren disponibles en todas las zonas geográficas.
Este producto contiene componentes clasificados como sigue de conformidad con el Reglamento (CE) nº 1272/2008:
Tabla 3. Información de riesgos
|
Riesgo |
Código |
Declaración |
|
Advertencia
|
H317 |
Puede provocar una reacción alérgica en la piel. |
|
H412 |
Perjudicial para los organismos acuáticos con efectos nocivos duraderos. |
|
|
P261 |
Evite inhalar la niebla o los vapores. |
|
|
P273 |
Evitar su emisión al medio ambiente. |
|
|
P280 |
Llevar guantes de protección. |
|
|
P333 + P313 |
En caso de irritación o erupción cutánea: Consultar a un médico. |
|
|
P362 + P364 |
Quitarse las prendas contaminadas y lavarlas antes de volver a usarlas. |
|
|
P501 |
Eliminar el contenido/el recipiente en una planta de eliminación de residuos aprobada. |
Este producto contiene CAS nº 55965-84-9, una masa de reacción de 5-cloro-2-metil-2H- isotiazol-3-ona y 2-metil-2H-isotiazol-3-ona (3:1).
MATERIAL SUMINISTRADO:
VENTANA PD-L1 (SP263) ASSAY incluye reactivo suficiente para realizar 50 pruebas.
Un dispensador de 5 mL de VENTANA PD-L1 (SP263) ASSAY contiene aproximadamente 8 μg de anticuerpo monoclonal de conejo.
El anticuerpo se diluye en Tris-HCl con una proteína transportadora y ProClin 300 al 0.10%, un conservante.
La concentración específica del anticuerpo es aproximadamente de 1.6 μg/mL. No se ha observado ninguna reactividad del anticuerpo no específica conocida en este producto.
VENTANA PD-L1 (SP263) ASSAY es un anticuerpo monoclonal recombinante de conejo producido como sobrenadante de un cultivo celular purificado.
Consulte la hoja de datos del kit de detección VENTANA correspondiente para obtener descripciones detalladas de los siguientes aspectos: Principio del procedimiento, Material y métodos, Recogida y preparación de muestras para análisis, Procedimientos de control de calidad, Resolución de problemas, Interpretación de los resultados y Limitaciones.
MATERIALES NECESARIOS PERO NO SUMINISTRADOS:
No se suministran reactivos de tinción, como kits de detección VENTANA, ni componentes auxiliares, incluyendo portaobjetos de control de tejido negativos y positivos.
Puede que no todos los productos que aparecen en la hoja de datos estén disponibles en todos los lugares. Consulte al representante local de asistencia técnica de Roche.
No se suministran los reactivos y materiales siguientes, pero pueden ser necesarios para la tinción:
1. Tejido de control recomendado
2. Portaobjetos para microscopio con carga positiva
3. Rabbit Monoclonal Negative Control Ig (nº de cat. 790-4795 / 06683380001)
4. OptiView DAB IHC Detection Kit (nº de cat. 760-700 / 06396500001)
5. EZ Prep Concentrate (10X) (nº cat. 950-102 / 05279771001)
6. Reaction Buffer Concentrate (10X) (nº cat. 950-300 / 05353955001)
7. ULTRA LCS (Predilute) (nº cat. 650-210 / 05424534001)
8. LCS (Predilute) (n.º cat. 650-010 / 05264839001)
9. ULTRA Cell Conditioning Solution (ULTRA CC1) (nº cat. 950-224 / 05424569001)
10. Cell Conditioning Solution (CC1) (nº cat. 950-124 / 05279801001)
11. Hematoxylin II (nº cat. 790-2208 / 05277965001)
12. Bluing Reagent (nº de cat. 760-2037 / 05266769001)
12. Medio de montaje permanente
14. Cubreobjetos de cristal o cinta adhesiva
15. Cubreobjetos automático o manual
16. Equipo de laboratorio de uso general
17. Instrumento BenchMark IHC/ISH
CONTROL DE REACTIVOS NEGATIVO:
Se debe utilizar el correspondiente portaobjetos de control de reactivo negativo en cada muestra como ayuda para la interpretación de los resultados. Rabbit Monoclonal Negative Control Ig es el anticuerpo de control de reactivo negativo correspondiente para este ensayo y se utiliza en lugar del anticuerpo primario para evaluar una tinción no específica. El procedimiento de tinción del control de reactivo negativo debe ser idéntico al anticuerpo primario. El uso de un reactivo de control negativo diferente o no usar el reactivo de control negativo recomendado puede dar como resultado una falsa interpretación del portaobjetos con tinción en el ensayo.
CONTROL DE TEJIDO POSITIVO:
Se debe incluir un control tisular en cada sesión de tinción. Esto contribuye a identificar fallos al aplicar los reactivos al portaobjetos. El tejido de control debe ser una muestra de autopsia reciente, biopsia o cirugía preparada o fijada con la mayor brevedad con un proceso idéntico al de las secciones de prueba. Estos tejidos se utilizan para hacer un seguimiento de todos los pasos que conlleva el proceso, desde la preparación del tejido hasta la tinción.
Se puede utilizar un tejido placentario humano normal cualificado como control tisular para VENTANA PD-L1 (SP263) ASSAY. Una muestra de tejido de placenta que sirva de control tisular debe presentar el patrón de tinción que se considera aceptable según la Tabla 6. El tejido placentario contiene elementos de tinción positivos y negativos para la proteína PD-L1 y, por tanto, es adecuado para su uso como control tisular. La tinción correcta de los componentes de tejido placentario se describe en la Tabla 6 y en la guía de interpretación (P/N 1015317).
Los controles de tejido positivos conocidos solo se deben usar para monitorizar el comportamiento correcto de los reactivos y los instrumentos, y no como ayuda para establecer un diagnóstico específico de las muestras de prueba. Si los controles de tejido positivos no muestran una tinción positiva, los resultados de las muestras de la prueba se deben considerar no válidos.
CARACTERÍSTICAS DE RENDIMIENTO:
Rendimiento de análisis:
Se realizaron pruebas de tinción para evaluar la sensibilidad, especificidad y precisión y los resultados se indican a continuación.
Sensibilidad y especificidad:
Se tiñeron arrays que contenían una serie de tejidos normales con VENTANA PD-L1 (SP263) ASSAY y se evaluó cualquier presencia de tinción membranosa de PD-L1, tal y como se detalla en la Tabla 9. También se observa tinción adicional, como tinción de células inmunitarias o citoplasmáticas (véase la nota al pie de la Tabla 9).
Además, se evaluó la tinción de un array de células tumorales y de células inmunitarias de un conjunto de tejidos neoplásicos con VENTANA PD-L1 (SP263) ASSAY según se describe en la Tabla 10.
Tabla 9. La sensibilidad/especificidad del ensayo VENTANA PD-L1 (SP263) ASSAY se determinó analizando tejidos normales FFPE
|
Tejido |
Nº de casos positivos/total |
Tejido |
Nº de casos positivos/total |
|
Cerebro |
0/3 |
Mieloide (médula ósea) a,b |
0/4 |
|
Cerebelo |
0/3 |
Pulmón b |
0/3 |
|
Glándula suprarrenal a |
0/3 |
Corazón |
0/3 |
|
Ovario |
0/3 |
Esófago a,b |
1/3 |
|
Páncreas a |
0/3 |
Estómago a,b |
0/3 |
|
Glándula paratiroidea |
0/4 |
Intestino delgado b |
0/3 |
|
Glándula pituitaria a,b |
0/3 |
Colon b |
0/3 |
|
Testículos |
0/3 |
Hígado |
0/3 |
|
Tiroides a,b |
0/3 |
Glándula salival b |
0/3 |
|
Mama |
0/3 |
Ganglio linfático b |
0/3 |
|
Bazo b |
0/3 |
Riñón b |
0/3 |
|
Laringe b |
0/3 |
Próstata |
0/3 |
|
Amígdala b |
3/3 |
Cuello del útero |
0/3 |
|
Endometrio |
0/3 |
Vejiga |
0/3 |
|
Músculo esquelético |
0/3 |
Piel c |
0/4 |
|
Nervio (disperso) |
0/3 |
Mesotelio b |
0/3 |
|
Glándula timo b |
0/3 |
||
Tinción adicional observada: a Tinción citoplasmática, b Tinción de células inmunitarias, c Tinción de melanocitos.
El porcentaje de células inmunitarias presente sobre el fondo no se pudo evaluar en este estudio, dado que no existe un área tumoral en la que se puedan puntuar las células inmunitarias que se infiltran en el tumor.
Tabla 10. La sensibilidad/especificidad de VENTANA PD-L1 (SP263) ASSAY se determinó analizando una variedad de tejidos neoplásicos FFPE para evaluar la presencia de tinción de membrana de células tumorales e inmunitarias
|
Patología |
Nº de casos positivos/total |
|
|---|---|---|
|
Células tumorales |
Células inmunitarias |
|
|
Glioblastoma (cerebro) |
0/1 |
1/1 |
|
Meningioma (cerebro) |
0/1 |
0/1 |
|
Ependimoma (cerebro) |
0/1 |
1/1 |
|
Oligodendroglioma (cerebro) |
0/1 |
0/1 |
|
Adenocarcinoma seroso (ovario) |
0/1 |
1/1 |
|
Adenocarcinoma (ovario) |
1/1 |
0/1 |
|
Neoplasia neuroendocrina (páncreas) |
0/1 |
0/1 |
|
Adenocarcinoma (páncreas) |
0/1 |
1/1 |
|
Seminoma (testículos) |
0/1 |
0/1 |
|
Carcinoma embrionario (testículos) |
0/1 |
0/1 |
|
Carcinoma medular (tiroides) |
0/1 |
0/1 |
|
Carcinoma papilar (tiroides) |
1/1 |
0/1 |
|
Carcinoma ductal in situ (mama) |
0/1 |
1/1 |
|
Carcinoma ductal invasivo (mama) |
0/2 |
0/2 |
|
Linfoma de linfocitos B; sin especificar (bazo) |
0/1 |
1/1 |
|
Carcinoma de células pequeñas (pulmón) |
1/1 |
1/1 |
|
Carcinoma de células escamosas (pulmón) |
1/1 |
1/1 |
|
Adenocarcinoma (pulmón) |
0/1 |
0/1 |
|
Carcinoma neuroendocrino (esófago) |
0/1 |
0/1 |
|
Adenocarcinoma (esófago) |
0/1 |
0/1 |
|
Carcinoma de células en anillo de sello (Estómago) |
0/1 |
0/1 |
|
Adenocarcinoma (intestino delgado) |
0/1 |
0/1 |
|
Sarcoma estromal (intestino delgado) |
0/1 |
0/1 |
|
Adenocarcinoma (colon) |
0/1 |
1/1 |
|
Tumor estromal gastrointestinal (GIST) (colon) |
0/1 |
0/1 |
|
Adenocarcinoma (recto) |
0/1 |
0/1 |
|
Tumor estromal gastrointestinal (GIST) (recto) |
0/1 |
0/1 |
|
Carcinoma hepatocelular (hígado) |
0/1 |
0/1 |
|
Hepatoblastoma (hígado) |
0/1 |
0/1 |
|
Carcinoma de células claras (riñón) |
0/1 |
0/1 |
|
Adenocarcinoma (próstata) |
0/2 |
0/2 |
|
Leiomioma (útero) |
0/1 |
0/1 |
|
Adenocarcinoma (útero) |
0/1 |
0/1 |
|
Carcinoma de células claras (útero) |
1/1 |
0/1 |
|
Carcinoma de células escamosas (cuello uterino) |
0/2 |
2/2 |
|
Rabdomiosarcoma embrionario (músculo estriado) |
0/1 |
0/1 |
|
Melanoma (recto) |
0/1 |
0/1 |
|
Carcinoma de células basales (piel) |
0/1 |
0/1 |
|
Carcinoma de células escamosas (piel) |
0/1 |
0/1 |
|
Neurofibroma (espalda) |
0/1 |
1/1 |
|
Neuroblastoma (retroperitoneo) |
0/1 |
0/1 |
|
Mesotelioma (cavidad abdominal) |
0/1 |
0/1 |
|
Linfoma de linfocitos B; sin especificar (mediastino) |
1/1 |
1/1 |
|
Linfoma de Hodgkin (ganglio linfático) |
1/1 |
1/1 |
|
Linfoma de linfocitos B, sin especificar (ganglio linfático) |
1/1 |
1/1 |
|
Linfoma anaplásico de células grandes (cavidad pélvica) |
1/1 |
1/1 |
|
Leiomiosarcoma (vejiga) |
0/1 |
0/1 |
|
Osteosarcoma (hueso) |
0/1 |
1/1 |
|
Rabdomiosarcoma de células fusiformes (retroperitoneo) |
0/1 |
0/1 |
|
Leiomiosarcoma (músculo liso) |
0/1 |
0/1 |
|
Carcinoma urotelial (vejiga) |
1/1 |
1/1 |
Repetibilidad y precisión intermedia – Control tisular placentario:
Se ha evaluado la repetibilidad y precisión intermedia de VENTANA PD-L1 (SP263) ASSAY para tejido de placenta humana en el instrumento BenchMark ULTRA en combinación con OptiView DAB IHC Detection Kit.
Para la repetibilidad en el mismo día: se tiñeron 5 portaobjetos replicados de cada una de las 8 muestras de placenta con VENTANA PD-L1 (SP263) ASSAY en un solo instrumento BenchMark ULTRA en un único día.
Para evaluar la precisión entre días, se tiñeron dos portaobjetos replicados de las 8 muestras de placenta con VENTANA PD-L1 (SP263) ASSAY en un mismo instrumento BenchMark ULTRA a lo largo de cinco días no consecutivos en un periodo de, al menos, 20 días.
Para evaluar la precisión entre instrumentos, cada una de las 12 muestras de placenta se tiñieron con VENTANA PD-L1 (SP263) ASSAY en tres instrumentos BenchMark ULTRA.
Todos los portaobjetos se evaluaron mediante la guía de puntuación del tejido de control de placenta de VENTANA PD-L1 (SP263) ASSAY que figura en la Tabla 6.
El porcentaje de concordancia global de la reproducibilidad en el mismo día, entre días y entre instrumentos fue del 100%, del 100% y del 98.8%, respectivamente.
Reproducibilidad entre lotes: control tisular placentario:
La reproducibilidad entre lotes de VENTANA PD-L1 (SP263) ASSAY para tejido de control se evaluó en 12 muestras de tejido placentario humano único utilizando tres lotes de anticuerpos VENTANA PD-L1 (SP263). El índice de concordancia porcentual global entre los lotes de anticuerpos fue del 98.8%.
RENDIMIENTO CLÍNICO EN EL CÁNCER DE PULMÓN NO MICROCÍTICO:
Rendimiento clínico de IMFINZI (durvalumab) en el estudio PACIFIC:
Se investigó la utilidad clínica de VENTANA PD-L1 (SP263) ASSAY en el estudio PACIFIC (NCT02125461), un estudio multicéntrico, aleatorizado, doble ciego y controlado por placebo, que evaluó la eficacia y la seguridad de IMFINZI (durvalumab) frente al placebo en pacientes con NSCLC inoperable localmente avanzado. Los pacientes se aleatorizaron 2:1 para recibir 10 mg/kg IMFINZI (n= 476) o 10 mg/kg placebo (n= 237) después de haber completado al menos 2 ciclos de quimioterapia basada en platino definitiva con terapia de radiación y teniendo la enfermedad estable, respuesta parcial o respuesta completa. La aleatorización se desglosó por género, edad (< 65 años vs 65 años) y tabaquismo (fumador vs no fumador). Se inscribieron a los pacientes independientemente de su nivel de expresión tumoral de PD-L1. Cuando estuvieron disponibles, las muestras de tejidos tumorales de archivo tomadas antes de la radioquimioterapia se analizaron retrospectivamente para evaluar la expresión de PD-L1 en las células tumorales con VENTANA PD-L1 (SP263) ASSAY. De los 713 pacientes aleatorizados, el 63% de los pacientes proporcionó una muestra de tejido con niveles suficientes de calidad y cantidad para determinar la expresión PD-L1 y un 37% tuvo un estado de PD-L1 desconocido. De 451 pacientes en los que se encontraba disponible la expresión de PD-L1, el 67% presentó expresión de PD-L1 en ≥ 1% de las células tumorales y en el 33% se observó expresión de PD-L1 en < 1% de las células tumorales.
Los dos criterios de valoración primarios del estudio fueron la supervivencia libre de progresión (PFS), según se evaluó mediante la revisión central, independiente y enmascarada (BICR) con la Versión 1.1 de los Criterios de Evaluación de Respuesta en Tumores Sólidos (RECIST v1.1) y la supervivencia global (OS) de IMFINZI frente a placebo.
El estudio demostró una mejora estadísticamente significativa de la PFS [índice de riesgo (HR) = 0,52 (CI del 95%: 0.42, 0.65), p < 0.0001] y en la OS [HR = 0.68 (CI del 95%: 0.53, 0.87), p= 0.00251] en el grupo que había recibido el tratamiento con IMFINZI en comparación con el grupo que había recibido placebo entre todos los pacientes aleatorizados. Las mejoras en la PFS y la supervivencia global (OS) en favor de los pacientes a los que se administró IMFINZI en comparación con aquellos a los que se trató con placebo se observaron de manera uniforme en todos los subgrupos predefinidos, entre otros, los elaborados a partir del origen étnico, la edad, el sexo, el historial de tabaquismo, el estado de la mutación EGFR y la histología.
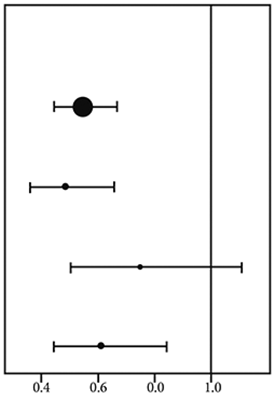
Se realizaron análisis del subgrupo exploratorio post-hoc adicionales para evaluar la eficacia mediante la expresión del tumor PD-L1 ≥ 1%, < 1% y para los pacientes cuyo estado de PD-L1 no pudo determinarse (PD-L1 desconocido). Los resultados de PFS y OS se resumen en la Figura 2 y la Figura 3.
Figura 2. Diagrama de bosque de OS según la expresión de PD-L1
|
Eventos/N (%) |
||||
|
IMFINZI |
Placebo |
HR (95% CI) |
||
|
Todos los pacientes |
|
183/476 (38.4%) |
116/237 (48.9%) |
0.68 (0.53, 0.87) |
|
PD-L1 tc ≥ 1% |
70/212 (33.0%) |
45/91 (49.5%) |
0.53 (0.36, 0.77) |
|
|
PD-L1 tc ≥ 1% |
41/90 (45.6%) |
19/58 (32.8%) |
1.36 (0.79, 2.34) |
|
|
PD-L1 Desconocido |
72/174 (41.4%) |
52/88 (59.1%) |
0.62 (0.43, 0.89) |
|
Figura 3. Diagrama de bosque de PFS según la expresión de PD-L1
|
Eventos/N (%) |
||||
|
IMFINZI |
Placebo |
HR (95% CI) |
||
|
Todos los pacientes |
|
214/476 (45.0%) |
157/237 (66.2%) |
0.52 (0.42, 0.65) |
|
PD-L1 tc ≥ 1% |
84/212 (39.6%) |
59/91 (64.8%) |
0.46 (0.33, 0.64) |
|
|
PD-L1 tc ≥ 1% |
49/90 (54.4%) |
40/58 (69.0%) |
0.73 (0.48, 1.11) |
|
|
PD-L1 Desconocido |
81/174 (46.6%) |
58/88 (65.9%) |
0.59 (0.42, 0.83) |
|
Rendimiento clínico de KEYTRUDA (pembrolizumab) en los estudios KEYNOTE-024, KEYNOTE-042 y KEYNOTE-010:
Resultados del ensayo clínico KEYNOTE-024: Se evaluaron la seguridad y la eficacia de KEYTRUDA (pembrolizumab) en KEYNOTE- 024 (NCT02142738), un estudio multicéntrico y controlado cuyo objetivo era el tratamiento del NSCLC metastásico sin tratamiento previo sin anomalías tumorales genómicas en EGFR o ALK. Los pacientes tuvieron una expresión de PD-L1 con una puntuación de la proporción del tumor (TPS) ≥ 50% según el PD-L1 IHC 22C3 pharmDx.9 Los pacientes se aleatorizaron (1:1) para recibir una dosis de 200 mg de KEYTRUDA cada tres semanas (n=154) o la quimioterapia con platino elegida por el investigador (n=151; incluyendo pemetrexed+carboplatino, pemetrexed+cisplatino, gemcitabina+cisplatino, gemcitabina+carboplatino o paclitaxel+carboplatino. Los pacientes con células no escamosas podrían recibir mantenimiento con pemetrexed).El criterio de valoración de la eficacia principal del estudio era la supervivencia libre de progresión (PFS) según la evaluación de una revisión central, independiente y enmascarada (BICR) con RECIST v1.1. Las medidas secundarias de resultado de eficacia fueron la supervivencia global (OS) y la tasa de respuesta objetiva (ORR), que se evaluaron según los criterios BICR con RECIST v1.1. Las conclusiones del ensayo demostraron una mejora estadísticamente significativa tanto en la PFS como en la OS en los pacientes aleatorizados que recibieron KEYTRUDA en comparación con los que recibieron quimioterapia. Consulte la Tabla 18 para ver un resumen de las principales medidas de eficacia en toda la población con intención de tratar (ITT). Los resultados en cuanto a la PFS y la ORR se presentaron a partir de un análisis provisional en una media de seguimiento de 11 meses. Los resultados en cuanto a la OS se presentaron a partir de un análisis final en una media de seguimiento de 25 meses.
Tabla 18. Resultados de la eficacia en el estudio KEYNOTE-024
|
Criterios |
KEYTRUDA 200 mg cada tres semanas n= 154 |
Quimioterapia n= 151 |
|
PFS* |
||
|
Número (%) de pacientes con evento |
73 (47%) |
116 (77%) |
|
Índice de riesgo a (CI del 95%) |
0,50 (0,37, 0,68) |
- |
|
Valor p b |
< 0.001 |
- |
|
Mediana en meses (CI del 95%) |
10.3 (6.7, NA) |
6,0 (4,2, 6,2) |
|
OS |
||
|
Número (%) de pacientes con evento |
73 (47%) |
96 (64%) |
|
Índice de riesgo a (CI del 95%) |
0,63 (0,47, 0,86) |
- |
|
Valor p b |
0,002 a |
- |
|
Mediana en meses (CI del 95%) |
30.0 |
14.2 |
|
(18,3, NA) |
(9,8, 19,0) |
|
|
Tasa de respuesta objetiva* |
||
|
Porcentaje de ORR (CI del 95%) |
45% (37, 53) |
28% (21, 36) |
|
Respuesta completa (%) |
4% |
1% |
|
Respuesta parcial (%) |
41% |
27% |
|
Duración de la respuesta c |
||
|
Media en meses (intervalo) |
No alcanzado (1.9+, 14.5+) |
6.3 (2,1+, 12,6+) |
|
% con duración ≥ 6 meses |
88% d |
59% e |
CI = Intervalo de confianza.
Evaluada mediante BICR según RECIST v1.1
a Índice de riesgo (KEYTRUDA en comparación con la quimioterapia) según el modelo de riesgo proporcional estratificado de Cox.
b Según la prueba del orden logarítmico estratificado.
c Basado en pacientes con una mejor respuesta general, confirmada mediante respuesta total o parcial o parcial.
d Según las estimaciones de Kaplan-Meier; se incluyen 43 pacientes con una respuesta de 6 meses o superior.
e Según las estimaciones de Kaplan-Meier; se incluyen 16 pacientes con una respuesta de 6 meses o superior.
NA= no disponible.
Resultados del ensayo clínico KEYNOTE-042:
Se evaluaron la seguridad y la eficacia de KEYTRUDA en KEYNOTE-042 (NCT02220894), un estudio multicéntrico y controlado cuyo objetivo era el tratamiento del NSCLC localmente avanzado o metastásico sin tratamiento previo. El diseño del estudio fue similar al de KEYNOTE-024, excepto en que los pacientes tenían una expresión PD- L1 con un TPS ≥ 1% basándose en el kit PD-L1 IHC 22C3 pharmDxTM. Los pacientes se aleatorizaron (1:1) para recibir una dosis de 200mg de KEYTRUDA cada tres semanas (n= 637) o la quimioterapia con platino elegida por el investigador (n= 637; entre otras, mantenimiento de pemetrexed, pemetrexed+carboplatino o paclitaxel+carboplatino). Ente los 1274 pacientes en KEYNOTE-042, 599 (47%) tenían tumores con expresión PD-L1 con un TPS ≥ 50% basándose en el kit PD-L1 IHC 22C3 pharmDxTM.
El criterio de valoración de la eficacia principal del estudio era OS. Las medidas secundarias de resultado de eficacia fueron la PFS y la ORR (que se evaluaron según los criterios BICR con RECIST v1.1). Las conclusiones del estudio demostraron una mejora estadísticamente significativa en la OS en aquellos pacientes cuyos tumores presentaban expresión de PD-L1 con una TPS ≥ 1% que se habían aleatorizado en el grupo de la monoterapia con KEYTRUDA en comparación con el grupo de la quimioterapia (HR 0.82; CI del 95% 0.71, 0.93 en el análisis final) y con los pacientes cuyos tumores presentaban expresión de PD-L1 con una TPS ≥ 50% aleatorizados en el grupo de la monoterapia con KEYTRUDA en comparación con el grupo de la quimioterapia. La Tabla 19 contiene un resumen de las medidas clave de eficacia para la población con TPS ≥ 50% en el análisis final realizado en un periodo de seguimiento medio de 15,4 meses.
Tabla 19. Resultados de eficacia (PD-L1 con una TPS ≥ 50%) en KEYNOTE-042
|
Criterios |
KEYTRUDA 200mg cada tres semanas n= 299 |
Quimioterapia n= 300 |
|
OS |
||
|
Número (%) de pacientes con evento |
180 (60%) |
220 (73%) |
|
Índice de riesgo CI del 95% a |
0,70 (0,58, 0,86) |
|
|
Valor p b |
0.0003 |
|
|
Media en meses (CI del 95%) |
20,0 (15,9, 24,2) |
12,2 (10,4, 14,6) |
|
PFS |
||
|
Número (%) de pacientes con evento |
238 (80%) |
250 (83%) |
|
Índice de riesgo CI del 95% a |
0,84 (0,70, 1,01) |
|
|
Media en meses (CI del 95%) |
6,5 (5,9, 8,5) |
6,4 (6,2, 7,2) |
|
Tasa de respuesta objetiva |
||
|
ORR% (CI del 95%) |
39% (34, 45) |
32% (27, 38) |
|
Respuesta completa (%) |
1% |
0,3% |
|
Respuesta parcial (%) |
38% |
32% |
|
Duración de la respuesta c |
||
|
Media en meses (intervalo) |
22.0 (2.1+, 36.5+) |
10.8 (1,8+, 30,4+) |
|
% con duración ≥ 18 meses |
57% |
34% |
a Índice de riesgo (KEYTRUDA en comparación con la quimioterapia) según el modelo de riesgo proporcional estratificado de Cox.
b Según la prueba del orden logarítmico estratificado.
c Basado en pacientes con una mejor respuesta objetiva, confirmada mediante respuesta total o parcial o parcial.
Resultados del ensayo clínico KEYNOTE-010:
Se investigaron la seguridad y la eficacia de KEYTRUDA en KEYNOTE-010 (NCT01905657), un estudio clínico multicéntrico, abierto y aleatorizado que se llevó a cabo con NSCLC tratado previamente con quimioterapia con contenido de platino.10 En los pacientes se observó una expresión de PD-L1 con una TPS ≥ 1% según una versión de ensayo clínico de PD-L1 IHC 22C3 pharmDx. Los pacientes con mutaciones activadoras de EGFR o translocalización de ALK también experimentaron la progresión de la enfermedad con el tratamiento aprobado para estas mutaciones antes de recibir KEYTRUDA. Los pacientes se aleatorizaron (1:1:1) y recibieron tratamiento con una dosis de 2mg/kg (n= 344) o 10mg/kg (n= 346) de KEYTRUDA cada tres semanas o una dosis de 75mg/m2 de docetaxel cada tres semanas (n= 343) hasta el avance de la enfermedad o la aparición de un nivel de toxicidad inaceptable. Las criterios de valoración principales fueron la OS y la PFS evaluadas por BICR usando RECIST v1.1.
A partir del ensayo del estudio clínico se aleatorizó a un total de 1033 pacientes con NSCLC en el estudio. Se analizaron de forma retrospectiva las muestras archivadas de 529 pacientes de ensayo clínico con PD-L1 IHC 22C3 pharmDx. De todas ellas, las muestras de 94 pacientes presentaban expresión de PD-L1 en < 1%, las muestras de 413 pacientes presentaban expresión de PD-L1 en ≥ 1% y las muestras de 163 pacientes presentaban expresión de PD-L1 en ≥ 50%.
El acuerdo de porcentaje positivo y negativo entre el ensayo clínico y PD-L1 IHC 22C3 pharmDx fueron: NPA= 94,5% (91,4%-96,6%); PPA=80,0% (76,9%-82,8%), en el umbral de 1% y NPA=98,3% (97,1%-99,0%); PPA=73,2% (67,9%-77,9%) en el umbral del 50%.
KEYTRUDA demostró beneficios clínicos duraderos en pacientes con NSCLC con expresión de PD-L1 (TPS ≥ 1%), que se incrementó en pacientes del subgrupo TPS ≥ 50%, tal y como se determinó con PD-L1 IHC 22C3 pharmDx. La magnitud del beneficio era comparable al del ensayo clínico global. En la Tabla 20 se resumen las medidas de eficacia claves en la población global con expresión de PD-L1 (TPS ≥ 1%) mediante CTA y entre los pacientes con expresión de PD-L1 (TPS ≥ 1%) mediante PD-L1 IHC 22C3 pharmDx. Los resultados de la eficacia fueron similares para los grupos tratados con 2 y 10mg/kg de KEYTRUDA. Los análisis de sensibilidad demostraron que los cálculos del índice de riesgo eran sólidos para determinar la posible repercusión de la falta de datos que procedía de los pacientes con expresión de PD-L1 (TPS ≥ 1%) según PD-L1 IHC 22C3 pharmDx, pero que podrían no haber presentado expresión de PD-L1 (TPS < 1%) según el ensayo del estudio clínico.
Tabla 20. Resultados de eficacia en KEYNOTE-010. Pacientes con resultados positivos en el estudio clínico global y el ensayo PD-L1 IHC 22C3 pharmDx: PD-L1 TPS ≥ 1%
|
Criterios |
KEYTRUDA 2 mg/kg bw cada tres semanas |
KEYTRUDA 10 mg/kg bw cada tres semanas |
Docetaxel 75 mg/m2 cada tres semanas |
|||
|---|---|---|---|---|---|---|
|
Ensayo clínico |
PD-L1 IHC 22C3 pharmDx |
Ensayo clínico |
PD-L1 IHC 22C3 pharmDx |
Ensayo clínico |
PD-L1 IHC 22C3 pharmDx |
|
|
Número de pacientes |
344 |
140 |
346 |
142 |
343 |
131 |
|
OS |
||||||
|
Muertes (%) |
284 (83%) |
109 (78%) |
264 (76%) |
106 (75%) |
295 (86%) |
111 (85%) |
|
Índice de riesgo* (CI del 95%) |
0,77 (0,66, 0,91) |
0,61 (0,46, 0,81) |
0,61 (0,52, 0,73) |
0,57 (0,43, 0,76) |
- |
- |
|
Valor p a |
0.00128 |
< 0.001 |
< 0.001 |
< 0.001 |
- |
- |
|
Mediana en meses (CI del 95%) |
10,4 (9,5, 11,9) |
12,5 (10,0, 17,1) |
13,2 (11,2, 16,7) |
12,3 (9,4, 20,5) |
8,4 (7,6, 9,5) |
7,6 (5,8, 9,6) |
|
PFS b |
||||||
|
Eventos (%) |
305 (89%) |
118 (84%) |
292 (84%) |
121 (85%) |
314 (92%) |
118 (90%) |
|
Índice de riesgo* (CI del 95%) |
0,88 (0,75, 1,04) |
0,62 (0,47, 0,82) |
0,75 (0,63, 0,89) |
0,75 (0,57, 0,98) |
- |
- |
|
Valor p a |
0.065 |
< 0.001 |
< 0.001 |
0.01776 |
- |
- |
|
Media en meses (CI del 95%) |
3.9 (3.1, 4.1) |
5.2 (4.1, 6.3) |
4.0 (2.7, 4.5) |
4.0 (2.2, 4.8) |
4.1 (3.8, 4.5) |
4.0 (2.3, 4.3) |
|
Tasa de respuesta general (ORR) b |
||||||
|
ORR% (CI del 95%) |
20% (16, 25) |
27% (20, 35) |
21% (17, 26) |
23% (17, 31) |
9% (6, 13) |
5% (2, 11) |
CI = Intervalo de confianza
* Índice de riesgo (KEYTRUDA en comparación con docetaxel) según el modelo de riesgo proporcional estratificado de Cox.
a Según la prueba de rango logarítmico estratificado.
b Evaluada mediante BICR según RECIST v1.1.
Rendimiento clínico del ensayo VENTANA PD-L1 (SP263) ASSAY en las poblaciones de NSCLC positivas en PD-L1 de KEYNOTE-024, KEYNOTE-042 y KEYNOTE-010: comparación de métodos de análisis publicado y análisis comparativo in silico:
Las muestras de tejido procedentes de los pacientes de cribado de los ensayos KEYNOTE-010, KEYNOTE-024 y KEYNOTE-042 no se encontraban disponibles para su análisis con el ensayo VENTANA PD-L1 (SP263) ASSAY. Por consiguiente, el rendimiento clínico de VENTANA PD-L1 (SP263) ASSAY se evaluó de forma indirecta a través de un estudio de rendimiento de análisis publicado y estudios comparativos in silico retrospectivos.
Estudio de rendimiento de análisis publicado:
Un estudio comparativo de método que llevó a cabo AstraZeneca comparaba VENTANA PD-L1 (SP263) ASSAY y PD-L1 IHC 22C3 pharmDx (que se habían empleado en los ensayos clínicos de KEYTRUDA).11
Aproximadamente 500 muestras de biopsia NSCLC adquiridas comercialmente que representan el rango dinámico de la expresión PD-L1 se tiñeron con VENTANA PD-L1 (SP263) ASSAY en el instrumento BenchMark ULTRA y con PD-L1 IHC 22C3 pharmDx en el Autostainer Link 48, en un laboratorio central único. Un anatomopatólogo formado en los ensayos VENTANA y Dako PD-L1 evaluó los portaobjetos con tinción. Ventana analizó posteriormente los datos de la expresión de PD-L1 de esta comparación de métodos para calcular los PPA y NPA de los umbrales del 1% y del 50%. El criterio de valoración primario del estudio era una estimación de punto del 85% o superior en PPA y NPA, con PD-L1 IHC 22C3 pharmDx como referencia. Tanto el PPA como el NPA fueron > 90% en ambos umbrales de expresión (consulte la Tabla 21).
Tabla 21. Comparación de métodos: índices de concordancia de VENTANA PD-L1 (SP263) ASSAY frente a PD-L1 IHC 22C3 pharmDx
|
Umbral del 1% |
PD-L1 IHC 22C3 pharmDx |
||
|
VENTANA PD-L1 (SP263) ASSAY |
Positiva |
Negativa |
Total |
|
Positiva |
256 |
21 |
277 |
|
Negativa |
24 |
199 |
223 |
|
Total |
280 |
220 |
500 |
|
n/N |
% (CI del 95%) |
||
|
Porcentaje de concordancia positiva |
256/280 |
91.4 (87.6-94.2) |
|
|
Porcentaje de concordancia negativa |
199/220 |
90.5 (85.8-93.7) |
|
|
Porcentaje de concordancia global |
455/500 |
91.0 (88.2-93.2) |
|
|
Umbral del 50% |
PD-L1 IHC 22C3 pharmDx |
||
|
VENTANA PD-L1 (SP263) ASSAY |
Positiva |
Negativa |
Total |
|
Positiva |
111 |
22 |
133 |
|
Negativa |
10 |
357 |
367 |
|
Total |
121 |
379 |
500 |
|
n/N |
% (CI del 95%) |
||
|
Porcentaje de concordancia positiva |
111/121 |
91.7 (85.5-95.4) |
|
|
Porcentaje de concordancia negativa |
357/379 |
94.2 (91.4-96.1) |
|
|
Porcentaje de concordancia global |
468/500 |
93.6 (91.1-95.4) |
|
CI = Intervalo de confianza.
Análisis comparativo in silico:
Para evaluar con mayor profundidad la eficacia de KEYTRUDA en los pacientes con NSCLC con expresión de PD-L1 en ≥ 1% de segunda línea (2L) (KEYNOTE-010) y en los pacientes de NSCLC con expresión de PD-L1 en ≥ 50% en primera línea (L1) (KEYNOTE-024 y KEYNOTE-042), según la identificación mediante VENTANA PD-L1 (SP263) ASSAY, se llevaron a cabo estudios de comparación in silico.
Los datos de un estudio comparativo de método patrocinado por Roche incluyendo una cohorte comercial de 850 muestras de NSCLC, que evaluaban la concordancia analítica entre VENTANA PD-L1 (SP263) ASSAY y PD-L1 IHC 22C3 pharmDx, se utilizaron para desarrollar un modelo predictivo para la imputación del estado PD-L1 SP263. El estado PD-L1 SP263 se imputó para todos los sujetos probados con PD-L1 IHC 22C3 pharmDx en estos tres ensayos. La eficacia de KEYTRUDA se evaluó posteriormente en las poblaciones con expresión de PD-L1 en ≥ 1% (KEYNOTE-010) y PD-L1 en ≥ 50% (KEYNOTE-024 y KEYNOTE-042) según la identificación de VENTANA PD-L1 (SP263) ASSAY que se había determinado mediante el método que se describe en Li (2015).14 Para el subgrupo de pacientes SP263+/22C3+, la eficacia se estimó directamente a partir de pacientes aleatorizados en los ensayos. En el subgrupo de pacientes SP263+/22C3- (que no se habían aleatorizado en los ensayos), se elaboró un modelo de un rango de hipótesis en el que se presuponía que la eficacia era la misma (hipótesis ideal) o se reducía con respecto al subgrupo SP263+/22C3+ (la hipótesis menos favorable presuponía un HR = 1). A continuación, se determinó la eficacia para los pacientes seleccionados SP263 combinando los grupos SP263+/22C3+ y SP263+/22C3- mediante ponderación como se indica en Li (2015).14 Para cada estudio, este cálculo se realizó en diversos resultados de ensayos simulados usando métodos de imputación y después se resumieron en diversas simulaciones de ensayos para comprender la solidez de las conclusiones.
Los resultados de los análisis comparativos in silico demuestran que el beneficio clínico de KEYTRUDA frente a la quimioterapia se mantienen en las poblaciones seleccionadas con expresión de PD-L1 (SP263). En particular, los efectos estimados del tratamiento son sólidos incluso para atenuar por completo el efecto de tratamiento entre los pacientes que son 22C3- y SP263+, lo que respalda el uso de la monoterapia KEYTRUDA en la configuración 1L para los pacientes con la expresión PD-L1 ≥ 50% TC y la configuración 2L para los pacientes con la expresión PD-L1 ≥ 1% TC, en la población de pacientes NSCLC identificados por medio de VENTANA PD-L1 (SP263) ASSAY.
Rendimiento Clínico de OPDIVO (nivolumab) en el estudio CHECKMATE-057:
Resultados del ensayo clínico CHECKMATE-057:
La seguridad y eficacia de OPDIVO se evaluaron en CHECKMATE-057 (NCT01673867), un estudio en fase 3, abierto y aleatorizado en sujetos adultos (≥ 18 años) con NSCLC avanzado o metastásico de células no escamosas tras el fracaso del tratamiento previo con biquimioterapia antineoplásica con platino.13 Los sujetos se aleatorizaron 1:1 para OPDIVO vs docetaxel y se estratificaron en función 1) del uso previo del tratamiento de mantenimiento frente a la ausencia de tratamiento de mantenimiento y 2) del tratamiento de segunda línea frente al tratamiento de tercera línea. Se recogieron muestras de tejido tumoral (referencia) previas al estudio antes de la aleatorización y antes del primer tratamiento para llevar a cabo análisis planificados previamente siguiendo los niveles de expresión de PD-L1 de referencia predefinidos (objetivo secundario).
Las muestras tumorales de archivo se evaluaron retrospectivamente para la expresión PD-L1 usando el ensayo PD-L1 IHC 28-8 pharmDx. En la población del ensayo, el 22% de los 582 pacientes obtuvo resultados que no era posible cuantificar. De los 455 pacientes restantes, el 46% presentaron negatividad en PD-L1, definida como la presencia de expresión de PD-L1 en < 1% de células tumorales, y el 54% presentaba expresión de PD-L1, definida como la presencia de expresión de PD-L1 en ≥ 1% de células tumorales. De entre los 246 pacientes con tumores con expresión de PD-L1, el 26% tenía ≥ 1% y < 5% de células tumorales con tinción positiva, el 7% tenía ≥ 5% y < 10% de células tumorales con tinción positiva, y el 67% tenía ≥ 10% de células tumorales con tinción positiva.
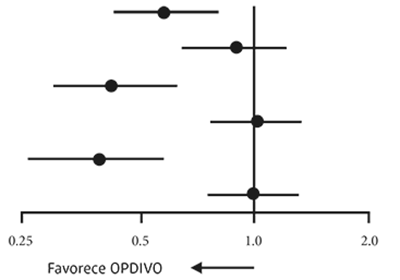
El criterio de valoración de la eficacia principal del estudio era OS. Los pacientes que presentaban expresión de PD-L1 según se había determinado mediante Dako PD-L1 IHC 28-8 pharmDx conforme a todos los niveles de expresión predefinidos en el grupo OPDIVO mostraron un mayor índice de supervivencia en comparación con aquellos que habían recibido tratamiento con docetaxel. No obstante, se observaron niveles de supervivencia similares a los de docetaxel en pacientes sin expresión de PD-L1. Se observaron diferencias significativas en la media de OS en OPDIVO sobre los subgrupos de docetaxel cuando se analizaron mediante el nivel de expresión PD-L1. La media de supervivencia global OS fue de 17.1, 18.2 y 19.4 meses en el caso de los pacientes que habían recibido tratamiento con OPDIVO en comparación con 9.0, 8.1 y 8.0 meses en aquellos que habían recibido tratamiento con docetaxel con niveles de expresión de PD- L1 ≥ 1%, ≥ 5% y ≥ 10%, respectivamente. No se observaron diferencias en OS entre los grupos de tratamiento en sujetos con niveles de expresión < 1%, < 5% y < 10%, con rangos de media de OS de 9,7 a 10,4 meses para OPDIVO y de 10,1 a 10,3 meses para docetaxel. Los índices de riesgo (HR) sin estratificar y la media de supervivencia global (OS) se muestran en la Figura 4.
Figura 4. Diagrama de bosque: supervivencia global (OS) según la expresión de PD-L1 en pacientes con NSCLC de células no escamosas: CHECKMATE-057
|
Media OS (meses) |
||||
|
PD- L1 Nivel de expresión |
HR sin estratificar |
opdivo |
Docetaxel |
|
|
≥ 1% (n= 246) |
|
0.59 |
17.1 |
9.0 |
|
< 1% (n= 209) |
0.90 |
10.4 |
10.1 |
|
|
≥ 5% (n=181) |
0.43 |
18.2 |
8.1 |
|
|
< 5% (n= 247) |
1.01 |
9.7 |
10.1 |
|
|
≥ 10% (n= 165 |
0.40 |
19.4 |
8.0 |
|
|
< 10% (n= 290) |
1.00 |
9.9 |
10.3 |
|
Nota: El índice de riesgo sin estratificar y el CI del 95% correspondiente se estimaron con un modelo de riesgo proporcional de Cox considerando el grupo aleatorizado como una sola covariable.
Rendimiento clínico de VENTANA PD-L1 (SP263) ASSAY en la población con NSCLC de CHECKMATE-057: comparación de métodos de análisis publicada y estudio comparativo de método externo:
Las muestras de tejido procedentes de los pacientes de cribado del ensayo CHECKMATE-057 no se encontraban disponibles para su análisis con el ensayo VENTANA PD-L1 (SP263) ASSAY. Por consiguiente, el rendimiento clínico de VENTANA PD-L1 (SP263) ASSAY se evaluó de forma indirecta a través de un estudio de rendimiento de análisis publicado y un estudio externo de concordancia entre VENTANA PD-L1 (SP263) ASSAY y Dako PD-L1 IHC 28-8 pharmDx.
Estudio de rendimiento de análisis publicado:
Un estudio comparativo de método que llevó a cabo AstraZeneca comparaba VENTANA PD-L1 (SP263) ASSAY y PD-L1 IHC 28-8 pharmDx (que se habían empleado en los ensayos clínicos de OPDIVO).11
Aproximadamente 500 muestras de biopsia NSCLC adquiridas comercialmente que representan el rango dinámico de la expresión PD-L1 se tiñeron con VENTANA PD-L1 (SP263) ASSAY en el instrumento BenchMark ULTRA y con PD-L1 IHC 28-8 pharmDx en el Autostainer Link 48, en un laboratorio central único. Un anatomopatólogo formado en los ensayos VENTANA y Dako PD-L1 evaluó los portaobjetos con tinción. Ventana analizó posteriormente los datos de la expresión de PD-L1 de esta comparación de métodos para calcular los PPA y NPA de las líneas de corte del 1%, del 5% y del 10%. El criterio de valoración primario del estudio era una estimación de punto del 85% o superior en PPA y NPA, con PD-L1 IHC 28-8 pharmDx como referencia. Tanto el PPA como el NPA fueron > 90% en los tres umbrales de expresión (consulte la Tabla 22).
Tabla 22. Comparación de métodos: Índices de concordancia de VENTANA PD-L1 (SP263) ASSAY frente a PD-L1 IHC 28-8 pharmDx
|
Umbral del 1% |
PD-L1 IHC 28-8 pharmDx |
||
|
VENTANA PD-L1 (SP263) ASSAY |
Positiva |
Negativa |
Total |
|
Positiva |
264 |
13 |
277 |
|
Negativa |
29 |
194 |
223 |
|
Total |
293 |
207 |
500 |
|
n/N |
% (CI del 95%) |
||
|
Porcentaje de concordancia positiva |
264/293 |
90.1 (86.1-93.0) |
|
|
Porcentaje de concordancia negativa |
194/207 |
93.7 (89.6-96.3) |
|
|
Porcentaje de concordancia global |
458/500 |
91.6 (88.8-93.7) |
|
|
Umbral del 5% |
PD-L1 IHC 28-8 pharmDx |
||
|
VENTANA PD-L1 (SP263) ASSAY |
Positiva |
Negativa |
Total |
|
Positiva |
225 |
9 |
234 |
|
Negativa |
21 |
245 |
266 |
|
Total |
246 |
254 |
500 |
|
n/N |
% (CI del 95%) |
||
|
Porcentaje de concordancia positiva |
225/246 |
91.5 (87.3-94.3) |
|
|
Porcentaje de concordancia negativa |
245/254 |
96.5 (93.4-98.1) |
|
|
Porcentaje de concordancia global |
470/500 |
94.0 (91.6-95.8) |
|
|
Umbral del 10% |
PD-L1 IHC 28-8 pharmDx |
||
|
VENTANA PD-L1 (SP263) ASSAY |
Positiva |
Negativa |
Total |
|
Positiva |
192 |
17 |
209 |
|
Negativa |
19 |
272 |
291 |
|
Total |
211 |
289 |
500 |
|
n/N |
% (CI del 95%) |
||
|
Porcentaje de concordancia positiva |
192/211 |
91.0 (86.4-94.2) |
|
|
Porcentaje de concordancia negativa |
272/289 |
94.1 (90.8-96.3) |
|
|
Porcentaje de concordancia global |
464/500 |
92.8 (90.2-94.8) |
|
CI = Intervalo de confianza.
Estudio comparativo de método externo:
Este estudio comprendía una cohorte de 850 muestras comerciales y evaluaba la concordancia del análisis entre los ensayos, siguiendo un diseño de no inferioridad con Dako PD-L1 IHC 28-8 pharmDx como referencia Li (2016).12 Cada caso se analizó dos veces con Dako PD-L1 IHC 28-8 pharmDx y una vez con VENTANA PD-L1 (SP263) ASSAY. Los criterios de valoración del estudio eran los porcentajes de concordancia positiva y negativa (PPA/NPA) entre los ensayos, que se deducían a partir de los PPA y NPA entre las dos réplicas del ensayo Dako PD-L1 IHC 28-8 pharmDx, que constituía la línea de base. Los análisis primarios del estudio se basaban en toda la cohorte de NSCLC incluyendo los subtipos histológicos de células escamosas y no escamosas.
Dado que el estudio CHECKMATE-057 únicamente incorporaba pacientes con NSCLC de células no escamosas, se llevó a cabo un análisis a posteriori para evaluar la concordancia entre los ensayos en el subgrupo con NSCLC de células no escamosas (N= 239) en los mismos umbrales de expresión de PD-L1 que se habían evaluado en el ensayo CHECKMATE-057. Las diferencias de PPA y NPA para el subgrupo de NSCLC de células no escamosas a los umbrales de expresión PD-L1 del 1%, 5% y 10% están por debajo del umbral de no inferioridad del 15% preespecificado para los análisis primarios (Tabla 23), lo que respalda el uso de VENTANA PD-L1 (SP263) ASSAY como dispositivo de seguimiento para OPDIVO en la misma población de pacientes objetivo.
Tabla 23. Concordancia entre VENTANA PD-L1 (SP263) ASSAY y Dako PD-L1 IHC 28-8 pharmDx usando una cohorte comercial de casos de NSCLC de células no escamosas
|
Prevalencia de la positividad de PD-L1 |
Umbral de la expresión |
Medida |
Estimación de punto |
Bilateral CI del 95% |
|
0.541 |
1% de TC |
Dif. PPA1 |
0,0% |
(-4.3, 4.3) |
|
Dif. PPA2 |
-5.5% |
(-10.9, 0.0) |
||
|
Dif. NPA1 |
8,4% |
(2.8, 14.5) |
||
|
Dif. NPA2 |
4,0% |
(-3.1, 11.4) |
||
|
0.398 |
5% DE TC |
Dif. PPA1 |
-2.8% |
(-7.7, 1.9) |
|
Dif. PPA2 |
-5.1% |
(-10.7, 0.4) |
||
|
Dif. NPA1 |
4.7% |
(-0.8, 10.6) |
||
|
Dif. NPA2 |
6.2% |
(1.2, 11.8) |
||
|
0.363 |
10% de TC |
Dif. PPA1 |
-2.0% |
(-7.0, 2.8) |
|
Dif. PPA2 |
-6.5% |
(-12.8, -0.4) |
||
|
Dif. NPA1 |
3.6% |
(-1.5, 8.9) |
||
|
Dif. NPA2 |
5.0% |
(0.5, 10.1) |
Notas: Dif. PPA1 y Dif. PPA2 = intraPPA (28-8 de la réplica 1 frente a 28-8 de la réplica 2) menos interPPA (SP263 frente a 28-8 de la réplica 1 o 2) PPA; Dif. NPA1 y Dif. NPA2 = intraNPA (28-8 de la réplica 1 frente a 28-8 de la réplica 2) menos interNPA (SP263 frente a 28-8 de la réplica 1 o 2) NPA.
Los CI se calculan mediante bootstrap con 100.000 nuevos muestreos.
El cálculo se lleva a cabo a partir de los 236 casos de NSCLC de células no escamosas que se pueden evaluar.
Rendimiento clínico de LIBTAYO (cemiplimab) en el estudio 1624:
Resultados del estudio 1624:
Se evaluaron la seguridad y la eficacia de LIBTAYO (cemiplimab) en el estudio 1624 (NCT03088540), un ensayo multicéntrico, abierto, aleatorizado y con control activo en pacientes con NSCLC localmente avanzado que no cumplían los criterios de idoneidad para una extirpación quirúrgica ni para un tratamiento con radioquimioterapia o con NSCLC metastásico.
El ensayo se diseñó para reclutar a pacientes cuyos tumores tenían una expresión alta de PD-L1 (puntuación de la proporción del tumor [TPS] ≥ 50% según se determinó mediante un ensayo de inmunohistoquímica usando el kit PD-L1 IHC 22C3 pharmDx) y que no habían recibido un tratamiento sistémico anterior para NSCLC metastásico. Se reclutaron a 710 pacientes en total (población con intención de tratar [mITT]) y se realizó un análisis en una población (n= 563) que tenía la expresión PD-L1 de TPS ≥ 50% usando PD-L1 IHC 22C3 pharmDx según el etiquetado aprobado (población 22C3+).
Los pacientes se aleatorizaron (1:1) para recibir LIBTAYO 350mg de forma intravenosa (IV) cada 3 semanas para un total de 108 semanas o un régimen de biquimioterapia de platino durante 4-6 ciclos seguido de un mantenimiento opcional con pemetrexed para aquellos pacientes con histología de células no escamosas que habían recibido un régimen con contenido de pemetrexed. La aleatorización se desglosó por histología (células no escamosas vs. escamosas) y región geográfica (Europa vs Asia vs el resto del mundo).
Los criterios de valoración de la eficacia principales del estudio fueron la supervivencia global (OS) y la supervivencia libre de progresión (PFS). Las conclusiones del ensayo demostraron, en la población con TPS ≥ 50% una mejora estadísticamente significativa en la OS y la PFS de los pacientes aleatorizados con tratamiento de LIBTAYO en comparación con los de la quimioterapia. Una eficacia parecida se observó en la población mITT (fecha de línea de corte de los datos clínicos: 1 de marzo de 2020).
Rendimiento clínico del ensayo VENTANA PD-L1 (SP263) ASSAY en el estudio 1624 en población con NSCLC con expresión de PD-L1 en ≥ 50% de TC: estudio comparativo clínico:
El rendimiento clínico del ensayo VENTANA PD-L1 (SP263) ASSAY se evaluó mediante muestras de archivo de ensayo clínico del estudio 1624. Se analizaron de forma retrospectiva 871 muestras de ensayo clínico con VENTANA PD-L1 (SP263) ASSAY, 481 procedentes de pacientes aleatorizados y 390 de un subgrupo aleatorio procedente de pacientes no aptos en el cribado en el umbral de expresión de PD-L1 del 50% en células tumorales (% de TC). Los coeficientes de aceptabilidad de la tinción para VENTANA PD- L1 (SP263) ASSAY se evaluaron a nivel de sujeto. El índice de aceptabilidad de la tinción final en la población del uso previsto (IU) fue del 92.8% (CI del 95%: 90.9, 94.4).
La concordancia del estado de PD-L1 entre los resultados de VENTANA PD-L1 (SP263) ASSAY y PD-L1 IHC 22C3 pharmDx se calculó tomando los resultados de PD-L1 IHC 22C3 pharmDx como referencia. Los resultados del análisis de concordancia se muestran en la Tabla 24.
Tabla 24. Concordancia del estado de PD-L1 entre los resultados de VENTANA PD-L1 (SP263) ASSAY y los resultados de PD-L1 IHC 22C3 pharmDx del estudio 1624.a
|
Estado de PD-L1 (SP263) c |
Estado de PD-L1 (22C3) b |
||
|
Positiva |
Negativa |
Total |
|
|
Positiva |
324 |
24 |
348 |
|
Negativa |
68 |
352 |
420 |
|
Total |
392 |
376 |
768 |
|
Tipos de concordancia d,e |
PPA = 82.7% (324/392) (CI del 95%: 78.6-86.1) |
NPA = 93.6% (352/376) (CI del 95%: 90.7-95.7) |
OPA = 88.0% (676/768) (CI del 95%: 85.5-90.1) |
a Todos los pacientes que presentaban resultados de PD-L1 IHC 22C3 pharmDx y VENTANA PD-L1 (SP263) ASSAY que se podían evaluar, salvo los pacientes cuyos resultados finales mediante VENTANA PD-L1 (SP263) ASSAY se asociaban con una variación del protocolo de diagnóstico.
b Realizado según el etiquetado aprobado. A efectos de los análisis, el resultado de la presencia de expresión de PD-L1 (22C3) con una TPS del ≥ 50% se consideraba positivo y el resultado de la presencia de expresión de PD-L1 (22C3) con una TPS del < 50% se consideraba negativo.
c A efectos de los análisis, el resultado de la presencia de expresión de PD-L1 (SP263) con una TPS del ≥ 50% se consideraba positivo y el resultado de la presencia de expresión de PD-L1 (SP263) con una TPS del < 50% se consideraba negativo.
d PPA= Porcentaje de concordancia positiva; NPA= Porcentaje de concordancia negativa; OPA= Porcentaje de concordancia global.
e Los intervalos de confianza (CI) del 95% bilaterales se calcularon mediante el método de puntuación de Wilson.
La eficacia de LIBTAYO se evaluó en una población que presentaba expresión de PD-L1 en ≥ 50% de células tumorales (TC) según la identificación mediante el ensayo VENTANA PD-L1 (SP263) ASSAY(SP263+) con el método que se describe en Li 2015.14 Para el subgrupo de pacientes con una expresión de SP263+ (PD-L1 [SP263] ≥ 50% TC)/ 22C3+ (TPS ≥ 50%), la eficacia se estimó basándose en los datos del estudio 1624. En el subgrupo de pacientes SP263+/22C3- (TPS < 50%) se consideraron diversos escenarios en los que se presuponía que la eficacia era la misma (hipótesis ideal) o se reducía con respecto al subgrupo SP263+/22C3+ (la hipótesis menos favorable presuponía un HR = 1). La eficacia para los pacientes SP263+ se determinó con la media ponderada de los grupos SP263+/22C3+ y SP263+/ 22C3- como se describe en Li 2015.14 Se realizaron análisis de sensibilidad para tener en cuenta el impacto de los resultados de VENTANA PD-L1 (SP263) ASSAY que faltaban realizando varias imputaciones. La eficacia del fármaco, teniendo en cuenta tanto los resultados de las pruebas de PD-L1 (SP263) disponibles e imputados, se calculó mediante los mismos métodos estadísticos empleados en el análisis de eficacia del estudio 1624.
Las características demográficas y basales eran similares entre los pacientes SP263+, los pacientes 22C3+ y los pacientes en la población de mITT.
La eficacia que se observó en los pacientes SP263+/22C3+ era parecida a la eficacia de los pacientes de 22C3+ (Tabla 25). Los resultados de los análisis comparativos, con o sin imputación y en todas las hipótesis que se habían tenido en cuenta, demostraron que el beneficio clínico de LIBTAYO frente a la quimioterapia se mantiene en la población de SP263+ con respecto a la población 22C3+, lo que respalda el uso de la monoterapia con LIBTAYO en la configuración 1L para pacientes con expresión PD-L1 ≥ 50% TC, en la población de pacientes NSCLC identificada por VENTANA PD-L1 (SP263) ASSAY.
Tabla 25. Eficacia clínica de Cemiplimab en pacientes con PD-L1 ≥ 50% determinada por PD-L1 IHC 22C3 PharmDx y VENTANA PD-L1 (SP263) ASSAY
|
Criterios de valoración |
22C3+ a,b (N = 563) |
22C3+, SP263+ c (N = 324) |
||
|
LIBTAYO n= 283 |
Quimioterapia n= 280 |
LIBTAYO n= 164 |
Quimioterapia n= 160 |
|
|
Supervivencia global |
||||
|
Número de muertes (%) |
70 |
105 |
38 |
56 |
|
(24.7) |
(37.5) |
(23.2) |
(35.0) |
|
|
Media en meses (95% CI) d |
NR |
14.2 |
22.1 |
15.5 |
|
(17.9, NE) |
(11.2, 17.5) |
(17.7, NE) |
(11.4, NE) |
|
|
Índice de riesgo (CI del 95%) e |
0.57 (0.42, 0.77) |
0.52 (0.34, 0.80) |
||
|
Valor p |
0.0002 |
0.0022 |
||
|
Supervivencia libre de progresión según BICR |
||||
|
Número de eventos (%) |
147 |
197 |
76 |
110 |
|
(51.9) |
(70.4) |
(46.3) |
(68.8) |
|
|
Media en meses (95% CI) d |
8.2 |
5.7 |
9.8 |
5.4 |
|
(6.1, 8.8) |
(4.5, 6.2) |
(8.1, 14.5) |
(4.2, 6.2) |
|
|
Índice de riesgo (CI del 95%) e |
0.54 (0.43, 0.68) |
0.43 (0.32, 0.59) |
||
|
Valor p |
< 0.0001 |
< 0.0001 |
||
BICR = Revisión central, independiente y enmascarada;
CI = Intervalo de confianza;
NE = Imposible de evaluar; NR= No alcanzado;
LIBTAYO = cemiplimab.
a 22C3+ hace referencia a un subgrupo de pacientes aleatorizados que presentan expresión de PD-L1 con una TPS ≥ 50% en células tumorales (TC) mediante PD-L1 IHC 22C3 pharmDx según el etiquetado aprobado.
b Del estudio 1624.
c 22C3+, SP263+ hace referencia a los pacientes con expresión de PD-L1 en ≥ 50% de TC con VENTANA PD-L1 (SP263) ASSAY y con una TPS ≥ 50% en células tumorales (TC) con IHC 22C3 de PD-L1.
d Según el método de Kaplan-Meier.
e Según el modelo de riesgo proporcional estratificado.
Rendimiento clínico de LIBTAYO (cemiplimab) en el estudio 16113 Parte 2:
El rendimiento clínico de VENTANA PD-L1 (SP263) ASSAY se evaluó en la Parte 2 del Estudio 16113 (NCT03409614), como un estudio aleatorizado global de Fase III para investigar la eficacia y la seguridad de LIBTAYO (cemiplimab) en combinación con biquimioterapia basada en platino como el tratamiento de primera línea para pacientes con NSCLC avanzado de células escamosas o no escamosas.
Se inscribieron en total a 466 pacientes (conjunto completo de análisis [FAS]). Los pacientes fueron aleatorizados (2:1) para recibir 350mg de cemiplimab más biquimioterapia basada en platino o placebo más biquimioterapia basada en platino intravenosamente cada 3 semanas durante 4 ciclos dependiendo de la tolerabilidad del paciente y la evaluación de la enfermedad. La aleatorización se desglosó por histología (células no escamosas vs. células escamosas) y por nivel de expresión de PD-L1 en TC (< 1%, 1% al 49%, ≥ 50%). A nivel global, el 57,1% de los pacientes presentó una histología de células no escamosas (los pacientes con histología de células escamosas se limitaron según el protocolo al 50%), y la mayoría (85,2%) de pacientes tuvieron una enfermedad metastásica (estadio IV) en la fase de cribaje vs, enfermedad localmente avanzada (14,8% [estadio IIIB: 10,7% o IIIC: 4.1%]).
Las muestras tumorales de 465 de los 466 pacientes incorporados al estudio se analizaron con VENTANA PD-L1 (SP263) ASSAY para determinar el nivel de expresión de PD-L1. El porcentaje de estos pacientes que tuvieron tumores con expresión de PD-L1 en ≥ 1% de las células tumorales (TC) determinado por VENTANA PD-L1 (SP263) ASSAY fue del 70.1%. El porcentaje de aceptabilidad de la tinción final entre los pacientes en la población de uso previsto de VENTANA PD-L1 (SP263) ASSAY fue del 100.0%.
La medida del criterio de valoración de la eficacia primaria de 16113 Parte 2 fue la supervivencia global (OS). La OS se definió como el periodo de tiempo desde el momento de la aleatorización hasta la muerte por cualquier motivo. El objetivo de eficacia primario fue comparar la OS de los pacientes que habían recibido cemiplimab y biquimioterapia basada en platino con la de los pacientes que habían recibido placebo y biquimioterapia basada en platino entre el FAS. Un criterio de valoración de la eficacia secundario fue la supervivencia libre de progresión (PFS) entre el FAS. La PFS se define como el periodo de tiempo desde la aleatorización hasta la fecha de la primera progresión del tumor documentada o la muerte por cualquier causa, lo que ocurra primero. La OS y la PFS también se evaluaron en pacientes con expresión de PD-L1 ≥ 1% TC según se define por VENTANA PD-L1 (SP263) ASSAY.
En el momento del análisis final (fecha de corte de datos clínicos: 14 de junio de 2021), se observó una mejora clínicamente significativa y estadísticamente importante en la OS en el grupo de cemiplimab/quimioterapia vs. el grupo de placebo/quimioterapia. En el FAS (N = 466), la OS media fue considerablemente mayor que en el grupo de cemiplimab/quimioterapia (21.9 meses [95% CI: 15.5 hasta NE]) en comparación con el grupo de placebo/quimioterapia (13.0 meses [95% CI: 11.9 a 16.1] (HR=0.706 [95% CI: 0.534 a 0.933], p= 0.0140).
El beneficio de OS se corroboró adicionalmente por una mejora significativa en la PFS. En el FAS (N = 466), la PFS media fue considerablemente mayor que en el grupo de cemiplimab/quimioterapia (8.2 meses [95% CI: 6.4, 9.3]) en comparación con el grupo de placebo/quimioterapia (5.0 meses [95% CI: 4.3, 6.2]) (HR=0.556 [95% CI: 0.442 a 0.699], p < 0.0001).
También se observó una mejora clínicamente significativa en la OS y la PFS en pacientes con PD-L1 ≥ 1% TC (n=327), lo que respalda el uso de LIBTAYO en combinación con la biquimioterapia basada en platino en la configuración 1L para pacientes NSCLC con una expresión de PD-L1 ≥ 1% TC identificada por VENTANA PD-L1 (SP263) ASSAY. Los resultados de la eficacia para pacientes con expresión de PD-L1 ≥ 1% TC o < 1% TC se recogen en la Tabla 26.
Tabla 26. Eficacia clínica de LIBTAYO en pacientes con expresión de PD-L1 ≥ 1% TC ov < 1% TC según se define por VENTANA PD-L1 (SP263) ASSAY
|
Criterios de valoración |
LIBTAYO + quimioterapia |
Placebo + quimioterapia |
|
Supervivencia global |
||
|
PD-L1 ≥ 1% |
(N = 217) |
(N = 110) |
|
Evento (%) |
78/217 (35.9%) |
55/ 110 (50.0%) |
|
Media (95% CI), (meses) a |
21.9 (17.3, NE) |
12.6 (10.3, 16.4) |
|
HR (CI del 95%) b |
0.552 (0.390, 0.781) |
|
|
PD-L1 < 1% |
(N = 95) |
(N = 44) |
|
Evento (%) |
54/95 (56.8%) |
27/44 (61.4%) |
|
Media (95% CI), (meses) a |
12.8 (9.6, 16.5) |
14.2 (9.1, 18.0) |
|
HR (CI del 95%) c |
1.006 (0.633, 1.600) |
|
|
Supervivencia libre de progresión |
||
|
PD-L1 ≥ 1% |
(N = 217) |
(N = 110) |
|
Evento (%) |
134/217 (61.8%) |
86/110 (78.2%) |
|
Media (95% CI), (meses) a |
8.5 (6.7, 10.7) |
5.5 (4.3, 6.2) |
|
HR (CI del 95%) b |
0.475 (0.361, 0.626) |
|
|
PD-L1 < 1% |
(N = 95) |
(N = 44) |
|
Evento (%) |
70/95 (73.7%) |
36/44 (81.8%) |
|
Media (95% CI), (meses) a |
6.2 (4.4, 8.3) |
4.4 (4.2, 6.2) |
|
HR (CI del 95%) c |
0.764 (0.509, 1.146) |
|
CI= intervalo de confianza; HR= índice de riesgo; NE= imposible de estimar.
LIBTAYO = cemiplimab.
a Según el método de Kaplan-Meier.
b Según el modelo de riesgo estratificado, estratificado por histología (células escamosas, células no escamosas).
c Según el modelo de riesgo no estratificado.
Rendimiento clínico de TECENTRIQ (atezolizumab) en el estudio IMpower010:
El rendimiento clínico de VENTANA PD-L1 (SP263) ASSAY se evaluó en IMpower010 (NCT02486718), un estudio en fase III, abierto y aleatorizado cuyo objetivo era investigar la eficacia y la seguridad de TECENTRIQ (atezolizumab) (anticuerpo anti-PD-L1) en comparación con la atención asistencial de calidad óptima (BSC) tras el tratamiento adyuvante con quimioterapia con cisplatino en pacientes con NSCLC en estadio IB-IIIA completamente extirpado.
Un total de 1280 pacientes inscritos tenían una resección completa del tumor y eran elegibles para recibir hasta 4 ciclos de quimioterapia basada en cisplatino. Un total de 1005 pacientes se aleatorizaron (1:1) y recibieron tratamiento con 1200mg de TECENTRIQ mediante infusión intravenosa cada tres semanas durante 16 ciclos, a menos que se presentara una recidiva de la enfermedad o un nivel de toxicidad inaceptable, o bien una atención asistencial de calidad óptima (BSC), tras la recuperación de la cirugía. La aleatorización se clasificó por género, estadio de la enfermedad, histología y expresión de PD-L1. Entre los pacientes aleatorizados, la enfermedad del 12% se encontraba en estadio IB, del 47% en estadio II y del 41% en estadio IIIA.
Las muestras tumorales de 1169 de los 1280 pacientes incorporados al estudio (incluidos los 985 de los 1005 pacientes aleatorizados) se analizaron con VENTANA PD-L1 (SP263) ASSAY para determinar el nivel de expresión de PD-L1. El porcentaje de estos pacientes que tuvieron tumores con expresión de PD-L1 en ≥ 1% o ≥ 50% de las células tumorales (TC) determinado por VENTANA PD-L1 (SP263) ASSAY fue del 55% y del 26%, respectivamente. El porcentaje de aceptabilidad de la tinción final entre los pacientes en la población de uso previsto de VENTANA PD-L1 (SP263) ASSAY fue del 99.3%.
La medida primaria de resultado de eficacia de IMpower010 fue la supervivencia sin enfermedad (DFS), según la evaluación del investigador. DFS se definió como el tiempo transcurrido entre la fecha de la aleatorización y la fecha del primero de cualquiera de los siguientes eventos: primera recidiva documentada de la enfermedad, NSCLC primario nuevo o fallecimiento por cualquier causa. El objetivo principal de la valoración de la eficacia era evaluar la DFS en la población de pacientes con expresión de PD-L1 en ≥ 1% de TC (mediante SP263) y estadio II-IIIA. Los objetivos de eficacia secundarios clave fueron evaluar DFS en la población de pacientes en estadio II-IIIA PD-L1 ≥ 50% de TC (mediante SP263) y la tasa de supervivencia global (OS) en la población ITT.
En el momento de realizar el análisis de DFS provisional (fecha de corte de datos clínicos: 21 de enero de 2021), el estudio cumplía con el criterio de valoración primario y se presentaron mejorías estadísticamente significativas en la DFS en el grupo de TECENTRIQ en comparación con el grupo de la BSC dentro de la población de pacientes con expresión de PD-L1 en ≥ 1% de TC en estadio II-IIIA (n= 476) (HR estratificado: 0.66, CI del 95%: 0.50, 0.88, valor p 0.004). El tiempo medio de seguimiento fue de unos 32 meses. En los análisis de objetivos secundarios a los que se sometieron los pacientes en estadio II-IIIA con expresión de PD-L1 en ≥ 50% de TC (n= 229), se demostraron mejorías con importancia clínica en la DFS con un HR estratificado de 0.43 (CI del 95%: 0.27, 0.68).
También se observó una mejora en la DFS con importancia clínica en pacientes en estadio II-IIIA con expresión de PD-L1 en ≥ 50% de TC sin mutaciones en EGFR o reorganizaciones en ALK (n= 209).
Los resultados de la eficacia en la población de pacientes en estadio II-IIIA con expresión de PD-L1 en ≥ 50% de TC (con y sin mutaciones de EGFR o reorganizaciones en ALK) se muestran en la Tabla 27.
Tabla 27. Resultados de eficacia de IMpower010 en pacientes con NSCLC en estadio II- IIIA con expresión de PD-L1 (SP263) ≥ 50% de TC
|
≥ 50% de TC |
≥ 50% de TC sin mutaciones en EGFR ni reorganizaciones en ALK |
|||
|
Grupo A (TECENTRIQ) n= 115 |
Grupo B (BSC) n= 114 |
Grupo A (TECENTRIQ) n= 106 |
Grupo B (BSC) n= 103 |
|
|
Eventos de DFS (%) |
28 (24.3%) |
52 (45.6%) |
24 (22.6%) |
45 (43.7%) |
|
Mediana de DFS, en meses (CI del 95%)a |
NE (42.3, NE) |
35.7 (29.7, NE) |
NE (NE, NE) |
37.3 (30.1, NE) |
|
Índice de riesgo (CI del 95%) b |
0.43 (0.27, 0.68) |
0.43 (0.26, 0.71) |
||
|
Valor p |
0.0002 |
0.0002 |
||
DFS= Supervivencia sin enfermedad; CI= Intervalo de confianza; NE= Imposible de estimar.
BSC= Atención asistencial de calidad óptima.
a El tiempo medio de seguimiento fue de unos 32 meses.
b Sin estratificar.
RENDIMIENTO DE ANÁLISIS EN EL CÁNCER DE PULMÓN NO MICROCÍTICO:
Sensibilidad: tejido de NSCLC:
La sensibilidad de VENTANA PD-L1 (SP263) ASSAY se evaluó en 733 casos únicos de muestras de NSCLC mediante lotes de producción fabricados de VENTANA PD-L1 (SP263) ASSAY. En la evaluación de la expresión de PD-L1 se observó tinción en un rango de tinción de células tumorales de entre el 0 y el 100%.
Repetibilidad y precisión intermedia: tejido de NSCLC
Se ha evaluado la repetibilidad y precisión intermedia de VENTANA PD-L1 (SP263) ASSAY en el instrumento BenchMark ULTRA en combinación con OptiView DAB IHC Detection Kit mediante la tinción de 24 casos únicos de NSCLC humano.
Para la repetibilidad en el mismo día: se tiñeron 5 portaobjetos replicados de cada una de las 8 muestras de NSCLC en un solo instrumento BenchMark ULTRA en un único día.
Para evaluar la precisión entre días, se tiñeron dos portaobjetos replicados de las muestras de NSCLC con VENTANA PD-L1 (SP263) ASSAY en un mismo instrumento BenchMark ULTRA a lo largo de cinco días no consecutivos en un periodo de, al menos, 20 días.
Para evaluar la precisión entre instrumentos, cada una de las muestras de NSCLC se tiñó con VENTANA PD-L1 (SP263) ASSAY en tres instrumentos BenchMark ULTRA, tres instrumentos BenchMark XT y tres instrumentos BenchMark GX. Todos los portaobjetos se enmascararon, aleatorizaron y evaluaron a través del algoritmo de puntuación de VENTANA PD-L1 (SP263) ASSAY que figura en la Tabla 7.
En la Tabla 11 se resumen los resultados.
Tabla 11. Estudio de repetibilidad y precisión intermedia de VENTANA PD-L1 (SP263) ASSAY en muestras individuales de tejido NSCLC: BenchMark ULTRA, BenchMark XT y BenchMark GX
|
Nivel de expresión de PD-L1 |
Expresión ≥ 1% |
Expresión ≥ 5% |
Expresión ≥ 10% |
Expresión ≥ 50% |
|
Repetibilidad y precisión |
Porcentaje de concordanci a global (CI del 95%) |
Porcentaje de concordanci a global (CI del 95%) |
Porcentaje de concordanci a global (CI del 95%) |
Porcentaje de concordanci a global (CI del 95%) |
|
Repetibilidad entre lotes (en un mismo día) |
100.0% (96.9-100.0)* |
99.2% (95.4-99.9)* |
98.3% (94.1-99.5)* |
100.0% (96.9-100.0)* |
|
Precisión entre días (5 días no consecutivos) |
100.0% (98.4-100.0)* |
97.9% (95.2-99.1)* |
98.8% (96.4-99.6)* |
100.0% (98.4-100)* |
|
Precisión entre plataformas (en 3 instrumentos BenchMark ULTRA) |
100% (99.4-100.0)* |
96.5% (94.7-97.6)* |
95.2% (93.3-96.6)* |
97.2% (94.6-99.2)** |
|
Precisión entre plataformas (en 3 instrumentos BenchMark XT) |
100.0% (94.0-100.0)* |
100.0% (94.0-100.0)* |
100.0% (94.0-100.0)* |
100.0% (94.0-100.0)* |
|
Precisión entre plataformas (en 3 instrumentos BenchMark GX i) |
100.0% (94.0-100.0)* |
100.0% (94.0-100.0)* |
100.0% (94.0-100.0)* |
100.0% (94.0-100.0)* |
* Los intervalos de confianza (CI) del 95% bilaterales se calcularon mediante el método de puntuación de Wilson.
** Los intervalos de confianza (CI) del 95% bilaterales se calcularon mediante el método bootstrap percentil de 2000 muestras bootstrap.
Reproducibilidad entre lotes: tejido de NSCLC:
La reproducibilidad entre lotes de VENTANA PD-L1 (SP263) ASSAY se determinó analizando tres lotes de VENTANA PD-L1 (SP263) ASSAY en 24 casos únicos de NSCLC en instrumentos BenchMark ULTRA con OptiView DAB IHC Detection Kit. Todos los casos se tiñeron con cada uno de los tres lotes del anticuerpo VENTANA PD-L1 (SP263). Los portaobjetos se enmascararon y aleatorizaron antes de evaluar la expresión de PD- L1, según indica el algoritmo de puntuación de VENTANA PD-L1 (SP263) ASSAY que figura en la Tabla 7. Los resultados se muestran en la Tabla 12 en forma de porcentajes de concordancia global, porcentajes de concordancia positiva y porcentajes de concordancia negativa para cada nivel de expresión.
Tabla 12. Índices de concordancia de la reproducibilidad entre lotes en muestras individuales de tejido de NSCLC
|
Reproducibilidad entre lotes |
Positiva Porcentaje Concordancia (CI del 95%) |
Negativa Porcentaje Concordancia (CI del 95%) |
Global Porcentaje Concordancia (CI del 95%) |
|
Promedio de las tres comparaciones entre lotes ≥ 1% de expresión |
100% (99.0-100.0)* |
100% (98.6-100.0)* |
100% (99.4-100.0)* |
|
Promedio de las tres comparaciones entre lotes Expresión ≥ 5% |
94.4% (91.4-96.5)* |
98.5% (96.4-99.3)* |
96.5% (94.7-97.6)* |
|
Promedio de las tres comparaciones entre lotes Expresión ≥ 10% |
97.5% (94.7-98.9)* |
93.8% (91.0-95.8)* |
95.2% (93.3-96.6)* |
|
Promedio de las tres comparaciones entre lotes Expresión ≥ 50% |
96.9% (91.9-99.7)** |
97.5% (94.9-99.5)** |
97.2% (94.4-99.1)** |
* Los intervalos de confianza (CI) del 95% bilaterales se calcularon mediante el método de puntuación de Wilson.
** Los intervalos de confianza (CI) del 95% bilaterales se calcularon mediante el método bootstrap percentil de 2000 muestras bootstrap.
Concordancia entre plataformas:
Se realizó un estudio entre plataformas que incluía cuatro indicaciones, 10 casos de NSCLC, 10 casos de SCCHN, 10 casos uroteliales, 10 casos de melanoma y 11 casos de placenta. Para este estudio, los portaobjetos se evaluaron a partir de estudios de precisión intermedia realizados previamente. La consistencia de la tinción se evaluó colectivamente valorando la intensidad de la tinción de concordancia así como la tinción promedio de PD-L1 en las células tumorales de varios tipos de tejido de cada una de las plataformas XT y GX con las muestras de referencia respectivas teñidas en la plataforma Benchmark ULTRA. Consulte los resultados en la Tabla 13.
Tabla 13. Concordancia entre plataformas del ensayo VENTANA PD-L1 (SP263)
|
Observación analizada |
Porcentaje de concordancia global (CI del 95%) |
|
|
BenchMark ULTRA: BenchMark XT |
Nivel de expresión de PD-L1 |
99.7 (99.1-100.0)a |
|
Intensidad de la tinción PL-L1 |
100.0 (98.5-10-0.0)b |
|
|
BenchMark ULTRA: BenchMark GX |
Nivel de expresión de PD- L1 |
99.4 (98.4-100.0)a |
|
Intensidad de la tinción PL-L1 |
98.4 (95.9- 99.4)b |
a Los intervalos de confianza (CI) del 95% bilaterales se calcularon mediante el método bootstrap percentil de 2000 muestras bootstrap.
b Los intervalos de confianza (CI) del 95% bilaterales se calcularon mediante el método de puntuación de Wilson.
Estudios de precisión del lector: tejido de NSCLC:
Para evaluar la precisión del lector y entre lectores, tres anatomopatólogos evaluaron un mínimo de 110 casos únicos. Los casos se enmascararon y aleatorizaron antes de evaluar la tinción IHC de PD-L1, según indica el algoritmo de puntuación de VENTANA PD-L1 (SP263) ASSAY que figura en la Tabla 7. Los resultados que aparecen en la Tabla 14 reflejan los porcentajes de precisión interlector e intralector para casos únicos de la cohorte del estudio.
Tabla 14. Resumen del estudio de precisión del lector y entre lectores de VENTANA PD- L1 (SP263) ASSAY en muestras individuales de tejido de NSCLC
|
Precisión del lector |
Promedio de concordancia positiva (CI del 95%)* |
Promedio de concordancia negativa (CI del 95%)* |
Porcentaje de concordancia global (CI del 95%)* |
|
Precisión interlectores; promedio de los tres lectores ≥ 1% de expresión |
94.3% (90.5-97.4) |
92.6% (87.8-96.5) |
93.5% (89.9-97.1) |
|
Precisión interlectores; promedio de los tres lectores Expresión ≥ 5% |
94.7% (90.7-97.8) |
94.7% (90.6-97.7) |
94.7% (91.1-97.7) |
|
Precisión interlectores; promedio de los tres lectores Expresión ≥ 10% |
93.0% (88.4-96.6) |
94.0% (90.0-97.1) |
93.5% (89.5-97.0) |
|
Precisión interlectores; promedio de los tres lectores Expresión ≥ 50% |
94.6% (90.6-97.8) |
95.0% (91.1-97.9) |
94.8% (91.2-97.8) |
|
Precisión del lector; promedio de los tres lectores ≥ 1% de expresión |
96.7% (94.7-98.3) |
95.6% (92.9-97.8) |
96.2% (94.1-98.0) |
|
Precisión del lector; promedio de los tres lectores Expresión ≥ 5% |
95.8% (92.7-98.2) |
96.0% (93.2-98.3) |
95.9% (93.3-98.2) |
|
Precisión del lector; promedio de los tres lectores Expresión ≥ 10% |
97.7% (95.9-99.2) |
98.1% (96.4-99.4) |
97.9% (96.2-99.4) |
|
Precisión del lector; promedio de los tres lectores Expresión ≥ 50% |
97.2% (95.2-98.8) |
97.3% (95.2-98.9) |
97.2% (95.4-98.8) |
* Los intervalos de confianza (CI) del 95% bilaterales se calcularon mediante el método bootstrap percentil de 2000 muestras bootstrap.
Estudio de reproducibilidad entre laboratorios del instrumento BenchMark ULTRA: tejido NSCLC:
Se llevó a cabo un estudio de reproducibilidad entre laboratorios de VENTANA PD-L1 (SP263) ASSAY con el objetivo de demostrar la reproducibilidad del ensayo a la hora de determinar la expresión de PD-L1 en casos de NSCLC mediante el análisis de 28 muestras de tejido a lo largo de cinco días no consecutivos durante un periodo de 20 días en tres laboratorios externos. Las muestras eran ciegas, se aleatorizaron y las evaluaron un total de seis lectores (2 lectores/sitio). Consulte los resultados en la Tabla 15.
Tabla 15. Reproducibilidad entre laboratorios: Índices de concordancia de VENTANA PD- L1 (SP263) ASSAY en muestras individuales de tejido de NSCLC
|
Reproducibilidad entre laboratorios |
Positiva Porcentaje Concordancia (CI del 95%)* |
Negativa Porcentaje Concordancia (CI del 95%)* |
Global Porcentaje Concordancia (CI del 95%)* |
|
En todos los casos ≥ 1% de expresión |
99.5% (98.6-100.0) |
100.0% (99.1-100.0) |
99.8% (99.3-100.0) |
|
En todos los casos Expresión ≥ 5% |
89.2% (85.9-91.9) |
93.7% (90.9-95.7) |
91.4% (89.3-93.2) |
|
En todos los casos Expresión ≥ 10% |
95.4% (92.6-97.2) |
94.0% (91.6-95.8) |
94.6% (92.8-95.9) |
|
En todos los casos Expresión ≥ 50% |
94.3% (90.2-98.1) |
90.1% (85.1-94.7) |
92.2% (89.0-95.2) |
* Nota: CI del 95% = Intervalo de confianza.
En el caso de los PPA/NPA/OPA, los datos de CI del 95% se calcularon mediante el método de puntuación de Wilson cuando la concordancia era del 100% o mediante el método bootstrap percentil a partir de 2000 muestras bootstrap en las concordancias inferiores al 100%.
Estudio de comparación de métodos entre los instrumentos BenchMark ULTRA PLUS y BenchMark ULTRA: tejido NSCLC:
Tres laboratorios de instituciones independientes de Estados Unidos participaron en un estudio de concordancia entre el instrumento BenchMark ULTRA PLUS y el instrumento BenchMark ULTRA. En el estudio se emplearon un total de 196 muestras de tejido de NSCLC únicas compradas, anonimizadas y que representaban todo el intervalo de tinción de VENTANA PD-L1 (SP263) ASSAY en las líneas de corte de 1% y 50%. La cohorte de tejidos contenía dos poblaciones de análisis superpuestas de 140 casos, en las que se había equilibrado la distribución entre los casos positivos y los negativos en PD-L1 de cada línea de corte, según lo determinado por la revisión de consenso de los anatomopatólogos de Roche Tissue Diagnostics (RTD). Para cada caso, se tiñeron dos portaobjetos de tejido con VENTANA PD-L1 (SP263) ASSAY y Rabbit Monoclonal Negative Control Ig en un instrumento BenchMark ULTRA mediante el protocolo de tinción recomendado en RTD, y se tiñeron dos portaobjetos de tejido más con VENTANA PD-L1 (SP263) ASSAY y Rabbit Monoclonal Negative Control Ig en un instrumento BenchMark ULTRA PLUS mediante el protocolo de tinción recomendado en uno de los tres laboratorios externos. En cada centro se tiñó aproximadamente 1/3 de los casos del estudio en un instrumento BenchMark ULTRA PLUS. Los portaobjetos del caso se aleatorizaron antes de su tinción en los instrumentos BenchMark ULTRA o BenchMark ULTRA PLUS. Dos anatomopatólogos de cada uno de los laboratorios externos y un anatomopatólogo de RTD, con la identidad de la muestra enmascarada, evaluaron por separado todos los portaobjetos teñidos en BenchMark ULTRA y todos los portaobjetos teñidos en BenchMark ULTRA PLUS con el algoritmo de puntuación VENTANA PD-L1 (SP263) ASSAY para NSCLC (Tabla 7) con un periodo de reposo de al menos dos semanas entre las lecturas. Se evaluó independientemente la equivalencia del rendimiento de VENTANA PD-L1 (SP263) ASSAY en el instrumento en fase de investigación (BenchMark ULTRA PLUS) en relación con el instrumento de referencia (BenchMark ULTRA) en las líneas de corte del 1% y el 50%. Los resultados se resumen en la Tabla 16.
Tabla 16. Concordancia combinada del estado de PD-L1 entre BenchMark ULTRA PLUS y BenchMark ULTRA en las líneas de corte del 1% y el 50%
|
Línea de corte del 1%a |
BenchMark ULTRA |
||
|
BenchMark ULTRA PLUS |
≥ 1% |
< 1% |
Total |
|
≥ 1% |
474 |
12 |
486 |
|
< 1% |
6 |
473 |
479 |
|
Total |
480 |
485 |
965 |
|
Concordanciac |
Porcentaje (n/N) (CI del 95%d) |
||
|
Porcentaje de concordancia positiva |
98,8 (474/480) (97,5-99,8) |
||
|
Porcentaje de concordancia negativa |
97,5 (473/485) (96,2-98,7) |
||
|
Porcentaje de concordancia global |
98,1 (947/965) (97,2-99,0) |
||
|
Línea de corte del 50%b |
BenchMark ULTRA |
||
|
BenchMark ULTRA PLUS |
≥ 50% |
< 50% |
Total |
|
≥ 50% |
478 |
21 |
499 |
|
< 50% |
5 |
464 |
469 |
|
Línea de corte del 50%b |
BenchMark ULTRA |
||
|
Total |
483 |
485 |
968 |
|
Concordanciac |
Porcentaje (n/N) (CI del 95%d) |
||
|
Porcentaje de concordancia positiva |
99,0 (478/483) (97,5-100,0) |
||
|
Porcentaje de concordancia negativa |
95,7 (464/485) (93,4-97,9) |
||
|
Porcentaje de concordancia global |
97.3 (942/968) (96.0-98.6) |
||
a Observaciones que se pudieron evaluar: n= 965.
b Observaciones que se pudieron evaluar: n= 968.
c La concordancia combinada agrupa todos los casos y los lectores.
d CI del 95%= Intervalo de confianza. Los intervalos de confianza del 95% bilaterales se calcularon mediante el método bootstrap percentil con 2000 réplicas estratificadas en grupos de puntuación para el diagnóstico (positiva, negativa, positiva límite, negativa límite).
Estudio de reproducibilidad entre laboratorios del instrumento BenchMark ULTRA PLUS: tejido NSCLC:
Se llevó a cabo un estudio de reproducibilidad entre laboratorios con la tinción de VENTANA PD-L1 (SP142) ASSAY en instrumentos BenchMark ULTRA PLUS para demostrar la reproducibilidad del ensayo a la hora de determinar la expresión de PD-L1 en muestras de tejido de NSCLC en las líneas de corte del 1% y del 50%. En el estudio, se emplearon un total de 38 muestras de tejido NSCLC únicas compradas con un intervalo de expresión de PD-L1. La cohorte de tejidos contenía dos poblaciones de análisis superpuestas de 28 casos, en las que se había equilibrado la distribución entre los casos positivos y los negativos en PD-L1 de cada línea de corte, según lo determinado por la revisión de consenso de los anatomopatólogos de Roche Tissue Diagnostics (RTD). Se tiñó cada caso en tres laboratorios externos en 5 días no consecutivos durante un periodo de, al menos, 20 días. Los portaobjetos del caso se aleatorizaron antes de su tinción en los instrumentos BenchMark ULTRA PLUS. En cada centro, dos anatomopatólogos evaluaron los portaobjetos con tinción por separado, con la identidad de la muestra enmascarada, mediante el algoritmo de puntuación VENTANA PD-L1 (SP263) ASSAY para NSCLC (Tabla 7). Los resultados se resumen en la Tabla 17.
Tabla 17. Reproducibilidad entre laboratorios de la tinción con VENTANA PD-L1 (SP142) ASSAY de muestras de NSCLC en instrumentos BenchMark ULTRA PLUS
|
Línea de corte |
Reproducibilidad entre laboratorios |
Porcentaje de concordancia (n/N) (CI del 95%)b |
|---|---|---|
|
≥ 1% a |
Concordancia global c (en comparación con la puntuación consensuada, en todos los sitios, días y lectores) |
PPA: 96,2 (404/420) (91,6-100,0) NPA: 100,0 (420/420) (99,1-100,0) OPA: 98,1 (824/840) (95,6-100,0) |
|
Concordancia entre sitios d (promedio de las comparaciones entre pares entre sitios) |
APA: 96,8 (7822/8080) (92,5-100,0) ANA: 97,0 (8462/8720) (93,0-100,0) OPA: 96,9 (8142/8400) (92,9-100,0) |
|
|
Concordancia entre lectores d (promedio de las comparaciones entre pares entre los lectores en cada sitio) |
APA: 97,0 (392/404) (93,2-100,0) ANA: 97,2 (424/436) (93,5-100,0) OPA: 97,1 (408/420) (93,6-100,0) |
|
|
≥ 50% a |
Concordancia global c (en comparación con la puntuación consensuada, en todos los sitios, días y lectores) |
PPA: 95,9 (374/390) (93,1-98,6) NPA: 98,9 (445/450) (96,9-100,0) OPA: 97,5 (819/840) (95,8-99,0) |
|
Concordancia entre sitios d (promedio de las comparaciones entre pares entre sitios) |
APA: 94,8 (7188/7580) (91,8-98,0) ANA: 95,7 (8828/9220) (92,6-98,4) OPA: 95,3 (8008/8400) (92,2-98,2) |
|
|
Concordancia entre lectores d (promedio de las comparaciones entre pares entre los lectores en cada sitio) |
APA: 94,5 (358/379) (91,1-97,8) ANA: 95,4 (440/461) (92,0-98,4) OPA: 95.0 (399/420), (91.7-98.1) |
APA = Promedio de concordancia positiva, ANA = Promedio de concordancia negativa, PPA = Porcentaje de concordancia positiva, NPA = Porcentaje de concordancia negativa, OPA = Porcentaje de concordancia global.
a n= 840 observaciones del portaobjetos PD-L1.
b Nota: CI del 95% = Intervalo de confianza.
c Resultados de concordancia del estudio con es estado modal de PD-L1 a nivel de caso.
d Índices de concordancia de pares.
Nota: En el caso de los PPA/NPA/OPA, los datos de CI del 95% se calcularon mediante el método de puntuación de Wilson cuando la concordancia era del 100% o mediante el método bootstrap percentil a partir de 2000 muestras bootstrap en las concordancias inferiores al 100%.
En el caso de los APA/ANA, los datos de CI del 95% se calcularon mediante el método de puntuación de Wilson cuando la concordancia era del 100% o mediante el método bootstrap percentil a partir de 2000 muestras bootstrap en las concordancias inferiores al 100%.
USO PREVISTO:
VENTANA PD-L1 (SP263) ASSAY está destinado a su uso en laboratorio para la detección cualitativa inmunohistoquímica del ligando de muerte programada 1 (PD-L1) mediante microscopía óptica en secciones de tejido fijado con formol y embebido en parafina (FFPE) que se indican en las tablas de más abajo teñidas con OptiView DAB IHC Detection Kit en un instrumento BenchMark IHC/ISH.
La expresión PD-L1 en cáncer de pulmón no microcítico (NSCLC) viene determinada por el porcentaje de células tumorales (% TC) con cualquier tinción de membrana sobre un fondo.
Consulte las tablas que aparecen a continuación para ver los tipos de tumor específicos y las aplicaciones clínicas. Consulte el etiquetado del correspondiente producto médico para ver las recomendaciones clínicas relativas a la expresión de PD-L1.
Figura 1. Tinción de cáncer de pulmón no microcítico con VENTANA PD-L1 (SP263) ASSAY
Tabla 1. Instrucciones de uso CDx
|
Tipo de tumor |
Nivel de expresión de PD-L1 |
Aplicación clínica |
|
NSCLC |
≥ 1% DE TC |
La expresión de PD-L1 en la membrana de las células tumorales (TC) que detecta el ensayo VENTANA PD-L1 (SP263) ASSAY en el NSCLC se indica para ayudar a identificar a los pacientes a los que se podría aplicar el tratamiento con IMFINZI™ (durvalumab). |
|
≥ 50% de TC ≥ 1% DE TC |
La expresión de PD-L1 en la membrana de las células tumorales (TC) que detecta el ensayo VENTANA PD-L1 (SP263) ASSAY en el NSCLC se indica para ayudar a identificar a los pacientes a los que se podría aplicar el tratamiento con KEYTRUDA® (pembrolizumab). |
|
|
≥ 50% de TC ≥ 1% DE TC |
La expresión de PD-L1 en la membrana de las células tumorales (TC) que detecta el ensayo VENTANA PD-L1 (SP263) ASSAY en el NSCLC se indica para ayudar a identificar a los pacientes a los que se podría aplicar el tratamiento con LIBTAYO® (cemiplimab). |
|
|
≥ 50% de TC |
La expresión de PD-L1 en la membrana de las células tumorales (TC) que detecta el ensayo VENTANA PD-L1 (SP263) ASSAY en el NSCLC se indica para ayudar a identificar a los pacientes en una fase temprana de NSCLC para el tratamiento adyuvante con TECENTRIQ® (atezolizumab). |
Para obtener más información sobre la interpretación de la tinción, consulte la guía de interpretación.
Tabla 2. Instrucciones de uso adicionales
|
Tipo de tumor |
Nivel de expresión de PD-L1 |
Aplicación clínica |
|
Carcinoma de células escamosas NSCLC |
≥ 1% de TC, ≥ 5% DE TC, y ≥ 10% de TC |
La expresión de PD-L1 en la membrana de las células tumorales (TC) que detecta el ensayo VENTANA PD-L1 (SP263) ASSAY en el NSCLC no escamoso puede asociarse a una mayor supervivencia con OPDIVO® (nivolumab). |
Para obtener más información sobre la interpretación de la tinción, consulte la guía de interpretación.
La interpretación de los resultados de VENTANA PD-L1 (SP263) ASSAY debe correr a cargo de un anatomopatólogo cualificado junto con un examen histológico, la información clínica pertinente y los controles adecuados.
Este producto está destinado para uso diagnóstico in vitro (IVD).
LIMITACIONES ESPECÍFICAS:
1. VENTANA PD-L1 (SP263) ASSAY se ha desarrollado para los instrumentos BenchMark IHC/ISH con el OptiView DAB IHC Detection Kit y no se ha aprobado para ningún otro instrumento de detección.
2. Se debe teñir un portaobjetos de la muestra del paciente con Rabbit Monoclonal Negative Control Ig. Otros reactivos de control negativos no son adecuados para este ensayo.
3. La prueba de isquemia fría de VENTANA PD-L1 (SP263) ASSAY que usó un modelo de tejido de xenoinjerto no estableció condiciones desde cero hasta 24 horas que no fueran favorables para el ensayo.
4. El ensayo no está validado para su uso con otros tipos de muestras citológicas (frotis, cepillados, lavados, irrigaciones o efusiones) o muestras óseas decalcificadas.
5. Los portaobjetos se deben secar y conservar a temperatura ambiente. Dado que se ha confirmado que los factores medioambientales influyen en la estabilidad de los antígenos en los portaobjetos con secciones, los laboratorios deben validar la estabilidad de los cortes de los portaobjetos en su propio entorno cuando se almacenan durante un periodo superior a los 45 días, si se considera oportuno.
6. Este ensayo no está validado para su uso con bloques celulares de FNA fijados en conservantes de citología.
7. Los bloques celulares FFPE de NSCLC procedentes de FNA no se han incluido en la comparación de métodos analíticos ni en los estudios de resultados clínicos que se recogen en esta hoja de datos.
8. Es posible que este ensayo no esté registrado en todos los instrumentos. Póngase en contacto con el representante local de servicio Roche para obtener más información.
PREPARACIÓN DE MUESTRAS:
Los tejidos FFPE que se procesan de forma habitual resultan adecuados para su uso con este anticuerpo primario cuando se utilizan con los kits de detección de VENTANA y los instrumentos BenchMark IHC/ISH.
Según los ensayos realizados con tejidos de la placenta y las amígdalas con expresión de PD-L1, el fijador de tejidos recomendado es una solución de formol tamponado neutro6 (NBF) al 10% durante un tiempo de fijación de entre al menos, 6 horas y 72 horas. Los fijadores aceptables para el uso con VENTANA PD-L1 (SP263) ASSAY son Zinc Formalin y los fijadores Z-5 cuando se utilizan con un tiempo de fijación mínimo de 6 horas. Otros fijadores, entre los que se incluyen el alcohol al 95%, AFA y PREFER, no se consideran aceptables para su uso con VENTANA PD-L1 (SP263) ASSAY. La cantidad usada de fijador es de entre 15 y 20 veces el volumen de tejido. La fijación se puede llevar a cabo a temperatura ambiente (entre 15 y 25°C).
Consulte la Guía de interpretación de VENTANA PD-L1 (SP263) ASSAY (P/N 1015317) para obtener información más detallada sobre la repercusión de la preparación de muestras en la tinción de PD-L1 con VENTANA PD-L1 (SP263) ASSAY. Las secciones se deben cortar con un grosor de 4-5μm y colocarse en portaobjetos cargados positivamente. Los portaobjetos deben teñirse inmediatamente, ya que la antigenicidad de los cortes de tejido puede disminuir con el tiempo.
Se recomienda analizar controles positivos y negativos a la vez que las muestras desconocidas.
PRINCIPIO DEL PROCEDIMIENTO:
VENTANA PD-L1 (SP263) ASSAY es un anticuerpo primario monoclonal de conejo que se une a PD-L1 en secciones de tejido FFPE. El anticuerpo puede visualizarse mediante OptiView DAB IHC Detection Kit (nº de cat. 760-700 / 06396500001). Consulte la hoja de datos correspondiente para obtener más información.
PROCEDIMIENTO DE TINCIÓN:
VENTANA PD-L1 (SP263) ASSAY se ha creado para su uso con los instrumentos BenchMark IHC/ISH junto con los kits de detección VENTANA y sus accesorios. Consulte las Tabla 4 y Tabla 5 para ver los protocolos de tinción recomendados.
Este anticuerpo se ha optimizado para tiempos de incubación específicos, pero el usuario debe validar los resultados obtenidos con este reactivo.
Los parámetros de los procedimientos automatizados se pueden visualizar, imprimir y editar según el procedimiento descrito en el Manual del usuario del instrumento. Consulte la hoja de datos del kit de detección VENTANA correspondiente para obtener más detalles sobre los procedimientos de tinción de inmunohistoquímica.
Tabla 4. Procedimientos de tinción de VENTANA PD-L1 (SP142) ASSAY en instrumentos BenchMark IHC/ISH
|
Plataforma del instrumento |
Procedimiento de tinción |
|
BenchMark GX |
GX VENTANA PD-L1 (SP263) ASSAY |
|
BenchMark XT |
XT VENTANA PD-L1 (SP263) ASSAY |
|
BenchMark ULTRA o BenchMark ULTRA PLUS |
ULTRA VENTANA PD-L1 (SP263) ASSAY |
Tabla 5. Protocolo de tinción recomendado para VENTANA PD-L1 (SP263) ASSAY junto con los instrumentos OptiView DAB IHC Detection Kit en BenchMark IHC/ISH
|
Paso del protocolo |
Entrada de parámetros |
|
Horneado |
Opcional |
|
Anticuerpo (Primario) |
VENTANA PD-L1 (SP263) seleccionado o Negative Control seleccionado |
|
Contratinción |
Hematoxylin II, 4 minutos |
RESUMEN Y EXPLICACIÓN:
VENTANA PD-L1 (SP263) ASSAY es un ensayo inmunohistoquímico que emplea un anticuerpo primario PD-L1 (SP263) monoclonal de conejo anti-PD-L1 (VENTANA PD-L1 (SP263)) para reconocer la proteína del ligando de muerte programada 1 (PD-L1).
PD-L1 es una proteína transmembrana que reduce las respuestas inmunitarias a través de la unión a sus dos receptores de muerte programada 1 (PD-1) y B7-1.1 PD-1 es un receptor de inhibición cuya expresión se observa en los linfocitos T tras su activación y se mantiene en estados de estimulación crónica, como las infecciones crónicas o el cáncer.2 La unión de PD-L1 con PD-1 inhibe la proliferación de los linfocitos T, la producción de citocinas y la actividad citolítica, dando lugar a la inactivación funcional o al agotamiento de los linfocitos T.2 B7.1 es una molécula cuya expresión se observa en células con antígeno y linfocitos T activados. La unión de PD-L1 a B7.1 en linfocitos T y células con antígeno puede participar en la reducción de las respuestas inmunitarias, como la inhibición de la activación de los linfocitos T y la producción de citocinas.3 La expresión de PD-L1 se ha observado en células inmunitarias y en células malignas.4,5 Se ha descrito que la expresión anómala de PD-L1 en células malignas y en células inmunitarias asociadas a tumores impide la inmunidad antitumoral, dando como resultado una evasión inmunológica.2,5 Por lo tanto, la interrupción de la vía PD-L1/PD-1 puede resultar una estrategia interesante para revitalizar la inmunidad específica frente a tumores de los linfocitos T que queda suprimida por la expresión de PD-L1 en el microentorno tumoral.
Se ha documentado la asociación entre la expresión de PD-L1 en células tumorales (TC) o en células inmunitarias que infiltran el tumor (IC) y el beneficio clínico de los inhibidores de la vía PD-L1/PD-1 en diferentes tipos de cáncer.
REFERENCIAS:
1. Keir ME, Butte MJ, Freeman GJ, et al. PD-1 and its ligands in tolerance and immunity. Annu Rev Immunol 2008; 26:677-704.
2. Blank C, Mackensen A. Contribution of the PD-L1/PD-1 pathway to T-cell exhaustion: an update on implications for chronic infections and tumor evasion. Cancer Immunol Immunother 2007; 56(5):739-745.
3. Butte MJ, Keir ME, Phamduy TB, et al. Programmed death-1 ligand 1 interacts specifically with the B7-1 costimulatory molecule to inhibit T-cell responses. Immunity 2007; 27(1):111-122.
4. Dong H, Zhu G, Tamada K, Chen L. B7-H1, a third member of the B7 family, co- stimulates T-cell proliferation and interleukin-10 secretion. Nat Med 1999; 5(12):1365-1369.
5. Massard C, Gordon MS, Sharma S, et al. Safety and efficacy of durvalumab (MEDI4736), an anti-programmed cell death ligand-1 immune checkpoint inhibitor, in patients with advanced urothelial bladder cancer. J Clin Oncol 2016; 34(26):3119- 3125.
6. Carson FL, Cappellano C. Histotechnology; A Self-Instructional Text, 5th edition. American Society for Clinical Pathology Press; 2020, 2022.
7. Occupational Safety and Health Standards: Occupational exposure to hazardous chemicals in laboratories. (29 CFR Part 1910.1450). Fed. Register.
8. Directive 2000/54/EC of the European Parliament and Council of 24 June 2020 on the protection of workers from risks related to exposure to biological agents at work.
9. Reck M, Rodriguez-Abreu D, Robinson AG, et al. Pembrolizumab versus chemotherapy for PD-L1-positive non-small cell lung cancer. The N Engl J Med 2016; 375(19):1823-1833.
10. Herbs RS, Baas P, Kim DW, et al. Pembrolizumab versus docetaxel for previously treated, PD-L1-positive, advanced non-small-cell lung cancer (KEYNOTE-010): a randomized controlled trial. Lancet 2016; 387:1540-1550.
11. Ratcliffe MJ, Sharpe A, Midha A, et al. Agreement between programmed cell death ligand-1 diagnostic assays across multiple protein expression cut-offs in non-small cell lung cancer. Clin Cancer Res 2017; 23(14):3585-3591.
12. Meijuan Li (2016) Statistical Methods for Clinical Validation of Follow-On Companion Diagnostic Devices via an External Concordance Study, Statistics in Biopharmaceutical Research, 8:3, 355-363, DOI: 10.1080/19466315.2016.1202859.
13. Borghaei H, Paz-Ares L, Horn L, et al. Nivolumab versus docetaxel in advanced non-squamous non–small cell lung cancer. N Engl J Med 2015; 373(17):1627-1639.
14. Li M. Statistical consideration and challenges in bridging study of personalized medicine. J Biopharm Stat 2015; 25(3):397-407.
Nota: En este documento se ha usado el punto como separador decimal para marcar el borde entre la parte entera y la parte fraccionaria de los numerales con decimales. No se han usado separadores para las unidades de millar.
El resumen de los aspectos de seguridad y rendimiento se puede ver a continuación: https://ec.europa.eu/tools/eudamed
Símbolos:
Ventana usa los siguientes símbolos y signos además de los indicados en la norma ISO 15223-1 (para USA: consulte elabdoc.roche.com/symbols para obtener más información).
|
GTIN |
Número mundial de artículo comercial |
|
Rx only |
Para USA: Precaución: Las normas nacionales restringen la venta de este dispositivo a médicos autorizados o por orden de estos. |
Propiedad intelectual:
VENTANA, BENCHMARK y OPTIVIEW son marcas comerciales de Roche. Todos los demás nombres de productos y marcas comerciales pertenecen a sus respectivos propietarios.
© 2024 Ventana Medical Systems, Inc.
For USA: Rx only
ROCHE DIAGNOSTICS GMBH
Sandhofer Strasse 116
68305 Mannheim Germany
+800 5505 6606