
VENTANA HER2 (4B5) RABBIT MONO CLONAL PRIMARY ANTIBODY RXDX
Para visualizar el contenido completo de la IPPA (información para prescribir amplia), deberá iniciar sesión.
INTERPRETACIÓN DE LAS TINCIONES Y RESULTADOS PREVISTOS:
El procedimiento de inmunotinción automatizada VENTANA hace que un producto de reacción de color marrón (DAB) se precipite en los sitios del antígeno localizados por el VENTANA HER2 (4B5) Assay. Un anatomopatólogo cualificado con experiencia en procedimientos de inmunohistoquímica debe evaluar los controles y calificar el producto con tinción antes de interpretar los resultados.
Controles positivos:
Debe examinarse el control de tejido positivo con tinción en primer lugar para comprobar que todos los reactivos han funcionado correctamente. La existencia de un producto de reacción con el color adecuado en las membranas de las células diana indica una reactividad positiva. En función de la duración de la incubación y de la potencia de la hematoxilina que se haya utilizado, la contratinción puede dar como resultado una coloración azul oscuro, más clara o más oscura, en los núcleos celulares. Una contratinción incompleta o excesiva puede comprometer la correcta interpretación de los resultados.
Si el control de tejido positivo no muestra una tinción positiva, los resultados de las muestras de la prueba se deben considerar no válidos.
Controles tisulares negativos:
El control tisular negativo se debe estudiar después del control de tejido positivo para comprobar el etiquetado específico del antígeno diana mediante el anticuerpo primario. La ausencia de una tinción específica en el control de tejido negativo confirma la falta de reactividad cruzada del anticuerpo con las células o los componentes celulares. Si se lleva a cabo una contratinción del tejido, es posible que se observe tinción alrededor de la parte exterior de la célula, como en los espacios intersticiales. Si se presenta una tinción específica en el control tisular negativo, deberán considerarse no válidos los resultados en la muestra del paciente.
Controles de reactivo negativo:
De presentarse tinción no especifica, tendrá una apariencia difusa. También es posible observar una ligera tinción esporádica en el tejido conjuntivo en aquellas secciones de tejido que se han fijado excesivamente con formol. Se deben utilizar células intactas para la interpretación de los resultados de tinción, ya que la tinción de las células necróticas o degeneradas suele ser no específica.
Tejido del paciente:
Las muestras del paciente se deben examinar en último lugar. La intensidad de la tinción positiva deberá evaluarse en contexto junto con la tinción de fondo del control de reactivo negativo. Como ocurre en todas las pruebas de inmunohistoquímica, un resultado negativo significa que no se ha detectado el antígeno en concreto, no necesariamente que el antígeno no esté presente en las células o en el tejido que se ha usado para el ensayo. Siempre que se vaya a interpretar un resultado de inmunohistoquímica debería examinarse también la morfología de cada muestra de tejido mediante una sección de tejido con tinción de hematoxilina y eosina. La interpretación de las conclusiones morfológicas del paciente y los datos clínicos pertinentes deben dejarse en manos de un anatomopatólogo cualificado.
Un anatomopatólogo cualificado con experiencia en procedimientos de inmunohistoquímica debe evaluar los controles positivos y negativos y calificar el producto con tinción antes de interpretar los resultados.
Convenciones de puntuación para la interpretación del anticuerpo VENTANA anti-HER2 (4B5) Antibody en carcinoma de mama:
En las tablas siguientes, se proporcionan los criterios de tinción. Consulte VENTANA HER2 (4B5) RABBIT MONOCLONAL PRIMARY ANTIBODY RXDX Guía de interpretación de la tinción del carcinoma de mama y del carcinoma gástrico (P/N 1021957ES) para obtener una descripción más detallada con imágenes de la tinción con VENTANA HER2 (4B5) Assay.
Tabla 5. Criterios de intensidad y patrón de tinción de membrana celular con VENTANA HER2 (4B5) Assay en carcinoma de mama cuando se utiliza el procedimiento de tinción indicado en la Tabla 1
|
Patrón de tinción |
Puntuación de HER2 (4B5) (para informar al facultativo responsable del tratamiento del paciente) |
Uso del procedimiento de tinción de la Tabla 1: Estado de notificación recomendado |
Aplicación clínica |
|
No se observa tinción de membrana o bien tinción difusa y parcial de la membrana en el 10% o porcentajes inferiores de células cancerosas |
0 |
Negativo en HER2 |
Ninguno |
|
Tinción difusa y parcial de la membrana en porcentajes superiores al 10% de células cancerosas |
1+ |
Bajo nivel de expresión de HER2 |
ENHERTU® (trastuzumab deruxtecan) |
|
Tinción débil a moderada completa de la membrana con un porcentaje de células cancerosas superior al 10% |
2+* Prueba condicionada: HER2 no amplificado |
Bajo nivel de expresión de HER2 |
|
|
2+* Prueba condicionada: HER2 amplificado |
Sobreexpresión/ positivo en HER2 |
HERCEPTIN (trastuzumab), PERJETA (pertuzumab), KADCYLA (trastuzumab emtansina) |
|
|
Tinción intensa completa de la membrana con un porcentaje de células cancerosas superior al 10% |
3+ |
Sobreexpresión/ positivo en HER2 |
* Se recomienda llevar a cabo una prueba condicionada para evaluar la amplificación del gen según las directrices de ASCO/CAP
Tabla 6. Criterios de intensidad y patrón de tinción de membrana celular con VENTANA HER2 (4B5) RxDx Assay en carcinoma de mama cuando se utiliza el procedimiento de tinción de membrana indicado en la Tabla 2
|
Patrón de tinción |
Puntuación de HER2 (4B5) (para informar al facultativo responsable del tratamiento del paciente) |
Uso del procedimiento de tinción de la Tabla 2: Estado de notificación recomendado |
Aplicación clínica |
|---|---|---|---|
|
No se observa tinción de membrana o bien tinción difusa y parcial de la membrana en el 10% o porcentajes inferiores de células cancerosas |
0 |
Negativo en HER2 |
Ninguno |
|
Tinción difusa y parcial de la membrana en porcentajes superiores al 10% de células cancerosas |
1+ |
Negativo en HER2 |
|
|
Tinción débil a moderada completa de la membrana con un porcentaje de células cancerosas superior al 10% |
2+* Prueba condicionada: HER2 no amplificado |
Negativo en HER2 |
|
|
2+* Prueba condicionada: HER2 amplificado |
Sobreexpresión/ positivo en HER2 |
HERCEPTIN (trastuzumab), PERJETA (pertuzumab), KADCYLA (trastuzumab emtansina) |
|
|
Tinción intensa completa de la membrana con un porcentaje de células cancerosas superior al 10% |
3+ |
Sobreexpresión/ positivo en HER2 |
* Se recomienda llevar a cabo una prueba condicionada para evaluar la amplificación del gen según las directrices de ASCO/CAP
Convenciones de puntuación para la interpretación del anticuerpo VENTANA anti-HER2 (4B5) en carcinoma gástrico:
Para que los carcinomas gástricos se consideren positivos en la sobreexpresión de proteína HER2, deben cumplir los criterios límite en cuanto a la intensidad y el patrón de la tinción de membrana (2+ o superior en una escala de 0 a 3+) y al porcentaje de células tumorales positivas. La tinción debe localizarse en la membrana celular pero no tiene que ser completamente circunferencial, ya que la tinción basolateral se observa con regularidad y debe considerarse para la puntuación. Podría observarse una tinción del citoplasma y/o el núcleo, pero esta tinción no debe incluirse en los criterios de establecimiento de la positividad. En el carcinoma gástrico, el porcentaje de células tumorales positivas depende de si la muestra procede de una biopsia (≥5 de células de cohesión) o de una extirpación quirúrgica de la muestra (≥10%).
A la hora de establecer las directrices para la puntuación de la inmunohistoquímica de HER2 en el cáncer gástrico, cabe destacar que, mientras que una tinción membranosa fuerte demuestra la sobreexpresión de proteína HER2 en las células neoplásicas, no tiene por qué ser una circunferencia completa.36
Se ha registrado el uso de tinción citoplasmática difusa con o sin tinción nuclear en cáncer gástrico.37 Solo debe usarse la tinción membranosa en la determinación de la sobreexpresión de proteína HER2 en el cáncer gástrico.
La tinción inmunohistoquímica con el clon 4B5 puede dar lugar a tinción citoplasmática y nuclear de la mucosa gástrica normal y, en reducidas ocasiones, de las células neoplásicas del carcinoma gástrico y del carcinoma de unión gastroesofágica. La naturaleza de esta tinción nuclear y tinción citoplasmática se desconoce actualmente. El patrón de tinción no debe confundirse con una tinción membranosa discontinua, dado que indica la positividad en HER2 en células neoplásicas.
Consulte la Guía de interpretación VENTANA HER2 (4B5) RABBIT MONOCLONAL PRIMARY ANTIBODY RXDX Guía de interpretación de la tinción del carcinoma de mama y del carcinoma gástrico (P/N 1021957ES) para obtener una descripción más detallada con microfotografías de la tinción con VENTANA HER2 (4B5) Assay.
Tabla 7. Criterios de intensidad y patrón de tinción de membrana celular con VENTANA HER2 (4B5) Assay en carcinoma gástrico cuando se utiliza el procedimiento de tinción indicado en la Tabla 3
|
Patrón de tinción: muestra de extirpación quirúrgica |
Patrón de tinción: muestra de biopsia |
Puntuación (para informar al facultativo responsable del tratamiento del paciente) |
Evaluación de la tinción HER2 |
Aplicación clínica |
|
No se presenta reactividad o reactividad membranosa en < 10% de las células tumorales |
No hay reactividad ni reactividad membranosa en ninguna célula tumoral |
0 |
Negativo en HER2 |
Ninguno |
|
Reactividad membranosa difusa o apenas perceptible en ≥ 10% de las células tumorales; las células son reactivas únicamente en parte de la membrana |
Grupo de células tumorales* con reactividad membranosa difusa o apenas perceptible independientemente del porcentaje de clústers tumorales con tinción |
1+ |
Negativo en HER2 |
|
|
Reactividad membranosa de débil a moderada, completa, basolateral o lateral en ≥ 10% de las células tumorales |
Grupo de clústers tumorales* con reactividad membranosa de débil a moderada, completa, basolateral o lateral independientemente del porcentaje de células tumorales con tinción |
2+** Prueba condicionada: HER2 no amplificado |
Negativo en HER2 |
|
|
2+** Prueba condicionada: HER2 amplificado |
Sobreexpresión/ positivo en HER2 |
HERCEPTIN (trastuzumab) |
||
|
Reactividad membranosa fuerte, completa, basolateral o lateral en ≥ 10% de las células tumorales |
Grupo de clústers tumorales* con reactividad membranosa fuerte, completa, basolateral o lateral independientemente del porcentaje de células tumorales con tinción |
3+ |
Sobreexpresión/ positivo en HER2 |
|
|
* ≥ 5 células de cohesión ** Se recomienda reflejar en ISH |
||||
RESOLUCIÓN DE PROBLEMAS:
1 Si el control positivo presenta una tinción más débil de lo previsto, compruebe el resto de la sesión de control positivo que se han analizado en la misma sesión y el mismo instrumento para establecer si el problema se debe al anticuerpo primario o a alguno de los reactivos secundarios comunes.
2 Si el control positivo es negativo, debe asegurarse de que el portaobjetos lleva la etiqueta de código de barras correcta. Si se ha etiquetado correctamente el portaobjetos, compruebe el resto de los controles positivos que se han analizado en la misma sesión y el mismo instrumento para establecer si el problema se debe al anticuerpo primario o a alguno de los reactivos secundarios comunes. Es posible que los tejidos se hayan recogido, fijado o desparafinado de forma incorrecta. Siga el procedimiento apropiado para llevar a cabo la recogida, la conservación y la fijación.
3 Si no se ha eliminado toda la parafina, es posible que no se presente la tinción. El procedimiento de desparafinado debería repetirse.
4 Si las secciones de tejido se pierden en el portaobjetos, debe comprobar que los portaobjetos tienen carga positiva.
5 Si se observan tinciones nucleares y citoplasmáticas en la mucosa normal cercana al área del tumor del carcinoma gástrico que generan confusión a la hora de interpretar la tinción de membrana, el caso se debe analizar mediante ISH.
6 Si necesita llevar a cabo acciones correctivas, consulte la sección Procedimiento de tinción, el Manual del usuario del instrumento o póngase en contacto con su representante local de asistencia técnica de Roche.
ALMACENAMIENTO Y ESTABILIDAD:
Tras la recepción y cuando no se utilice, consérvese de 2-8°C. No lo congele.
Para garantizar una dispensación adecuada del reactivo y la estabilidad del anticuerpo, vuelva a poner el tapón del dispensador después de cada uso y almacene inmediatamente el dispensador en la nevera, en posición vertical.
Todos los dispensadores de anticuerpos tienen una fecha de caducidad. Si se almacena correctamente, el reactivo se mantendrá estable hasta la fecha indicada en la etiqueta. No usar el reactivo después de la fecha de caducidad.
ADVERTENCIAS Y PRECAUCIONES:
1 Para uso diagnóstico in vitro (IVD).
2 Solo para uso profesional.
3 No utilizar por encima del número especificado de ensayos.
4 Los portaobjetos con carga positiva pueden verse afectados por presiones ambientales, dando lugar a una tinción incorrecta. Póngase en contacto con su representante de servicio de Roche para obtener más información sobre el uso de este tipo de portaobjetos.
5 Los materiales de origen animal o humano deben manipularse como materiales biopeligrosos y eliminarse con las precauciones adecuadas. En caso de exposición, deberán seguirse las directivas sanitarias de las autoridades responsables.30,31
6 Evite el contacto de los reactivos con los ojos y las membranas mucosas. Si los reactivos entran en contacto con zonas sensibles, lávelas con agua abundante.
7 Evite la contaminación microbiana de los reactivos, dado que podría dar lugar a resultados incorrectos.
8 Si se utiliza tal y como se indica en las instrucciones, este producto no se clasifica como una sustancia biopeligrosa. El conservante del reactivo es azida sódica. Los indicios de sobreexposición a esta sustancia pueden ser, entre otros, irritación en la piel y los ojos e irritación en las membranas mucosas y las vías respiratorias altas. La concentración de azida sódica que contiene este producto es 0.05% y no cumple los criterios necesarios para clasificarla como sustancia peligrosa. La acumulación de NaN3 puede reaccionar con los componentes de plomo y cobre de las tuberías y generar ácidas metálicas extremadamente explosivas. Cuando se vaya a eliminar, añada grandes cantidades de agua para evitar la acumulación de azidas en las tuberías.32 Es posible que se presenten reacciones alérgicas sistémicas en personas con mayor sensibilidad.
9 Para obtener más información sobre el uso de este producto, consulte el Manual del usuario del instrumento BenchMark IHC/ISH y las instrucciones de uso de todos los componentes necesarios que puede encontrar en navifyportal.roche.com.
10 Consultar a las autoridades locales y/o estatales sobre el método de eliminación recomendado.
11 El etiquetado de seguridad de los productos sigue principalmente las directrices del SGA de la UE. Está disponible bajo petición la hoja de datos de seguridad para los usuarios profesionales.
12 Para comunicar la sospecha de incidentes graves relacionados con este dispositivo, póngase en contacto con su representante local de servicio Roche y con las autoridades competentes del Estado o País Miembro de residencia del usuario.
MATERIAL SUMINISTRADO:
VENTANA HER2 (4B5) Assay contiene reactivo suficiente para 50 pruebas.
Un dispensador de 5 mL de anticuerpo VENTANA HER2 (4B5) Assay contiene aproximadamente 30 μg de anticuerpo monoclonal de conejo dirigido contra el antígeno HER2 humano.
El anticuerpo se diluye en un tampón salino formado por 0.05 M Tris, 0.01 M EDTA y Brij-35 al 0.05% con una proteína transportadora al 0.3% y azida sódica al 0.05%, un conservante. Existen trazas de aproximadamente un 0.25% de suero bovino fetal de la solución de partida.
La concentración del anticuerpo específico es aproximadamente de 6 μg/mL.
El anticuerpo VENTANA HER2 (4B5) es un IgG de conejo diluido de sobrenadantes de cultivo tisular.
Consulte la hoja de datos del kit de detección VENTANA correspondiente para obtener descripciones detalladas de los siguientes aspectos: Principio del procedimiento, Material y métodos, Recogida y preparación de muestras para análisis, Procedimientos de control de calidad, Resolución de problemas, Interpretación de los resultados y Limitaciones.
MATERIALES NECESARIOS PERO NO SUMINISTRADOS
No se suministran reactivos de tinción, como kits de detección VENTANA, ni componentes auxiliares, incluyendo portaobjetos de control de tejido negativos y positivos.
Puede que no todos los productos que aparecen en la hoja de datos estén disponibles en todos los lugares. Consulte al representante local de asistencia técnica de Roche.
No se suministran los reactivos y materiales siguientes, pero pueden ser necesarios para la tinción:
1 Tejido de control recomendado
2 Portaobjetos para microscopio con carga positiva
3 CONFIRM Negative Control Rabbit Ig (n.º cat. 760-1029 / 05266238001)
4 ultraView DAB Detection Kit (n.º cat. 760-500 / 05269806001)
5 EZ Prep Concentrate (10X) (n.º cat. 950-102 / 05279771001)
6 Reaction Buffer Concentrate (10X) (n.º cat. 950-300 / 05353955001)
7 LCS (Predilute) (n.º cat. 650-010 / 05264839001)
8 ULTRA LCS (Predilute) (n.º cat. 650-210 / 05424534001)
9 Cell Conditioning Solution (CC1) (n.º cat. 950-124 / 05279801001)
10 ULTRA Cell Conditioning Solution (ULTRA CC1) (n.º cat. 950-224 / 05424569001)
11 Hematoxylin II (n.º cat. 790-2208 / 05277965001)
12 Bluing Reagent (n.º de cat. 760-2037 / 05266769001)
13 Equipo de laboratorio de uso general
14 Instrumento BenchMark IHC/ISH
CONTROL TISULAR POSITIVO:
Se debe analizar un tejido de control positivo que se haya fijado y procesado de la misma forma que las muestras del paciente con cada conjunto de condiciones de la prueba y en cada uno de los procedimientos de tinción con VENTANA HER2 (4B5) Assay que se lleven a cabo. La práctica de laboratorio óptima consiste en incluir una sección de control positivo en el mismo portaobjetos que la muestra de la prueba. Esto contribuye a identificar fallos al aplicar los reactivos al portaobjetos. Un tejido con una tinción débil positiva es más adecuado para el control de calidad. El tejido de control puede contener elementos de tinción tanto positiva como negativa y ambos sirven como control positivo y negativo. El tejido de control debe ser una muestra de autopsia reciente, biopsia o cirugía, preparada o fijada con la mayor brevedad con un proceso idéntico al de las secciones de prueba.
Estos tejidos se utilizan para hacer un seguimiento de todos los pasos que conlleva el proceso, desde la preparación del tejido hasta la tinción. El uso de una sección de tejido fijada o procesada de forma diferente a la muestra de la prueba actúa como control en todos los pasos de reactivo y del método, salvo en los de fijación y preparación de tejidos. La opción más idónea es elegir un tejido que se caracterice por una tinción débil pero positiva, para garantizar que el sistema es sensible a pequeños niveles de degradación de reactivos o a problemas con la metodología IHC. Sin embargo, habitualmente los tejidos neoplásicos positivos en HER2 suelen dar como resultado una tinción positiva fuerte dada la naturaleza de la patología (sobreexpresión).
Los controles de tejido positivos conocidos solo se deben usar para monitorizar el comportamiento correcto de los reactivos y los instrumentos, y no como ayuda para establecer un diagnóstico específico de las muestras de prueba. Si los controles de tejido positivos no muestran una tinción positiva, los resultados de las muestras de la prueba se deben considerar no válidos.
Como ejemplo de tejido de control positivo para VENTANA HER2 (4B5) Assay se encuentra el tejido de carcinoma de mama invasivo positivo o las muestras de carcinoma gástrico con positividad débil conocida en HER2. Los componentes de tinción positiva del tejido (tinción membranosa de células neoplásicas) sirven para comprobar que el anticuerpo se ha aplicado y el instrumento ha funcionado correctamente.
Control tisular negativo:
El mismo portaobjetos que se utiliza como control de tejido positivo (carcinoma ductal o lobulillar invasivo de mama o carcinoma gástrico) puede servir como control tisular negativo. En los componentes sin tinción (como el estroma, las células linfáticas y los vasos sanguíneos circundantes) debería observarse una absoluta ausencia de tinción específica y deberían ofrecer una indicación sobre la tinción de fondo específica (falso positivo) con el anticuerpo primario. Utilice un tejido negativo conocido, fijado, procesado y embebido de forma idéntica a la muestra del paciente.
Control de reactivo negativo:
Se debe utilizar el control de reactivo negativo de cada muestra en cada sesión como ayuda para la interpretación de los resultados. Para evaluar la tinción no especifica se utiliza un control de reactivo negativo en lugar del anticuerpo primario. La tinción del portaobjetos debería realizarse con CONFIRM Negative Control Rabbit Ig. El periodo de incubación del control de reactivo negativo debe ser idéntico al del periodo de incubación del anticuerpo primario.
Discrepancias no explicadas:
Las discrepancias no explicadas en los controles deberían comunicarse al representante local de asistencia técnica de Roche de forma inmediata. Si los resultados de los controles de calidad no cumplen las especificaciones, los resultados del paciente no serán válidos. Consulte la sección sobre Resolución de problemas. Identifique el problema y corríjalo; a continuación, repita las muestras del paciente.
Verificación del ensayo:
Antes de comenzar a utilizar un anticuerpo o un sistema de tinción en un procedimiento diagnóstico, se debe comprobar la especificidad del anticuerpo mediante pruebas en una serie de tejidos que contengan características de rendimiento en inmunohistoquímica conocidas y que reflejen tejidos positivos y negativos conocidos (consulte la sección Procedimientos de control de calidad que se ha mencionado anteriormente y que se encuentra en la hoja de datos del producto y las recomendaciones sobre control de calidad de College of American Pathologists Laboratory Accreditation Program, Anatomic Pathology Checklist34, CLSI Approved Guideline35 o todos ellos). Estos procedimientos de control de calidad deben repetirse para cada nuevo lote de anticuerpo nuevo o cuando se produzca un cambio en los parámetros del ensayo. Los tejidos de cáncer de mama y gástrico con un estado de HER2 conocido son adecuados para la verificación del ensayo.
PROCEDIMIENTOS DE CONTROL DE CALIDAD:
Controles de líneas celulares:
VENTANA pone a su disposición, por separado, cuatro controles de líneas celulares fijados con formol y embebidos en parafina, cortados y colocados en un único portaobjetos cargado. PATHWAY HER-2 4 in 1 Control Slides (con n.º de referencia 781-2991) pueden resultar útiles a la hora de llevar a cabo la validación preliminar del instrumento o del método de procesamiento que se utiliza para la tinción de los portaobjetos con VENTANA HER2 (4B5). Estos cuatros controles de líneas celulares se caracterizan por la hibridación in situ para el número de copia del gen, Tabla 4. Cuando se procesan y se tiñen adecuadamente, la tinción de las líneas celulares debería aparecer tal y como se describe en la hoja de datos de PATHWAY HER-2 4 in 1 Control Slide. Si la tinción indicada no es evidente en las secciones correspondientes, concretamente en los controles 1+ y 2+, debería repetirse la tinción de los tejidos.
Tabla 4. Características de PATHWAY HER-2 4 in 1 Control Slides
|
Puntuación IHC de HER2 |
Estirpe celular |
Proporción HER2/Chr17* |
|
0 |
MDA-MB-231 |
1.11 |
|
1+ |
T47D |
1.12 |
|
2+ |
MDA-MB-453 |
2.66 |
|
3+ |
BT-474 |
5.53 |
* La proporción HER2/Chr17 es la media de tres lotes de PATHWAY HER-2 4 in 1 Control Slides obtenida mediante hibridación in situ con fluorescencia (FISH).
CARACTERÍSTICAS DE RENDIMIENTO:
Se realizaron pruebas de tinción para evaluar la sensibilidad, especificidad y precisión y los resultados se indican a continuación.
RENDIMIENTO CLÍNICO:
Cáncer de mama con nivel bajo de HER2
Estudio de resultados clínicos: DESTINY-BREAST04
DESTINY-BREAST04 es un estudio en fase III, multicéntrico, aleatorizado, abierto y con control activo que se llevó a cabo para evaluar la seguridad y la eficacia de trastuzumab deruxtecan (ENHERTU®) en pacientes con cáncer de mama inoperable y/o metastásico con bajos niveles de expresión de HER2.
Para poder ser elegibles a la inclusión en el estudio, los tumores tenían que demostrar bajos niveles de la expresión HER2 determinada usando IHC con el anticuerpo anti-HER2 (4B5).
Se consideró que un tumor con una puntuación IHC de HER2 de 1+ para indicar un estado de HER2 bajo. El tumor también se consideraba indicativo de bajos niveles de HER2 si su puntuación de IHC de HER2 era de 2+ y la prueba condicionada con el ensayo INFORM HER2 Dual ISH señalaba la ausencia de amplificación del gen HER2 (ISH-). Los pacientes inscritos fueron aleatorizados en una proporción 2:1 para el tratamiento con trastuzumab deruxtecan (ENHERTU®) o con el tratamiento de quimioterapia a elección del facultativo. La puntuación de nivel bajo de HER2 (IHC 1+ or IHC 2+/ISH-), obtenida de forma centralizada, era uno de los tres factores de estratificación que se emplearon para la aleatorización de los pacientes en el estudio.
Los análisis de eficacia se llevaron a cabo en el conjunto completo de análisis y en la población positiva al receptor hormonal (positivos en receptor de estrógeno y/o receptor de progesterona).
En el análisis primario se analizó la supervivencia libre de progresión (PFS) basada en la evaluación de la revisión central, independiente y enmascarada (BICR) en el subconjunto positivo en el receptor hormonal con estratificación según la puntuación/el estado de nivel bajo de HER2 (IHC 1+ o IHC 2+/ISH-) evaluado de forma centralizada, la cantidad de líneas previas de quimioterapia (1 o 2) y el tratamiento anterior con inhibidores dependientes de la ciclina (CDK) 4/6 (sí o no). El tratamiento con trastuzumab deruxtecan (ENHERTU®) se asoció a un aumento estadísticamente significativo y con importancia clínica en el PFS, así como una supervivencia global (OS) en esta población en comparación con el tratamiento de elección del médico.
Tabla 39. PFS y OS según BIRC en la población con receptor de hormona positiva y conjunto completo de análisis (DESTINY-BREAST04)
|
Parámetro |
Población positiva en el receptor hormonal |
Conjunto completo de análisis |
||
|
Trastuzumab deruxtecan (ENHERTU®) N = 331 |
Tratamiento seleccionado por el facultativo N = 163 |
Trastuzumab deruxtecan (ENHERTU®) N = 373 |
Tratamiento seleccionado por el facultativo N = 184 |
|
|
PFS medio a (CI del 95%) |
10.1 (9.5, 11.5) |
5.4 (4.4, 7.1) |
9.9 (9.0, 11.3) |
5.1 (4.2, 6.8) |
|
Índice de riesgo b [CI del 95%] |
0.51 (0.40, 0.64) |
0.50 (0.40, 0.63) |
||
|
Valor p c |
< 0.0001 |
< 0.0001 |
||
|
Supervivencia global (OS) |
||||
|
OS media a (CI del 95%) |
23.9 (20.8, 24.8) |
17.5 (15.2, 22.4) |
23.4 (20.0, 24.8) |
16.8 (14.5, 20.0) |
|
Índice de riesgo b [CI del 95%] |
0.64 (0.48, 0.86) |
0.64 (0.49, 0.84) |
||
|
Valor p c |
0.0028 |
0.0010 |
||
CI = Intervalo de confianza, PFS = Supervivencia libre de progresión, OS = Supervivencia global.
a Las medias de PFS y OS son estimaciones a partir del análisis Kaplan-Meier. Los CI del 95% bilaterales de la media de PFS y OS se calcularon con el método Brookmeyer- Crowley.
b Según el modelo de riesgo proporcional estratificado de Cox. Los factores de estratificación fueron la puntuación de nivel bajo de HER2, la cantidad de líneas previas de quimioterapia y el tratamiento anterior con inhibidores de la quinasa 4/6 dependientes de la ciclina (en el caso del conjunto de análisis completo y de los casos positivos en el receptor hormonal) o bien del estado del receptor hormonal/quinasa dependiente de la ciclina (en el caso del conjunto completo de análisis).
c Valor p de la prueba del orden logarítmico estratificado.
Cáncer de mama con positividad en HER2:
Estudios comparativos del anticuerpo monoclonal de conejo del clon 4B5 con el anticuerpo monoclonal de ratón PATHWAY anti-HER2 (CB11) Mouse Monoclonal Antibody en cáncer de mama:
Se llevó a cabo un estudio comparativo del método para evaluar la relación del clon 4B5 con el anticuerpo PATHWAY anti-HER2 (CB11) Mouse Monoclonal Antibody (anticuerpo PATHWAY anti-HER2 (CB11)) y PathVysion HER2 FISH, siendo ambos pruebas diagnósticas homologadas previamente. Seis investigadores participaron en el estudio. Al estudio se incorporaron dos cohortes independientes de muestras de cáncer de mama invasivo: una con 178 muestras procedentes de Cleveland Clinic Foundation (Cohorte 1) y otra con 144 muestras extraídas por IMPATH Predictive Oncology procedentes de distintos sitios internacionales (Cohorte 2). Dos conjuntos de tres investigadores diferentes evaluaron las dos cohortes independientes (Cohorte 1:n = 178, Cohorte 2:n = 144) mediante casos conocidos de cáncer de mama teñidos con el anticuerpo PATHWAY anti-HER2 (CB11) y el clon 4B5. Los datos FISH se obtuvieron del historial del paciente. Se creó una puntuación de consenso entre los tres lectores en cada anticuerpo para evitar la conocida variabilidad del lector que se produce en la puntuación de HER2.42,43,44 Se evaluaron un total de 322 casos. Los portaobjetos teñidos con el anticuerpo PATHWAY anti-HER2 (CB11) se procesaron y tiñieron siguiendo las instrucciones del fabricante especificadas en la hoja de datos del anticuerpo PATHWAY anti-HER2 (CB11). Había transcurrido un periodo medio de aproximadamente un año entre la tinción y la lectura de los portaobjetos con tinción del anticuerpo PATHWAY anti- HER2 (CB11) A continuación se exponen los resultados con importancia clínica (positivo/negativo) del ensayo de IHC de HER2 (4B5) y del ensayo de IHC PATHWAY anti-HER2 (CB11) en las dos cohortes:
Tabla 40. Puntuaciones con importancia clínica: Ensayos IHC de la Cohorte 1
|
Anticuerpo HER2 (4B5) |
Anticuerpo PATHWAY anti-HER2 (CB11) |
||
|---|---|---|---|
|
Positiva |
Negativa |
Total |
|
|
Positiva |
86 |
5 |
91 |
|
Negativa |
7 |
80 |
87 |
|
Total |
93 |
85 |
178 |
|
n/N |
Porcentaje (%) (Intervalo de confianza del 95%) |
||
|
Porcentaje de concordancia positiva |
86/93 |
92.5 (85.2-96.9) |
|
|
Porcentaje de concordancia negativa |
80/85 |
94.1 (86.8-98.1) |
|
|
Porcentaje de concordancia global |
166/178 |
93.3 (88.5-96.4) |
|
|
Los resultados con importancia clínica se consideraban positivos por IHC (2+ y 3+) y negativos (0+ y 1+) |
|||
Tabla 41. Puntuaciones con importancia clínica: Ensayos IHC de la Cohorte 2
|
Anticuerpo HER2 (4B5) |
Anticuerpo PATHWAY anti-HER2 (CB11) |
||
|
Positiva |
Negativa |
Total |
|
|
Positiva |
69 |
22 |
91 |
|
Negativa |
0 |
53 |
53 |
|
Total |
69 |
75 |
144 |
|
n/N |
Porcentaje (%) (Intervalo de confianza del 95%) |
||
|
Porcentaje de concordancia positiva |
69/69 |
100 % (97.5-100 %) |
|
|
Porcentaje de concordancia negativa |
53/75 |
70.6 (58.5-80.1) |
|
|
Porcentaje de concordancia global |
122/144 |
84.7 (78.2-90.0) |
|
|
Los resultados con importancia clínica se consideraban positivos por IHC (2+ y 3+) y negativos (0+ y 1+) |
|||
Los resultados con base en IHC del ensayo IHC de HER2 (4B5) se compararon también con los resultados del ensayo PathVysion HER2 FISH. Los resultados positivos por IHC se corresponden con las puntuaciones de IHC 2+ o 3+ y los resultados positivos por FISH se corresponden con los casos que han presentado la amplificación del gen HER2. A continuación se recogen los datos de concordancia del ensayo de IHC con el clon 4B5 en comparación con los resultados por FISH de las dos cohortes:
Tabla 42. Concordancia con importancia clínica: IHC con FISH de la Cohorte 1
|
Anticuerpo HER2 (4B5) |
n/N |
Porcentaje (%) (Intervalo de confianza del 95%) |
|
Porcentaje de concordancia positiva |
83/93 |
89.2 (82.5-95.1) |
|
Porcentaje de concordancia negativa |
77/85 |
90.6 (84.0-96.4) |
|
Porcentaje de concordancia global |
160/178 |
90.0 (85.4-93.6) |
Tabla 43. Concordancia con importancia clínica: IHC con FISH de la Cohorte 2
|
Anticuerpo HER2 (4B5) |
n/N |
Porcentaje (%) (Intervalo de confianza del 95%) |
|
Porcentaje de concordancia positiva |
80/86 |
93.0 (87.9-96.3) |
|
Porcentaje de concordancia negativa |
47/58 |
81.0 (73.4-86.0) |
|
Porcentaje de concordancia global |
127/144 |
88.2 (82.1-92.2) |
Conclusiones: Este estudio demuestra que existe una concordancia de importancia clínica (concordancia global entre los resultados positivos/negativos) entre el ensayo del clon 4B5 y el ensayo PATHWAY anti-HER2 (CB11), probando en consecuencia que el ensayo con el anticuerpo VENTANA HER2 (4B5) RxDx es una alternativa aceptable a PATHWAY anti-HER2 (CB11) Assay que sirve como ayuda en la evaluación de las pacientes con cáncer de mama para las que se está planteando la posibilidad de administrar un tratamiento con trastuzumab (Herceptin). Este estudio también demuestra que los resultados de la expresión HER2 obtenidos del ensayo IHC del clon 4B5 son comparables a los resultados del estado del gen HER2 determinados por el análisis FISH.
Comparación con el ensayo de inscripción en los estudios de PERJETA (pertuzumab) y KADCYLA (trastuzumab emtansina) en carcinoma de mama:
La concordancia con los ensayos de inscripción en las cohortes de los estudios de PERJETA y KADCYLA se determinó mediante la tinción de las muestras del ensayo con VENTANA HER2 (4B5) Assay. Se evaluó un total de 2753 muestras para el ensayo PERJETA y 99 muestras evaluadas para el ensayo KADCYLA se tiñeron con VENTANA HER2 (4B5) Assay. Se determinaron tipos de concordancia (PPA, NPA y OPA). Los CI del 95% (intervalos de confianza del 95% bilaterales) se calcularon mediante el método de puntuación.
Tabla 44. Concordancia de los ensayos del clon 4B5 y Dako en cuanto al estado de HER2 en todos los sujetos HER2 cuya evaluación fue posible. Los sujetos que pudieron someterse a evaluación mediante IHC obtuvieron un estado de HER2 Positivo o Negativo que se determinó mediante el clon 4B5 y el ensayo de IHC de inscripción
|
Estudio |
Puntuación del clon 4B5 b |
Estado Dako HER2 a,b |
||
|
Positiva |
Negativa |
Total |
||
|
Perjeta y kadcyla |
3+ |
2380 |
15 |
2395 |
|
2+ |
140 |
122 |
262 |
|
|
0/1+ |
38 |
135 |
173 |
|
|
Total |
2558 |
272 |
2830 |
|
|
Porcentaje de concordancia positiva Porcentaje, n/N (CI del 95%) |
2380/2558 (93.0) (92.0-94.0) |
|||
|
Porcentaje de concordancia negativa Porcentaje, n/N (CI del 95%) |
257/272 (94.5) (91.1-96.6) |
|||
|
Porcentaje de concordancia global Porcentaje, n/N (CI del 95%) |
2637/2830 (93.2) (92.2-94.1) |
|||
a Positiva = IHC positiva y/o ISH amplificada. Negativa = IHC negativa y ISH no amplificada o ISH no amplificada y IHC no positiva.
b IHC: Positiva = 3+; Negativa = 0, 1+ o 2+.
Tabla 45. Concordancia de los ensayos del clon 4B5 y Dako en cuanto al estado de IHC en todos los sujetos cuya evaluación mediante IHC fue posible. Los sujetos que pudieron someterse a evaluación mediante IHC obtuvieron un estado de HER2 Positivo o Negativo que se determinó mediante el clon 4B5 y el ensayo de IHC de inscripción
|
Estudio |
Estado del clon 4B5 a |
Estado Dako HercepTest a |
||
|
Positiva |
Negativa |
Total |
||
|
PERJETA y KADCYLA |
Positiva |
2330 |
65 |
2395 |
|
Negativa |
21 |
414 |
435 |
|
|
Total |
2351 |
479 |
2830 |
|
|
Porcentaje de concordancia positiva Porcentaje, n/N (CI del 95%) |
2330/2351 (99.1) (98.6-99.4) |
|||
|
Porcentaje de concordancia negativa Porcentaje, n/N (CI del 95%) |
414/479 (86.4) (83.1-89.2) |
|||
|
Porcentaje de concordancia global Porcentaje, n/N (CI del 95%) |
2744/2830 (97.0) (96.3-97.5) |
|||
|
PERJETA |
Positiva |
2267 |
63 |
2330 |
|
Negativa |
10 |
399 |
4090 |
|
|
Total |
2277 |
462 |
2739 |
|
|
Porcentaje de concordancia positiva Porcentaje, n/N (CI del 95%) |
2267/2277 (99.6) (99.2-99.8) |
|||
|
Porcentaje de concordancia negativa Porcentaje, n/N (CI del 95%) |
399/462 (86.4) (82.9-89.2) |
|||
|
Porcentaje de concordancia global Porcentaje, n/N (CI del 95%) |
2666/2739 (97.3) (96.7-97.9) |
|||
|
KADCYLA |
Positiva |
63 |
2 |
65 |
|
Negativa |
11 |
15 |
26 |
|
|
Total |
74 |
17 |
91 |
|
|
Porcentaje de concordancia positiva Porcentaje, n/N (CI del 95%) |
63/74 (85.1) (75.3-91.5) |
|||
|
Porcentaje de concordancia negativa Porcentaje, n/N (CI del 95%) |
15/17 (88.2) (65.7-96.7) |
|||
|
Porcentaje de concordancia global Porcentaje, n/N (CI del 95%) |
78/91 (85.7) (77.1-91.5) |
|||
a Positiva = 3+; Negativa = 0,1+ o 2+.
Tabla 46. Concordancia de los ensayos del clon 4B5 y Dako en cuanto a la puntuación de IHC en todos los sujetos cuya evaluación mediante IHC fue posible. Los sujetos que pudieron someterse a evaluación mediante IHC obtuvieron un estado de HER2 Positivo o Negativo que se determinó mediante el clon 4B5 y el ensayo de IHC de inscripción
|
Estudio |
Puntuación del clon 4B5 |
Estado Dako HercepTest a |
|||
|---|---|---|---|---|---|
|
3+ |
2+ |
0/1+ |
Total |
||
|
PERJETA y KADCYLA |
3+ |
2330 |
64 |
1 |
2395 |
|
2+ |
12 |
235 |
15 |
262 |
|
|
0/1+ |
9 |
26 |
138 |
173 |
|
|
Total |
2351 |
479 |
2830 |
||
|
Porcentaje de concordancia global n/N (%) (CI del 95%) |
2703/2830 (95.5) (94.7-96.2) |
||||
|
PERJETA |
3+ |
2267 |
62 |
1 |
2330 |
|
2+ |
9 |
226 |
13 |
248 |
|
|
0/1+ |
1 |
24 |
135 |
161 |
|
|
Total |
2277 |
312 |
150 |
2736 |
|
|
Porcentaje de concordancia global n/N (%) (CI del 95%) |
2629/2739 (96.0) (95.2-96.7) |
||||
|
KADCYLA |
3+ |
63 |
2 |
0 |
65 |
|
2+ |
3 |
9 |
2 |
14 |
|
|
0/1+ |
8 |
2 |
2 |
12 |
|
|
Total |
74 |
13 |
4 |
91 |
|
|
Porcentaje de concordancia global n/N (%) (CI del 95%) |
74/91 (81.3) (72.1-88.0) |
||||
Tabla 47. Aceptabilidad de la tinción del clon 4B5. Sujetos sometidos a pruebas de IHC. La tinción IHC se considera aceptable si se puede asignar una puntuación de IHC válida (0,1+, 2+ o 3+). Entro los motivos que pueden convertir la tinción en inaceptable figuran un control negativo inaceptable, la pérdida del tejido, cantidad de tumor insuficiente, tinción de fondo inaceptable y una morfología inaceptable
|
Parámetro |
PERJETA |
KADCYLA |
PERJETA y KADCYLA |
|
Cantidad de pruebas IHC iniciales |
2753 |
99 |
2852 |
|
Aceptabilidad de la tinción inicial n/N (%) (CI del 95%) |
2708/2753 (98.4) (97.8, 98.8) |
92/99 (92.9) (86.1, 96.5) |
2800/2852 (98.2) (97.6, 98.6 ) |
|
Cantidad de pruebas IHC repetidas |
40 |
0 |
40 |
|
Aceptabilidad de la tinción final n/N (%) (CI del 95%) |
2746/2753 (99.7) (99.5, 99.9) |
92/99 (92.9) (86.1, 96.5) |
2838/2852 (99.5) (99.2, 99.7 ) |
Estudio de los resultados clínicos: KATHERINE
Se investigó el rendimiento del clon 4B5 de HER2 y de INFORM HER2 Dual ISH DNA Probe Cocktail (ensayo INFORM HER2 Dual ISH) en KATHERINE (BO27938), un estudio clínico multicéntrico, abierto y aleatorizado de fase III para evaluar la eficacia y la seguridad de trastuzumab emtansina (KADCYLA) frente a trastuzumab (Herceptin) como tratamiento adyuvante en pacientes con cáncer de mama primario positivo en HER2 que padecían un tumor residual patológico presente en la mama o los ganglios linfáticos axilares tras un tratamiento preoperatorio (NCT01772472).
Las muestras del paciente se tiñeron con el clon 4B5 y/o INFORM HER2 Dual ISH y se evaluaron para la aceptabilidad de la tinción y el estado HER2. En general, la mayoría de muestras consistían en una biopsia previa al tratamiento (80.9%), se recopilaron principalmente como una biopsia (75.3%) o por métodos quirúrgicos (24.3%). La mayor parte de las muestras presentaban subtipo neoplásico ductal (95.4%) y la mayoría no se habían obtenido de una muestra metastásica (96.2%).
En el estudio KATHERINE se incluyeron 1486 pacientes cuyos resultados mostraban positividad en HER2, con cáncer de mama en etapa temprana con un tumor invasivo residual en la mama o en los ganglios linfáticos axilares y que habían llevado a cabo un tratamiento con taxano y trastuzumab como parte del régimen neoadyuvante antes de la incorporación al ensayo. Los pacientes recibieron radioterapia y/o terapia hormonal simultáneamente al tratamiento del estudio según las directrices locales. Las muestras de tumor de mama debían presentar sobreexpresión de HER2, definida como una puntuación 3+ en IHC o una proporción de amplificación ≥ 2.0 de ISH, que se había establecido en un laboratorio central. Se aleatorizaron los pacientes (1:1) para recibir trastuzumab o KADCYLA. La aleatorización se estratificó por la fase clínica de su presentación, por el estado del receptor de hormona, por la terapia preoperatoria dirigida contra HER2 (trastuzumab, trastuzumab con otros agentes dirigidos contra HER2) y por el estado patológico nodal evaluados con posterioridad al tratamiento preoperatorio.
El criterio de valoración de la eficacia del estudio KATHERINE fue la supervivencia sin cáncer invasivo (IDFS). IDFS se definió como el periodo de tiempo desde la fecha de la aleatorización a la primera presentación de una recidiva de un tumor ipsilateral invasivo de mama, una recidiva del cáncer de mama invasivo ipsilateral local o regional, una metástasis a distancia, un cáncer de mama contralateral invasivo o la muerte por cualquier motivo.
Se observó una mejoría clínica y estadísticamente significativa en la IDFS en los pacientes cuyas muestras de cáncer de mama se identificaron como positivas en HER2 con el ensayo IHC del clon 4B5, que recibieron trastuzumab emtansina (KADCYLA) en comparación con trastuzumab (Herceptin) (HR = 0.43, CI del 95% [0.32, 0.58]), lo que corresponde a una reducción del 57% del riesgo de un evento de IDFS. Los resultados de eficacia para el subgrupo positivo mediante IHC se muestran en la Tabla 48.
El análisis de los datos también demuestra que, con o sin el ajuste por muestreo diferencial en la población del estudio debido a la preselección local de las pruebas, las estimaciones de eficacia del fármaco son similares.
Tabla 48. Resultados de eficacia de KATHERINE en el subgrupo positivo mediante IHC
|
KADCYLA N= 573 |
Trastuzumab N= 559 |
|
|
Criterio de valoración primario |
Supervivencia sin cáncer invasivo (IDFS) a |
|
|
Número (%) de pacientes con evento |
64 (11.2%) |
130 (23.3%) |
|
HR [CI del 95%] |
0.43 [0.32, 0.58] |
|
|
Índice de casos sin eventos en 3 años (%) b |
89.0 |
75.7 |
a Datos recogidos del primer análisis provisional
b El índice de casos sin eventos en 3 años se calculó mediante estimaciones Kaplan- Meier
Los datos del estudio KATHERINE indican que el tratamiento adyuvante con trastuzumab emtansina (KADCYLA) demostró ser claramente beneficioso en comparación con el tratamiento con trastuzumab (Herceptin) en pacientes con cáncer de mama en etapa temprana positivos a HER2 y enfermedad residual tras la finalización de un tratamiento neoadyuvante. El clon 4B5 de HER2 y los ensayos INFORM HER2 Dual ISH resultan útiles para identificar a pacientes que podrían beneficiarse del tratamiento con trastuzumab emtansina (KADCYLA).
Cáncer gástrico:
Comparación del clon 4B5 con HercepTest en cáncer gástrico humano:
Se llevó a cabo un estudio externo y enmascarado para comparar el rendimiento de la tinción con el clon 4B5 en un instrumento BenchMark XT con la del ensayo Dako HercepTest. Se estudiaron dos cohortes de muestras, (1) microarrays de tejido de nueva construcción (TMAs) que contienen 248 casos de cáncer gástrico (posteriormente se encontraron seis casos duplicados y se eliminaron), y (2) un subgrupo de 183 muestras de ensayos clínicos de trastuzumab para el ensayo de cáncer gástrico (ToGA) que investigaban el estado HER2 y el resultado clínico en pacientes tratados con Herceptin (trastuzumab). El laboratorio tiñó los casos con el clon 4B5 y HercepTest. Se tiñeron un total de 431 casos con ambos ensayos y (una vez que se habían retirado los casos duplicados) se incorporaron 398 casos únicos a la comparación. Un anatomopatólogo asignó la puntuación a los casos con una escala de 0/1+, 2+ y 3+. Los casos positivos debían tener una puntuación de 2+ y 3+ y los negativos, de 0 y 1+. Los índices de concordancia entre el clon 4B5 y HercepTest, para las dos cohortes en estudio, se recogen en la tabla que aparece a continuación.
Tabla 49. Datos de concordancia del clon 4B5 (IHC) frente a HercepTest en carcinoma gástrico
|
Origen del tejido |
Porcentaje de concordancia global (CI del 95%) |
Porcentaje de concordancia positiva (CI del 95%) |
Porcentaje de concordancia negativa (CI del 95%) |
|
TMA a y ToGA b |
91.0 (87.7-93.4) |
82.1 (70.2-90.0) |
92.4 (89.1-94.8) |
|
n |
362 / 398 |
46 / 56 |
316 / 342 |
Los resultados de IHC se consideraban positivos en anticuerpo (2+ y 3+) y negativos (0+ y 1+).
a TMA: muestras de tejido de microarray
b ToGA: muestras de ensayo clínico del ensayo ToGA
RENDIMIENTO DE ANÁLISIS:
Sensibilidad y especificidad:
La sensibilidad y especificidad de VENTANA HER2 (4B5) Assay se determinó mediante un estudio que presentaba tinción de membrana no específica en la mayor parte de los tejidos normales. Los resultados de tinción se muestran en la Tabla 8. La sensibilidad y especificidad de VENTANA HER2 (4B5) Assay también se determinó mediante un estudio que presentaba tinción de membrana no específica en la mayor parte de los tejidos neoplásicos. Los resultados de tinción se muestran en la Tabla 9. La tinción para comprobar la sensibilidad y la especificidad se llevó a cabo con el protocolo iVIEW DAB Detection Kit en un instrumento BenchMark XT o con el protocolo ultraView Universal DAB Detection Kit en el caso del instrumento BenchMark ULTRA.
La tinción positiva en el epitelio tonsilar, epitelio esofágico, próstata, vejiga, nervio periférico, paratiroideo, cáncer de mama, adenocarcinoma de estómago, colon y cáncer ovárico coincide con la documentación publicada sobre la expresión de HER2.
Tabla 8. La sensibilidad/especificidad de VENTANA HER2 (4B5) Assay se determinó analizando tejidos normales FFPE
|
Tejido |
N.º de casos positivos/ total |
Tejido |
N.º de casos positivos/ total |
|---|---|---|---|
|
Cerebro |
0/6 |
Intestino delgado |
0/6 |
|
Cerebelo |
0/6 |
Colon |
0/46 |
|
Glándula suprarrenal |
0/6 |
Hígado |
0/6 |
|
Ovario |
0/6 |
Glándula salival |
0/3 |
|
Páncreas |
0/6 |
Lengua |
0/3 |
|
Ganglio linfático |
0/12 |
Riñón |
0/6 |
|
Glándula pituitaria |
0/5 |
Próstata |
1/6 |
|
Testículos |
0/6 |
Vejigab |
3/3 |
|
Tiroides |
0/6 |
Recto |
0/6 |
|
Mama |
0/14 |
Glándula paratiroideac |
4/6 |
|
Bazo |
0/6 |
Endometrio |
0/3 |
|
Amígdalaa |
3/6 |
Útero |
0/3 |
|
Timo |
0/5 |
Cuello del útero |
0/5 |
|
Médula ósea |
0/3 |
Endocérvix |
0/1 |
|
Pulmón |
0/6 |
Músculo esquelético |
0/6 |
|
Corazón |
0/5 |
Piel |
0/6 |
|
Pericardio |
0/3 |
Nervio |
2/6 |
|
Esófago |
1/6 |
Mesotelio |
0/3 |
|
Estómago |
0/11 |
N/A |
N/A |
a Tinción focal de la superficie de las células epiteliales
b Tinción membranosa de las células superficiales en paraguas
c Tinción focal de membrana
Tabla 9. La sensibilidad/especificidad de VENTANA HER2 (4B5) Assay se determinó analizando una variedad de tejidos neoplásicos FFPE
|
Patología |
N.º de casos positivos/total |
|---|---|
|
Glioblastoma (cerebro) |
0/2 |
|
Meningioma (cerebro) |
0/1 |
|
Oligodendroglioma (cerebro) |
0/1 |
|
Adenocarcinoma seroso (ovario) |
0/2 |
|
Carcinoma, sin especificar (ovario) |
1/2 |
|
Neoplasia neuroendocrina (páncreas) |
0/1 |
|
Adenocarcinoma (páncreas) |
0/1 |
|
Carcinoma, sin especificar (páncreas) |
0/3 |
|
Seminoma (testículos) |
0/1 |
|
Carcinoma embrionario (testículos) |
0/1 |
|
Carcinoma medular (tiroides) |
0/1 |
|
Carcinoma papilar (tiroides) |
0/1 |
|
Carcinoma, sin especificar (tiroides) |
0/3 |
|
Carcinoma ductal microinvasivo (mama) |
2/2 |
|
Carcinoma ductal invasivo (mama) |
44/98 |
|
Carcinoma, sin especificar (mama) |
1/4 |
|
Carcinoma de células pequeñas (pulmón) |
0/1 |
|
Carcinoma de células escamosas (pulmón) |
0/1 |
|
Carcinoma, sin especificar (pulmón) |
0/2 |
|
Adenocarcinoma (pulmón) |
0/1 |
|
Carcinoma de células escamosas (esófago) |
0/1 |
|
Adenocarcinoma (esófago) |
0/1 |
|
Adenocarcinoma mucinoso (estómago) |
0/4 |
|
Adenocarcinoma (estómago) |
8/88 |
|
Carcinoma de células en anillo de sello (Estómago) |
0/4 |
|
Carcinoma, sin especificar (estómago) |
0/3 |
|
Adenocarcinoma (intestino delgado) |
0/1 |
|
Tumor estromal gastrointestinal (intestino delgado) |
0/1 |
|
Adenocarcinoma (colon) |
0/32 |
|
Tumor estromal gastrointestinal (colon) |
0/1 |
|
Carcinoma, sin especificar (colon) |
1/3 |
|
Adenocarcinoma (recto) |
1/5 |
|
Tumor estromal gastrointestinal (recto) |
0/1 |
|
Melanoma (recto) |
0/1 |
|
Carcinoma hepatocelular (hígado) |
0/3 |
|
Hepatoblastoma (hígado) |
0/1 |
|
Carcinoma, sin especificar (hígado) |
0/3 |
|
Carcinoma de células claras (riñón) |
0/1 |
|
Carcinoma, sin especificar (riñón) |
0/5 |
|
Adenocarcinoma (próstata) |
0/2 |
|
Carcinoma, sin especificar (próstata) |
0/3 |
|
Leiomioma |
0/3 |
|
Adenocarcinoma (útero) |
0/1 |
|
Carcinoma de células claras (útero) |
0/1 |
|
Carcinoma de células escamosas (cuello uterino) |
0/2 |
|
Rabdomiosarcoma embrionario (músculo estriado) |
0/1 |
|
Carcinoma de células basales (piel) |
0/1 |
|
Carcinoma de células escamosas (piel) |
1/1 |
|
Neurofibroma (lumbar) |
0/1 |
|
Neuroblastoma (retroperitoneo) |
0/1 |
|
Mesotelioma (peritoneo) |
0/1 |
|
Rabdomiosarcoma polimorfo (peritoneo) |
0/1 |
|
Linfoma, sin especificar |
0/3 |
|
Linfoma de linfocitos B, sin especificar (bazo) |
0/1 |
|
Linfoma de linfocitos B, sin especificar (ganglio linfático) |
0/2 |
|
Linfoma de Hodgkin (ganglio linfático) |
0/1 |
|
Carcinoma urotelial (vejiga) |
1/1 |
|
Leiomiosarcoma (vejiga) |
0/1 |
|
Osteosarcoma (hueso) |
0/1 |
|
Leiomiosarcoma (músculo liso) |
0/1 |
|
Adenocarcinoma rectal (metastásico) |
0/1 |
|
Adenocarcinoma de colon (metastásico) |
0/7 |
|
Adenocarcinoma mucinoso de colon (metastásico) |
0/1 |
|
Melanoma |
0/2 |
|
Neoplasia neuroendocrina, sin especificar |
0/2 |
|
Sarcoma, sin especificar |
0/2 |
|
Carcinoma no diferenciado, sin especificar |
0/1 |
Rendimiento de análisis en cáncer de mama con nivel bajo de HER2
Repetibilidad y precisión intermedia de HER2 bajo en el instrumento BenchMark ULTRA:
• En el estudio de repetibilidad y precisión intermedia se incluyeron veinticuatro casos de carcinoma de mama que reflejaban todo el intervalo de tinción de IHC de HER2. El diseño del estudio verificaba la precisión de la tinción de tejidos de carcinoma de mama teñidos con el anticuerpo VENTANA HER2 (4B5) Assay.
• Tres lotes de anticuerpo VENTANA HER2 (4B5) (entre lotes de anticuerpo) Tres lotes de ultraView DAB IHC Detection Kit (entre lotes de kits de detección).
• En tres días diferentes (entre días).
• Tres instrumentos BenchMark ULTRA (entre instrumentos).
• En todas las condiciones de precisión intermedia (en una misma sesión).
• Se asignó un modo a cada caso en función de las muestras consolidadas por condiciones de la prueba: entre lotes de anticuerpo, entre lotes de kits de detección, entre instrumentos y entre días. En el caso de la condición en una misma sesión, cada caso se comparó con sus muestras duplicadas en cada sesión de prueba. Se enmascararon ya aleatorizaron todos los portaobjetos y, a continuación, se evaluaron siguiendo los criterios de intensidad y patrón de tinción de membrana celular con la tinción VENTANA HER2 (4B5) Assay (Tabla 5). Los resultados se resumen en la Tabla 10.
Tabla 10. Repetibilidad y precisión intermedia del anticuerpo VENTANA HER2 (4B5) Assay en tejidos de cáncer de mama con una baja puntuación de HER2
|
Repetibilidad/ Precisión |
Concordancia |
|||
|---|---|---|---|---|
|
Tipo |
n/N |
% |
CI del 95% |
|
|
Entre lotes de anticuerpo |
PPA |
96/96 |
100.0 |
(96.2, 100.0) |
|
NPA |
48/48 |
100.0 |
(92.6, 100.0) |
|
|
OPA |
144/144 |
100.0 |
(97.4, 100.0) |
|
|
Entre kits de detección |
PPA |
93/96 |
96.9 |
(92.2, 100.0) |
|
NPA |
48/48 |
100.0 |
(92.6, 100.0) |
|
|
OPA |
141/144 |
97.9 |
(94.4, 100.0) |
|
|
Entre instrumentos (BenchMark ULTRA) |
PPA |
95/96 |
99.0 |
(96.7, 100.0) |
|
NPA |
48/48 |
100.0 |
(92.6, 100.0) |
|
|
OPA |
143/144 |
99.3 |
(97.9, 100.0) |
|
|
Entre días |
PPA |
94/96 |
97.9 |
(93.3, 100.0) |
|
NPA |
48/48 |
100.0 |
(92.6, 100.0) |
|
|
OPA |
142/144 |
98.6 |
(95.8 100.0) |
|
|
En la misma sesión |
PPA |
142/144 |
98.6 |
(96.5, 100.0) |
|
NPA |
72/72 |
100.0 |
(94.9, 100.0) |
|
|
OPA |
214/216 |
99.1 |
(97.7, 100.0) |
|
Nota: Porcentaje de concordancia positiva (PPA), porcentaje de concordancia negativa (NPA) y porcentaje de concordancia global (OPA).
Nota: El intervalo de confianza (CI) del 95% bilateral se calculó utilizando el método bootstrap percentil de 2000 muestras bootstrap. Los CI del 100% de los PPA, NPA y OPA se calcularon mediante el método de puntuación de Wilson.
Nota: A efectos del análisis del estudio, las puntuaciones de HER2 0 y 3+ se agruparon como casos negativos dado que ninguno de esos casos cumplía con los criterios de idoneidad para recibir tratamientos dirigidos a nivel bajo de HER2 según el diseño del ensayo clínico y las puntuaciones de HER2 de 1+ y 2+ se agruparon como casos positivos porque cumplían con los criterios de idoneidad para recibir los tratamientos dirigidos a nivel bajo de HER2 según el diseño del ensayo.
Estudio de comparación entre los instrumentos BenchMark ULTRA frente a Benchmark XT y Benchmark GX para HER2 bajo:
En el estudio de precisión intermedia se incluyeron diez casos de carcinoma de mama que reflejaban todo el intervalo de tinción de IHC de HER2. El diseño del estudio verificaba la precisión de la tinción de tejidos de carcinoma de mama teñidos con VENTANA HER2 (4B5) Assay en varios instrumentos y diferentes plataformas.
Cada muestra se asignó a un modo en función de las muestras consolidadas por condiciones de la prueba. Cada muestra se comparó con sus muestras duplicadas en cada sesión de prueba. Se enmascararon ya aleatorizaron todos los portaobjetos y, a continuación, se evaluaron siguiendo los criterios de intensidad y patrón de tinción de membrana celular con la tinción VENTANA HER2 (4B5) Assay (Tabla 5). Los resultados se resumen en la Tabla 11.
Tabla 11. Repetibilidad y precisión intermedia del anticuerpo VENTANA HER2 (4B5) Assay en tejidos de cáncer de mama con una baja puntuación de HER2
|
Repetibilidad/ Precisión |
Concordancia |
|||
|
Tipo |
n/N |
% |
CI del 95% |
|
|
Entre plataformas (ULTRA/GX/XT) |
PPA |
90/90 |
100.0 |
(95.9, 100.0) |
|
NPA |
90/90 |
100.0 |
(95.9, 100.0) |
|
|
OPA |
180/180 |
100.0 |
(97.9, 100.0) |
|
|
Entre instrumentos (BenchMark ULTRA) |
PPA |
30/30 |
100.0 |
(88.6, 100.0) |
|
NPA |
30/30 |
100.0 |
(88.6, 100.0) |
|
|
OPA |
60/60 |
100.0 |
(94.0, 100.0) |
|
|
Entre instrumentos (BenchMark GX) |
PPA |
30/30 |
100.0 |
(88.6, 100.0) |
|
NPA |
30/30 |
100.0 |
(88.6, 100.0) |
|
|
OPA |
60/60 |
100.0 |
(94.0, 100.0) |
|
|
Entre instrumentos (BenchMark XT) |
PPA |
30/30 |
100.0 |
(88.6, 100.0) |
|
NPA |
30/30 |
100.0 |
(88.6, 100.0) |
|
|
OPA |
60/60 |
100.0 |
(94.0, 100.0) |
|
Nota: Porcentaje de concordancia positiva (PPA), porcentaje de concordancia negativa (NPA) y porcentaje de concordancia global (OPA).
Nota: El intervalo de confianza (CI) del 95% bilateral se calculó utilizando el método bootstrap percentil de 2000 muestras bootstrap. Los CI del 100% de los PPA, NPA y OPA se calcularon mediante el método de puntuación de Wilson.
Nota: A efectos del análisis del estudio, las puntuaciones de HER2 0 y 3+ se agruparon como casos negativos dado que ninguno de esos casos cumplía con los criterios de idoneidad para recibir tratamientos dirigidos a nivel bajo de HER2 según el diseño del ensayo clínico y las puntuaciones de HER2 de 1+ y 2+ se agruparon como casos positivos porque cumplían con los criterios de idoneidad para recibir los tratamientos dirigidos a nivel bajo de HER2 según el diseño del ensayo.
Precisión del lector para HER2 bajo en el instrumento BenchMark ULTRA:
Se evaluaron la precisión del lector y entre lectores mediante la evaluación de la concordancia del estado de nivel bajo de HER2 entre tres lectores y con tres lectores de forma individual. En el estudio se incluyeron 100 casos de carcinoma de mama que reflejaban todo el intervalo de tinción de IHC de HER2. Se enmascararon y aleatorizaron las muestras antes de su evaluación para determinar el estado de HER2 bajo según el patrón de tinción de membrana celular con la tinción con VENTANA HER2 (4B5) Assay (Tabla 5). Los lectores puntuaron todas las muestras dos veces, con un mínimo de dos semanas entre lecturas. La concordancia de la precisión entre lectores y del lector se resumen en la Tabla 12.
Tabla 12. Precisión del lector y entre lectores de VENTANA HER2 (4B5) Assay con puntuaciones de HER2 bajo
|
Precisión |
Concordancia |
|||
|
Tipo |
n/N |
% |
CI del 95% |
|
|
Del lector |
APA |
312/333 |
93.7 |
(90.9, 96.4) |
|
ANA |
246/267 |
92.1 |
(88.0, 95.6) |
|
|
OPA |
279/300 |
93.0 |
(90.0, 96.0) |
|
|
Entre lectores |
APA |
300/332 |
90.4 |
(85.8, 94.3) |
|
ANA |
236/268 |
88.1 |
(82.1, 93.0) |
|
|
OPA |
268/300 |
89.3 |
(84.7, 94.0) |
|
Nota: Promedio de concordancia positiva (APA), promedio de concordancia negativa (ANA) y porcentaje de concordancia global (OPA).
Nota: El intervalo de confianza (CI) del 95% bilateral se calculó utilizando el método bootstrap percentil de 2000 muestras bootstrap.
Nota: A efectos del análisis del estudio, las puntuaciones de HER2 0 y 3+ se agruparon como casos negativos dado que ninguno de esos casos cumplía con los criterios de idoneidad para recibir tratamientos dirigidos a nivel bajo de HER2 según el diseño del ensayo clínico y las puntuaciones de HER2 de 1+ y 2+ se agruparon como casos positivos porque cumplían con los criterios de idoneidad para recibir los tratamientos dirigidos a nivel bajo de HER2 según el diseño del ensayo.
Reproducibilidad entre laboratorios: Estudio para HER2 bajo en el instrumento BenchMark ULTRA Se llevó a cabo un estudio de reproducibilidad entre laboratorios con VENTANA HER2 (4B5) Assay para demostrar la reproducibilidad den ensayo a la hora de determinar el estado de bajo nivel de HER2 en los casos de carcinoma de mama. El estudio incluía 28 muestras de tejido de carcinoma de mama FFPE archivadas y no identificadas que se tomaron en tres instrumentos BenchMark ULTRA en cada uno de los cinco días no consecutivos durante 20 días en tres laboratorios externos. Las muestras presentaron el intervalo de tinción de VENTANA HER2 (4B5) Assay.
Cada uno de los cinco conjuntos de portaobjetos que se tiñeron por muestra y por día de tinción se aleatorizaron y se evaluaron posteriormente por parte de seis lectores (dos lectores por sitio) para establecer su estado de nivel bajo de HER2. Los resultados del estado de bajo nivel de HER2 de todos los lectores, los sitios y los días de las muestras se combinaron y se analizaron en comparación con los modos de lector de los mismos casos para determinar la reproducibilidad global del estado de nivel bajo de HER2. El resumen de índices de concordancia en todas las observaciones evaluables usando los modos del lector de nivel de muestra para HER2 bajo como referencia puede consultarse en la Tabla 13.
Tabla 13. Reproducibilidad entre laboratorios de todos los índices de concordancia global de VENTANA HER2 (4B5) Assay con puntuación de HER2 baja
.
|
Reproducibilidad entre laboratorios |
Concordancia |
|||
|---|---|---|---|---|
|
Tipo |
n/N |
% |
CI del 95% |
|
|
Global |
PPA |
407/416 |
97.8 |
(96.2, 99.3) |
|
NPA |
416/418 |
99.5 |
(98.8, 100.0) |
|
|
OPA |
823/834 |
98.7 |
(97.7, 99.4) |
|
|
En el mismo sitio |
PPA |
407/416 |
97.8 |
(96.2, 99.3) |
|
NPA |
416/418 |
99.5 |
(98.8, 100.0) |
|
|
OPA |
823/834 |
98.7 |
(97.7, 99.4) |
|
|
Del lector |
PPA |
407/416 |
97.8 |
(96.2, 99.3) |
|
NPA |
416/418 |
99.5 |
(98.8, 100.0) |
|
|
OPA |
823/834 |
98.7 |
(97.7, 99.4) |
|
Nota: Porcentaje de concordancia positiva (PPA), porcentaje de concordancia negativa (NPA) y porcentaje de concordancia global (OPA).
Nota: Los intervalos de confianza del 95% bilaterales se calcularon utilizando el método bootstrap percentil con 2000 replicados.
Nota: A efectos del análisis del estudio, las puntuaciones de HER2 0 y 3+ se agruparon como casos negativos dado que ninguno de esos casos cumplía con los criterios de idoneidad para recibir tratamientos dirigidos a nivel bajo de HER2 según el diseño del ensayo clínico y las puntuaciones de HER2 de 1+ y 2+ se agruparon como casos positivos porque cumplían con los criterios de idoneidad para recibir los tratamientos dirigidos a nivel bajo de HER2 según el diseño del ensayo.
Además, se llevaron a cabo comparaciones entre sitios, lectores y días para HER2 bajo. En la Tabla 14 se resumen los resultados. Los datos indican una reproducibilidad del ensayo en 5 días, 3 sitios y 6 lectores.
Tabla 14. Reproducibilidad entre laboratorios de todos los porcentajes de concordancia global del anticuerpo VENTANA HER2 (4B5) Assay con puntuación de bajo nivel de HER2
|
Reproducibilidad entre laboratorios |
Concordancia |
|||
|
Tipo |
n/N |
% |
CI del 95% |
|
|
Entre sitios |
APA |
7884/8102 |
97.3 |
(95.4, 98.8) |
|
ANA |
8240/8458 |
97.4 |
(95.7, 98.8) |
|
|
OPA |
8062/8280 |
97.4 |
(95.5, 98.8) |
|
|
Entre lectores |
APA |
398/409 |
97.3 |
(95.4, 98.8) |
|
ANA |
414/425 |
97.4 |
(95.6, 98.8) |
|
|
OPA |
406/417 |
97.4 |
(95.5, 98.8) |
|
|
Entre días |
APA |
1580/1620 |
97.5 |
(95.9, 98.9) |
|
ANA |
1652/1692 |
97.6 |
(96.2, 98.9) |
|
|
OPA |
1616/1656 |
97.6 |
(96.1, 98.9) |
|
Nota: Promedio de concordancia positiva (APA), promedio de concordancia negativa (ANA) y porcentaje de concordancia global (OPA).
Nota: Los intervalos de confianza del 95% bilaterales se calcularon utilizando el método bootstrap percentil con 2000 replicados.
Nota: A efectos del análisis del estudio, las puntuaciones de HER2 0 y 3+ se agruparon como casos negativos dado que ninguno de esos casos cumplía con los criterios de idoneidad para recibir tratamientos dirigidos a nivel bajo de HER2 según el diseño del ensayo clínico y las puntuaciones de HER2 de 1+ y 2+ se agruparon como casos positivos porque cumplían con los criterios de idoneidad para recibir los tratamientos dirigidos a nivel bajo de HER2 según el diseño del ensayo.
Rendimiento de análisis en casos de mama con nivel de HER2 bajo:
Concordancia entre los instrumentos BenchMark ULTRA y BenchMark ULTRA PLUS para HER2 bajo:
Tres laboratorios participaron en un estudio de concordancia para evaluar la equivalencia de rendimiento entre el instrumento BenchMark ULTRA y el instrumento BenchMark ULTRA PLUS. Para el análisis estadístico de HER2 bajo, se seleccionaron previamente 160 de los casos (80 positivos y 80 negativos, incluidos 16 casos límite) para el análisis antes de las lecturas de los anatomopatólogos. Los portaobjetos de tejido de todos los casos se tiñeron con un control de reactivo negativo y con VENTANA HER2 (4B5) Assay en un laboratorio interno de Roche en un instrumento BenchMark ULTRA y utilizando el protocolo de tinción recomendado. Los portaobjetos con tejido sin tinción de todos los casos se aleatorizaron y se distribuyeron de forma equitativa para su tinción en un instrumento BenchMark ULTRA PLUS con el protocolo de tinción recomendado de VENTANA HER2 (4B5). Con el estado de casos enmascarados, lectura de un lector por sitio y los portaobjetos teñidos con el instrumento BenchMark ULTRA PLUS de su sitio, se determinó el estado de HER2 (4B5). Los resultados se analizaron en Roche. Los resultados se resumen en la Tabla 15.
Tabla 15. Concordancia combinada del estado de HER2 bajo de los casos teñidos con VENTANA HER2 (4B5) Assay en el instrumento BenchMark ULTRA frente al instrumento BenchMark ULTRA PLUS
|
BenchMark ULTRA PLUS |
BenchMark ULTRA |
||||
|
Lector de Roche = positiva, lector externo = positiva |
Lector de Roche = positiva, lector externo= negativa |
Lector de Roche = negativa, lector externo= positiva |
Lector de Roche = negativa, lector externo= negativa |
Total |
|
|
Positiva |
272 |
13 |
25 |
11 |
321 |
|
Negativa |
8 |
9 |
2 |
298 |
317 |
|
Total |
280 |
22 |
27 |
309 |
638 |
|
Porcentaje de positiva % (n/N) |
97.1 (272/280) |
59.1 (13/22) |
92.6 (25/27) |
3.6 (11/309) |
N/A |
|
n/N |
% (CI del 95%) |
||||
|
PPA |
285/302 |
94.4 (91.6, 96.8) |
|||
|
NPA |
300/336 |
89.3 (84.3, 94.4) |
|||
|
OPA |
585/638 |
91.7 (88.5, 94.6) |
|||
Nota: Porcentaje de concordancia positiva (PPA); Porcentaje de concordancia negativa (NPA); Porcentaje de concordancia global (OPA).
Nota: Los intervalos de confianza del 95% bilaterales se calcularon mediante el método bootstrap percentil con 2000 réplicas estratificadas en grupos de puntuación de cualificación mediante IHC.
Nota: La concordancia combinada incluyó todos los casos y los lectores ULTRA PLUS.
Nota: A los efectos del análisis del estudio, las puntuaciones de HER2 0 y 3+ se agruparon como casos negativos dado que ninguno de esos casos cumplía con los criterios de idoneidad del ensayo clínico que investigaba el cáncer de mama con nivel bajo de HER2. Las puntuaciones de HER2 de 1+ y 2+ se agruparon como casos positivos porque cumplían con los criterios de idoneidad o eran potencialmente elegibles para el ensayo clínico.
Estudio de reproducibilidad entre laboratorios de BenchMark ULTRA PLUS para HER2 bajo:
Se llevó a cabo un estudio de reproducibilidad entre laboratorios con VENTANA HER2 (4B5) Assay para demostrar la reproducibilidad den ensayo a la hora de determinar el estado de bajo nivel de HER2 en los casos de carcinoma de mama.
Este estudio incluía 28 muestras de tejido de carcinoma de mama FFPE archivadas y no identificadas que se tomaron en tres instrumentos BenchMark ULTRA PLUS en cada uno de los cinco días no consecutivos durante 20 días en tres laboratorios externos. Las muestras presentaron el intervalo de tinción de VENTANA HER2 (4B5) Assay.
Cada uno de los 5 conjuntos de portaobjetos teñidos por muestra y por día de tinción se aleatorizó y evaluó con 6 lectores en total (2 lectores por sitio) para determinar un estado de HER2 bajo. Los resultados del estado de bajo nivel de HER2 de todos los lectores, los sitios y los días de las muestras se combinaron y se analizaron en comparación con los modos de lector de los mismos casos para determinar la reproducibilidad global del estado de nivel bajo de HER2. El resumen de los índices de concordancia en todas las observaciones evaluables usando los modos del lector de nivel de muestra para HER2 bajo como referencia puede consultarse en la Tabla 16.
Tabla 16. Reproducibilidad entre laboratorios de los índices de concordancia global para VENTANA HER2 (4B5) Assay en carcinoma de mama con puntuación de HER2 baja
|
Reproducibilidad entre laboratorios |
Concordancia |
|||
|
Tipo |
n/N |
% |
CI del 95% |
|
|
Análisis primario/global |
PPA |
407/420 |
96.9 |
(93.6, 99.3) |
|
NPA |
405/420 |
96.4 |
(92.2, 100.0) |
|
|
OPA |
812/840 |
96.7 |
(94.0, 98.9) |
|
|
Estratificado por sitio |
PPA |
407/420 |
96.9 |
(93.6, 99.3) |
|
NPA |
405/420 |
96.4 |
(92.2, 100.0) |
|
|
OPA |
812/840 |
96.7 |
(94.0, 98.9) |
|
|
Estratificado por lector |
PPA |
412/425 |
96.9 |
(94.8, 98.7) |
|
NPA |
405/415 |
97.6 |
(94.9, 100.0) |
|
|
OPA |
817/840 |
97.3 |
(95.2, 98.9) |
|
Nota: Los intervalos de confianza del 95% bilaterales se calcularon mediante el método bootstrap percentil con 2000 réplicas estratificadas en grupos de puntuación de cualificación de casos.
Nota: A los efectos del análisis del estudio, las puntuaciones de HER2 0 y 3+ se agruparon como casos negativos dado que ninguno de esos casos cumplía con los criterios de idoneidad del ensayo clínico que investigaba el cáncer de mama con nivel bajo de HER2. Las puntuaciones de HER2 de 1+ y 2+ se agruparon como casos positivos porque cumplían con los criterios de idoneidad o eran potencialmente elegibles para el ensayo clínico.
Además, se llevaron a cabo comparaciones entre pares del estado de HER2 (4B5) entre sitios, entre lectores y entre días. Tal y como se recoge en la Tabla 17, los datos indican una reproducibilidad del ensayo en 5 días, 3 sitios y 6 lectores.
Tabla 17. Reproducibilidad entre laboratorios de los índices de concordancia entre pares para VENTANA HER2 (4B5) Assay en carcinoma de mama con puntuación de HER2 baja
|
Reproducibilidad entre laboratorios |
Concordancia |
|||
|
Tipo |
n/N |
% |
CI del 95% |
|
|
Entre sitios |
APA |
7982/8440 |
94.6 |
(90.3, 97.9) |
|
ANA |
7902/8360 |
94.5 |
(90.6, 98.0) |
|
|
OPA |
7942/8400 |
94.5 |
(90.5, 98.0) |
|
|
Entre lectores |
APA |
402/422 |
95.3 |
(91.7, 98.1) |
|
ANA |
398/418 |
95.2 |
(91.8, 98.1) |
|
|
OPA |
400/420 |
95.2 |
(91.9, 98.1) |
|
|
Entre días |
APA |
1608/1688 |
95.3 |
(91.5, 98.2) |
|
ANA |
1592/1672 |
95.2 |
(91.8, 98.2) |
|
|
OPA |
1600/1680 |
95.2 |
(91.8, 98.2) |
|
Nota: Los intervalos de confianza del 95% bilaterales se calcularon mediante el método bootstrap percentil con 2000 réplicas estratificadas en grupos de puntuación de cualificación de casos.
Nota: A los efectos del análisis del estudio, las puntuaciones de HER2 0 y 3+ se agruparon como casos negativos dado que ninguno de esos casos cumplía con los criterios de idoneidad del ensayo clínico que investigaba el cáncer de mama con nivel bajo de HER2. Las puntuaciones de HER2 de 1+ y 2+ se agruparon como casos positivos porque cumplían con los criterios de idoneidad o eran potencialmente elegibles para el ensayo clínico.
Rendimiento de análisis en casos de mama con HER2 positivo:
Concordancia entre los instrumentos BenchMark ULTRA y BenchMark ULTRA PLUS para HER2 positivo:
Tres laboratorios participaron en un estudio de concordancia para evaluar la equivalencia de rendimiento entre el instrumento BenchMark ULTRA y el instrumento BenchMark ULTRA PLUS. En los análisis de HER2 positivo, la puntuación HER2 IHC de 2+ o 3+ se define como HER2 positivo y una puntuación de 0 o 1+ se define como HER2 negativo. Para el análisis estadístico de HER2 positivo, se seleccionaron previamente 160 de los casos (80 positivos y 80 negativos, incluidos 16 casos límite) para el análisis antes de las lecturas de los anatomopatólogos. Los portaobjetos de tejido de todos los casos se tiñeron con un control de reactivo negativo y con VENTANA HER2 (4B5) Assay en un laboratorio interno de Roche en un instrumento BenchMark ULTRA y utilizando el protocolo de tinción recomendado. Los portaobjetos con tejido sin tinción de todos los casos se aleatorizaron y se distribuyeron de forma equitativa para su tinción en un instrumento BenchMark ULTRA PLUS con el protocolo de tinción recomendado de VENTANA HER2 (4B5). Con el estado de casos enmascarados, lectura de un lector por sitio y los portaobjetos teñidos con el instrumento BenchMark ULTRA PLUS de su sitio, se determinó el estado de HER2 (4B5). Los resultados se analizaron en Roche. Los resultados se resumen en la Tabla 18.
Tabla 18. Concordancia combinada del estado de HER2 positivo de los casos teñidos con VENTANA HER2 (4B5) Assay en el instrumento BenchMark ULTRA frente a BenchMark ULTRA PLUS
|
BenchMark ULTRA PLUS |
BenchMark ULTRA |
||||
|---|---|---|---|---|---|
|
Lector de Roche = positiva, lector externo= positiva |
Lector de Roche = positiva, lector externo= negativa |
Lector de Roche = negativa, lector externo= positiva |
Lector de Roche = negativa, lector externo= negativa |
Total |
|
|
Positiva |
245 |
18 |
11 |
13 |
287 |
|
Negativa |
6 |
13 |
5 |
327 |
351 |
|
Total |
251 |
31 |
16 |
340 |
638 |
|
Porcentaje de positiva % (n/N) |
97.6 (245/251) |
58.1 (18/31) |
68.8 (11/16) |
3.8 (13/340) |
N/A |
|
n/N |
% (CI del 95%) |
||||
|
PPA |
(263/282) |
93.3 (89.3, 96.6) |
|||
|
NPA |
(332/356) |
93.3 (89.7, 96.5) |
|||
|
OPA |
(595/638) |
93.3 (90.8, 95.6) |
|||
Nota: Los intervalos de confianza del 95% bilaterales se calcularon mediante el método bootstrap percentil con 2000 réplicas estratificadas en grupos de puntuación de cualificación mediante IHC (0,1+, 2+, 3+).
Nota: La concordancia combinada incluyó todos los casos y los lectores ULTRA PLUS.
Nota: Porcentaje de concordancia positiva (PPA); Porcentaje de concordancia negativa (NPA); Porcentaje de concordancia global (OPA).
Estudio de reproducibilidad entre laboratorios de BenchMark ULTRA PLUS para HER2 positivo:
Se llevó a cabo un estudio de reproducibilidad entre laboratorios con VENTANA HER2 (4B5) Assay para demostrar la reproducibilidad del ensayo a la hora de determinar el estado de positividad de HER2 en los casos de carcinoma de mama. Este estudio incluía 28 muestras de tejido de carcinoma de mama FFPE archivadas y no identificadas que se tomaron en tres instrumentos BenchMark ULTRA PLUS en cada uno de los cinco días no consecutivos durante 20 días en tres laboratorios externos. Las muestras presentaron el intervalo de tinción de VENTANA HER2 (4B5) Assay.
Cada uno de los 5 conjuntos de portaobjetos teñidos por muestra y por día de tinción se aleatorizó y evaluó con 6 lectores en total (2 lectores por sitio) para determinar un estado de HER2 positivo. Los resultados del estado de HER2 positivo de todos los lectores, los sitios y los días de las muestras se combinaron y analizaron en comparación con los modos de lector de los mismos casos para determinar la reproducibilidad global del estado de HER2 positivo. El resumen de los índices de concordancia en todas las observaciones evaluables usando los modos del lector de nivel de muestra para HER2 positivo como referencia puede consultarse en la Tabla 19.
Tabla 19. Reproducibilidad entre laboratorios de los índices de concordancia global para VENTANA HER2 (4B5) Assay en carcinoma de mama con puntuación de HER2 positivo
|
Reproducibilidad entre laboratorios |
Concordancia |
|||
|
Tipo |
n/N |
% |
CI del 95% |
|
|
Análisis primario/global |
PPA |
411/420 |
97.9 |
(95.7, 99.5) |
|
NPA |
410/420 |
97.6 |
(94.3, 100.0) |
|
|
OPA |
821/840 |
97.7 |
(96.0, 99.3) |
|
|
Estratificado por sitio |
PPA |
411/420 |
97.9 |
(95.7, 99.5) |
|
NPA |
410/420 |
97.6 |
(94.3, 100.0) |
|
|
OPA |
821/840 |
97.7 |
(96.0, 99.3) |
|
|
Estratificado por lector |
PPA |
413/420 |
98.3 |
(96.9, 99.5) |
|
NPA |
412/420 |
98.1 |
(95.8, 100.0) |
|
|
OPA |
825/840 |
98.2 |
(96.9, 99.4) |
|
Nota: Los intervalos de confianza del 95% bilaterales se calcularon mediante el método bootstrap percentil con 2000 réplicas estratificadas en grupos de puntuación de cualificación de casos.
Nota: A los efectos del análisis del estudio, las puntuaciones de HER2 IHC de 0 y 1+ se agruparon como casos negativos y las puntuaciones de HER2 IHC de 2+ y 3+ se agruparon como casos positivos.
Además, se llevaron a cabo comparaciones entre pares del estado de HER2 (4B5) entre sitios, entre lectores y entre días. Tal y como se recoge en la Tabla 20, los datos indican una reproducibilidad del ensayo en 5 días, 3 sitios y 6 lectores.
Tabla 20. Reproducibilidad entre laboratorios de los índices de concordancia entre pares para VENTANA HER2 (4B5) Assay en carcinoma de mama con tinción positiva de HER2
|
Reproducibilidad entre laboratorios |
Concordancia |
|||
|
Tipo |
n/N |
% |
CI del 95% |
|
|
Entre sitios |
APA |
8074/8420 |
95.9 |
(92.8, 98.6) |
|
ANA |
8034/8380 |
95.9 |
(92.5, 98.7) |
|
|
OPA |
8054/8400 |
95.9 |
(92.7, 98.6) |
|
|
Entre lectores |
APA |
402/421 |
95.5 |
(92.0, 98.6) |
|
ANA |
400/419 |
95.5 |
(91.6, 98.6) |
|
|
OPA |
401/420 |
95.5 |
(91.9, 98.6) |
|
|
Entre días |
APA |
1634/1684 |
97.0 |
(95.0, 98.9) |
|
ANA |
1626/1676 |
97.0 |
(94.8, 98.9) |
|
|
OPA |
1630/1680 |
97.0 |
(95.0, 98.9) |
|
Nota: Los intervalos de confianza del 95% bilaterales se calcularon mediante el método bootstrap percentil con 2000 réplicas estratificadas en grupos de puntuación de cualificación de casos.
Nota: A los efectos del análisis del estudio, las puntuaciones de HER2 IHC de 0 y 1+ se agruparon como casos negativos y las puntuaciones de HER2 IHC de 2+ y 3+ se agruparon como casos positivos.
Características de rendimiento en el instrumento BenchMark ULTRA con iVIEW DAB Detection Kit o ultraView Universal DAB Detection Kit:
Tinción entre laboratorios con el instrumento BenchMark ULTRA y reproducibilidad entre días: Tres laboratorios, pertenecientes a instituciones diferentes de EE. UU., participaron en el estudio de reproducibilidad entre laboratorios. Se distribuyeron portaobjetos con secciones de 48 casos de carcinoma de mama invasivo FFPE [12 unidades de cada una de las categorías de agrupamiento HER2 (0, 1+, 2+, 3+)] y un par de PATHWAY HER-2 4 in 1 Control Slides en cada una de las 12 sesiones de tinción para estudiar la tinción en los sitios con un instrumento BenchMark ULTRA utilizando el protocolo de tinción recomendado y ultraView Universal DAB Detection Kit. Entre los controles figuraban PATHWAY HER-2 4 in 1 Control Slides y un segundo portaobjetos de cada caso teñido con el reactivo Ig negativo. Los anatomopatólogos, con los estados de casos enmascarados, evaluaron los portaobjetos y facilitaron una puntuación clínica (como 0, 1+, 2+ y 3+). Los resultados se analizaron en Ventana. Con la nomenclatura estándar para las tablas de 2x2, el promedio de concordancia positiva (APA) en todos los sitios se calculó a través de la fórmula [2a/(2a+b+c)] y el promedio de concordancia negativa (ANA) se calculó a través de la fórmula [2d/(2d+b+c)]. En todos los centros, el APA entre centros, con base en la evaluación clínica (positivo/negativo), fue del 90.0% (108/120) y el ANA fue del 92.9% (156/168). En las comparaciones entre pares de los sitios, se calculó el APA mediante a/(a+c) y el ANA mediante d/(b+d). Los porcentajes de APA entre sitios fueron del 93.0% (40/43), del 87.2% (34/39) y del 89.5% (34/38) entre el sitio A y el sitio B, entre el sitio A y el sitio C y entre el sitio B y el sitio C, respectivamente. Los porcentajes de ANA entre sitios fueron del 94.3% (50/53), del 91.2% (52/57) y del 93.1% (54/58) entre el sitio A y el sitio B, entre el sitio A y el sitio C y entre el sitio B y el sitio C, respectivamente.
Las tablas que aparecen a continuación son presentaciones de 3x3 de los resultados de cada lector con base en la puntuación clínica, en la que se separaron 2+ y 3+.
Tabla 21. Análisis 3x3 de los índices de concordancia entre laboratorios entre el sitio A y el sitio B: clon 4B5 en un instrumento BenchMark ULTRA con ultraView Universal DAB Detection Kit
|
Sitio A |
Sitio B |
|||
|---|---|---|---|---|
|
3+ |
2+ |
0, 1+ |
Total |
|
|
3+ |
12 |
2 |
0 |
14 |
|
2+ |
0 |
6 |
2 |
8 |
|
0, 1+ |
0 |
1 |
25 |
26 |
|
Total |
12 |
9 |
27 |
48 |
|
Porcentaje de concordancia global: (OPA): n/N (%) (CI del 95%) |
43/48 (89.6) (77.8-95.5) |
|||
Tabla 22. Análisis 3x3 de los índices de concordancia entre laboratorios entre el sitio A y el sitio C: clon 4B5 en un instrumento BenchMark ULTRA con ultraView Universal DAB Detection Kit
|
Sitio A |
Sitio C |
|||
|
3+ |
2+ |
0, 1+ |
Total |
|
|
3+ |
12 |
1 |
1 |
14 |
|
2+ |
0 |
4 |
4 |
8 |
|
0, 1+ |
0 |
0 |
26 |
26 |
|
Total |
12 |
5 |
31 |
48 |
|
Porcentaje de concordancia global: (OPA): n/N (%) (CI del 95%) |
42/48 (87.5) (75.3-94.1) |
|||
Tabla 23. Análisis 3x3 de los índices de concordancia entre laboratorios entre el sitio B y el sitio C: clon 4B5 en un instrumento BenchMark ULTRA con ultraView Universal DAB Detection Kit
|
Sitio B |
Sitio C |
|||
|
3+ |
2+ |
0, 1+ |
Total |
|
|
3+ |
12 |
0 |
0 |
12 |
|
2+ |
0 |
5 |
4 |
9 |
|
0, 1+ |
0 |
0 |
27 |
27 |
|
Total |
12 |
5 |
31 |
48 |
|
Porcentaje de concordancia global: (OPA): n/N (%) (CI del 95%) |
44/48 (91.7) (80.4-96.7) |
|||
Reproducibilidad entre días de la tinción con el instrumento BenchMark ULTRA:
En la parte del estudio correspondiente a la reproducibilidad entre días (IDR) se incluyeron 12 casos con una distribución prevista de aproximadamente tres (3) casos de cada puntuación clínica (0,1+, 2+ y 3+). En total se llevó a cabo la parte del estudio correspondiente a la IDR mediante cinco sesiones en un instrumento BenchMark ULTRA en un solo centro (Sitio C) y en un periodo mínimo de 20 días, de forma que las tinciones no se realizaran en dos días consecutivos. Los porcentajes de APA y ANA de la IDR, con base en la evaluación clínica de la tinción con el clon 4B5 en el centro C en todos los días, fue en ambos casos del 100%. Los índices de los porcentajes de concordancia global (OPA) de las comparaciones entre días, con base en las puntuaciones clínicas, fueron del 100% en cada una de las comparaciones entre días y en la combinación de todos los días.
Estudio de comparación entre el instrumento BenchMark ULTRA y el instrumento BenchMark XT:
Dos laboratorios de tinción únicos y tres sitios de lectura de EE. UU. participaron en el estudio de comparación de plataformas. Se distribuyeron aleatoriamente portaobjetos con secciones de 280 casos de carcinoma de mama invasivo FFPE [aproximadamente 70 casos de cada una de las categorías de agrupamiento HER2 (0,1+, 2+, 3+)] a dos sitios de tinción (140 casos a cada uno) para que se llevara a cabo la tinción en un instrumento BenchMark XT y un instrumento BenchMark ULTRA con los respectivos protocolos de tinción recomendados y ultraView Universal DAB Detection Kit. Entre los controles figuraban PATHWAY HER-2 4 in 1 Control Slides y un segundo portaobjetos de cada caso teñido con el reactivo Ig negativo. Los casos con tinción del Sitio 1 y el Sitio 2 se dividieron en cuatro conjuntos de portaobjetos que se facilitaron, de uno en uno, a los tres lectores cualificados (anatomopatólogos), un lector en el Sitio 1, otro en el Sitio 2 y, el último, en el Sitio 3. Tanto los estados de los casos como la plataforma de tinción estaban enmascarados y los anatomopatólogos evaluaron en estas condiciones los cuatro conjuntos de portaobjetos y proporcionaron una puntuación clínica en cada caso (es decir, 0,1+, 2+, 3+). Los resultados se analizaron en Ventana. Los porcentajes de PPA (y el límite inferior de los intervalos de confianza del 95% bilateral) de la tinción con el anticuerpo del clon 4B5 en el instrumento BenchMark ULTRA frente al instrumento BenchMark XT, basados en la evaluación clínica (positiva, negativa), fueron del 91.6% (85.9), 91.2% (85.3) y del 94.9% (89.3) en el caso del Lector A, B y C, respectivamente. Los porcentajes de NPA (y el límite inferior de los intervalos de confianza del 95% bilateral) de la tinción con el anticuerpo del clon 4B5 en el instrumento BenchMark ULTRA frente al instrumento BenchMark XT, basados en la evaluación clínica (positiva, negativa), fueron del 91.9 (85.8), 93.8% (88.3), y 99.3 (96.3) para el Lector A, B y C, respectivamente. El OPA entre la tinción del clon 4B5 usando el instrumento BenchMark ULTRA frente al instrumento BenchMark XT, basados en el análisis 2x2 de la evaluación clínica (positiva, negativa), fueron del 91.8%, 92.5% y 97.4% en el caso del Lector A, B y C, respectivamente. En las tablas que aparecen a continuación se muestra la presentación 3x3 de la concordancia entre las plataformas para cada lector, con base en las puntuaciones clínicas (0/1+, 2+, 3+):
Tabla 24. Análisis 3x3 de los índices de concordancia entre plataformas del instrumento BenchMark ULTRA frente al instrumento BenchMark XT: Lector A
|
Instrumento BenchMark ULTRA |
Instrumento BenchMark XT |
|||
|
Lector A |
3+ |
2+ |
0, 1+ |
Total |
|
3+ |
84 |
11 |
1 |
96 |
|
2+ |
8 |
28 |
9 |
45 |
|
0, 1+ |
4 |
8 |
114 |
126 |
|
Total |
96 |
47 |
124 |
267 |
|
Porcentaje de concordancia global: n/N (%) (CI del 95%) |
226/267 (84.6) (79.8-88.5) |
|||
Tabla 25. Análisis 3x3 de los índices de concordancia entre plataformas del instrumento BenchMark ULTRA frente al instrumento BenchMark XT: Lector B
|
Instrumento BenchMark ULTRA |
Instrumento BenchMark XT |
|||
|
Lector B |
3+ |
2+ |
0, 1+ |
Total |
|
3+ |
64 |
2 |
1 |
67 |
|
2+ |
3 |
56 |
7 |
66 |
|
0, 1+ |
2 |
10 |
122 |
134 |
|
Total |
69 |
68 |
130 |
267 |
|
Porcentaje de concordancia global: n/N (%) (CI del 95%) |
242/267 (90.6) (86.5-93.6) |
|||
Tabla 26. Análisis 3x3 de los índices de concordancia entre plataformas del instrumento BenchMark ULTRA frente al instrumento BenchMark XT: Lector C
|
Instrumento BenchMark ULTRA |
Instrumento BenchMark XT |
|||
|
Lector C |
3+ |
2+ |
0, 1+ |
Total |
|
3+ |
64 |
1 |
0 |
65 |
|
2+ |
2 |
45 |
1 |
48 |
|
0, 1+ |
0 |
6 |
148 |
154 |
|
Total |
66 |
52 |
149 |
267 |
|
Porcentaje de concordancia global: n/N (%) (CI del 95%) |
257/267 (96,3) (93.2-98.0) |
|||
Reproducibilidad entre anatomopatólogos de las muestras del estudio de comparación de instrumentos:
Los índices de concordancia positiva y negativa con intervalos de confianza del 95% bilateral se calcularon con base en las seis posibles comparaciones entre pares de todos los lectores en cada plataforma.
En el instrumento BenchMark ULTRA, los porcentajes de PPA de los lectores A frente a B, A frente a C, B frente a C, B frente a A, C frente a A y C frente a B fueron del 94.7% (126/133), del 98.2% (111/113), del 98.2% (111/113), del 89.4% (126/141), del 78.7% (111/141) y del 83.5% (111/133) respectivamente. Los porcentajes NPA para el Lector A frente a B, A frente a C, B frente a C, B frente a A, C frente a A, y C frente a B fueron del 88,8% (119/134), 80,5% (124/154), 85,7% (132/154), 94,4% (119/126), 98,4% (124/126),
y 98,5% (132/134), respectivamente. El porcentaje de OPA más elevado se dio entre los lectores A y B (91.8%) y algo más bajo entre los lectores B y C (91.0%) y los lectores A y C (88.8%).
En el instrumento BenchMark XT, los porcentajes de PPA de los lectores A frente a B, A frente a C, B frente a C, B frente a A, C frente a A y C frente a B fueron del 94.9 % (130/137), del 98.3% (116/118), del 98.3% (116/118), del 90.9% (130/143), del 81.1% (116/143) y del 84.7% (116/137) respectivamente. Los porcentajes NPA para el Lector A frente a B, A frente a C, B frente a C, B frente a A, C frente a A, y C frente a B fueron del 90.0% (117/130), 81.9% (122/149), 85.9% (128/149), 94.4% (117/124), 98.4% (122/124), y 98.5% (128/130), respectivamente. El porcentaje de OPA más elevado se dio entre los lectores A y B (92.5%) y algo más bajo entre los lectores B y C (91.4%) y los lectores A y C (89.1%).
Estudio de comparación entre iVIEW DAB Detection Kit y ultraView Universal DAB Detection Kit:
En el Sitio 1, se utilizó una cohorte de 140 casos de carcinoma de mama invasivo FFPE [aproximadamente 35 casos de cada categoría de agrupamiento HER-2 (0,1+, 2+, 3+)] para llevar a cabo el estudio comparativo entre el kit de detección iVIEW DAB y ultraView Universal DAB Detection Kit de las tinciones con el clon 4B5 con un instrumento BenchMark ULTRA. Un único laboratorio de tinción y tres centros de lectura de EE. UU. participaron en el estudio de comparación de detección. En el caso de la tinción con el anticuerpo del clon 4B5 en el instrumento BenchMark ULTRA, los porcentajes de PPA entre los resultados obtenidos mediante los métodos iVIEW DAB Detection Kit y ultraView Universal DAB Detection Kit, con base en la evaluación clínica (positiva, negativa), fueron del 95.8% (68/71), del 96.9% (63/65) y del 96.5% (55/57) en el caso de los lectores A, B y C, respectivamente, y los porcentajes de NPA entre los métodos de detección fueron del 90.8% (59/65), del 91.5% (65/71) y del 97.5% (77/79) en el caso de los lectores A, B y C, respectivamente. Los porcentajes de OPA ente los kits de detección fueron del 93.4% (127/136), 94.1% (128/136) y 97.1% (132/136) para los Lectores A, B y C. En las tablas que aparecen a continuación se muestra la presentación 3x3 de los índices de concordancia de la comparación de la detección de cada lector, con base en las puntuaciones clínicas (0/1+, 2+, 3+).
Tabla 27. Análisis 3x3 de los índices de concordancia entre iVIEW DAB Detection Kit frente a ultraView Universal DAB Detection Kit del Lector A: tinción con el clon 4B5 en un instrumento BenchMark ULTRA
|
iVIEW DAB Detection Kit |
ultraView Universal DAB Detection Kit |
|||
|
Lector A |
3+ |
2+ |
0, 1+ |
Total |
|
3+ |
43 |
5 |
0 |
48 |
|
2+ |
3 |
17 |
6 |
26 |
|
0, 1+ |
0 |
3 |
59 |
62 |
|
Total |
46 |
25 |
65 |
136 |
|
Porcentaje de concordancia global: n/N (%) (CI del 95%) |
119/136 (87.5) (80.9-92.0) |
|||
Tabla 28. Análisis 3x3 de los índices de concordancia entre iVIEW DAB Detection Kit frente a ultraView Universal DAB Detection Kit del Lector B: tinción con el clon 4B5 en un instrumento BenchMark ULTRA
|
iVIEW DAB Detection Kit |
ultraView Universal DAB Detection Kit |
|||
|
Lector B |
3+ |
2+ |
0, 1+ |
Total |
|
3+ |
32 |
0 |
0 |
32 |
|
2+ |
0 |
31 |
6 |
37 |
|
0, 1+ |
1 |
1 |
65 |
67 |
|
Total |
33 |
32 |
71 |
136 |
|
Porcentaje de concordancia global: n/N (%) (CI del 95%) |
128/136 (94,1) (88.8-97.0) |
|||
Tabla 29. Análisis 3x3 de los índices de concordancia entre iVIEW DAB Detection Kit frente a ultraView Universal DAB Detection Kit del Lector C: tinción con el clon 4B5 en un instrumento BenchMark ULTRA
|
iVIEW DAB Detection Kit |
ultraView Universal DAB Detection Kit |
|||
|---|---|---|---|---|
|
Lector C |
3+ |
2+ |
0, 1+ |
Total |
|
3+ |
32 |
0 |
0 |
32 |
|
2+ |
0 |
23 |
2 |
25 |
|
0, 1+ |
0 |
2 |
77 |
79 |
|
Total |
32 |
25 |
79 |
136 |
|
Porcentaje de concordancia global: n/N (%) (CI del 95%) |
132/136 (97.1) (92.7-98.9) |
|||
Reproducibilidad entre anatomopatólogos de las muestras del estudio de comparación de detección:
Los índices de concordancia positiva y negativa con intervalos de confianza del 95% bilateral se calcularon con base en las seis posibles comparaciones entre pares de todos los lectores con cada método.
En el caso de iVIEW DAB Detection Kit, los porcentajes de PPA de los lectores A frente a B, A frente a C, B frente a C, B frente a A, C frente a A y C frente a B fueron del 100.0% (69/69), del 98.2% (56/57), del 96.5 % (55/57), del 93.2% (69/74), del 75.7% (56/74) y del 79.7% (55/69) respectivamente. Los porcentajes NPA para el Lector A frente a B, A frente a C, B frente a C, B frente a A, C frente a A, y C frente a B fueron del 92.5% (62/67), 77.2% (61/79), 82.3% (65/79), 100.0% (62/62), 98.4% (61/62) y 97.0% (65/67), respectivamente. El porcentaje de concordancia global más elevado se dio entre los lectores A y B (96.3%) y algo más bajo entre los lectores B y C (86.0%) y los lectores A y C (88.2%).
En el caso de ultraView Universal DAB Detection Kit, los porcentajes de PPA de los lectores A frente a B, A frente a C, B frente a C, B frente a A, C frente a A y C frente a B fueron del 96.9% (63/65), del 98.2% (56/57), del 98.2% (56/57), del 88.7% (63/71), del 78.9% (56/71) y del 86.2% (56/65) respectivamente. Los porcentajes NPA para el Lector A frente a B, A frente a C, B frente a C, B frente a A, C frente a A, y C frente a B fueron del 88.7% (63/71), 81.0% (64/79), 88.6% (70/79), 96.9% (63/65), 98.5% (64/65), y 98.6% (70/71), respectivamente. Los índices de concordancia global fueron parecidos entre cada par de lectores, con resultados de 92.6% (126/136), 88.2% (120/136) y de 92.6% (126/136) en el caso de los lectores A y B, los lectores A y C y los lectores B y C, respectivamente.
Rendimiento de análisis en los casos gástricos:
Estudios de precisión del instrumento BenchMark ULTRA y el instrumento BenchMark XT:
Se llevó a cabo un estudio de repetibilidad entre sesiones con el instrumento BenchMark XT en cinco sesiones que se realizaron durante un periodo de cinco días (no consecutivos). Cinco portaobjetos que contenían tres casos de tejido gástrico con puntuaciones de la expresión de HER2 de 0,1+, 2+ y 3+ demostraron una concordancia del 100% en el valor positivo o negativo de cada tejido.
Se llevó a cabo un estudio de repetibilidad entre sesiones con el instrumento BenchMark XT en 28 portaobjetos que contenían tres casos de tejido gástrico con puntuaciones de la expresión de HER2 de 0,1+, 2+ y 3+. Todos los casos obtuvieron una puntuación equivalente en el valor positivo o negativo de cada tipo de tejido.
Se evaluó la repetibilidad entre plataformas en tres instrumentos BenchMark XT. En estas sesiones, los 30 portaobjetos de dos bloques diferentes con varios tejidos que contenían tres casos de tejido gástrico con puntuaciones de la expresión de HER2 de 0, 1+, 2+ y 3+ obtuvieron una puntuación equivalente en el valor positivo o negativo de cada tipo de tejido.
Se evaluó la repetibilidad entre plataformas en tres instrumentos BenchMark ULTRA En estas sesiones, los 15 portaobjetos de un bloque de varios tejidos obtuvieron una puntuación equivalente en el valor positivo o negativo de cada tipo de tejido.
Se evaluó la repetibilidad entre plataformas en tres instrumentos BenchMark XT y tres instrumentos BenchMark ULTRA. En estas sesiones, los 30 portaobjetos de un bloque de varios tejidos obtuvieron una puntuación equivalente en el valor positivo o negativo de cada tipo de tejido.
Comparativa entre iVIEW DAB Detection Kit y ultraView Universal DAB Detection Kit con casos gástricos:
El clon 4B5 se utilizó para llevar a cabo una prueba comparativa entre kits de detección en dos instrumentos (el instrumento BenchMark XT y el instrumento BenchMark ULTRA) con iVIEW DAB Detection Kit y ultraView Universal DAB Detection Kit. Para llevar a cabo la prueba, se usaron dos cuentos diez casos de tejido. Los portaobjetos con tinción se evaluaron para asignar una puntuación clínica positiva o negativa.
Los índices de aceptabilidad de la morfología y del fondo fueron del 100% tanto en los kit de detección como en los instrumentos. En las tablas que aparecen a continuación se presentan las comparaciones directas de las evaluaciones clínicas positivas y negativas entre los kits de detección en cada instrumento.
Tabla 30. Evaluación clínica de ultraView Universal DAB Detection Kit frente a iVIEW DAB Detection Kit en el instrumento BenchMark XT
|
ultraView Universal DAB Detection Kit |
iVIEW DAB Detection Kit |
||
|
Positiva |
Negativa |
Total |
|
|
Positiva |
21 |
0 |
21 |
|
Negativa |
0 |
189 |
189 |
|
Total |
21 |
189 |
210 |
|
n/N |
% (CI del 95%) |
||
|
Porcentaje de concordancia positiva |
21/21 |
100 (84.5-100) |
|
|
Porcentaje de concordancia negativa |
189/189 |
100 (98.0-100) |
|
|
Porcentaje de concordancia global |
210/210 |
100 (98.2-100) |
|
Tabla 31. Comparación de la evaluación clínica en los instrumentos BenchMark XT y BenchMark ULTRA con ultraView Universal DAB Detection Kit
|
Instrumento BenchMark XT con ultraView Universal DAB Detection Kit |
Instrumento BenchMark ULTRA con ultraView Universal DAB Detection Kit |
||
|
Positiva |
Negativa |
Total |
|
|
Positiva |
20 |
1 |
21 |
|
Negativa |
0 |
189 |
189 |
|
Total |
20 |
190 |
210 |
|
n/N |
% (CI del 95%) |
||
|
Porcentaje de concordancia positiva |
20/20 |
100 (83.9-100) |
|
|
Porcentaje de concordancia negativa |
189/190 |
99.5 (97.1-99.9) |
|
|
Porcentaje de concordancia global |
209/210 |
99.5 (97.4-99.9) |
|
Reproducibilidad entre laboratorios del clon 4B5 en el carcinoma gástrico:
El estudio se llevó a cabo en tres sitios de prueba. Las muestras se seleccionaron para su incorporación al estudio en función de la puntuación clínica de IHC del clon 4B5, de forma que hubiera más o menos la misma cantidad de casos positivos (3+) que negativos (0,1+). Además, se estudiaron un máximo de cuatro casos de cáncer gástrico cualificados con 2+.
En los tres sitios se emplearon un instrumento BenchMark XT y un instrumento BenchMark ULTRA para llevar a cabo cuatro sesiones de tinción en cada instrumento. Se aleatorizó la tinción de los casos mediante un proceso de aleatorización estratificada que asignaba los casos de forma que cada sesión contuviera casos que representasen todas las categorías de puntuación de HER2 en el cáncer gástrico. Las sesiones de cada instrumento en cada uno de los sitios contenían los mismos casos. En cada sitio, se tiñó un portaobjetos de cada caso con el clon 4B5 y otro portaobjetos del mismo caso con CONFIRM Negative Control Rabbit Ig en un instrumento BenchMark ULTRA. Un segundo par de portaobjetos del mismo caso se tiñó de forma similar en el instrumento BenchMark XT en cada portaobjetos. Un lector cualificado de cada sitio asignó la puntuación a los portaobjetos del caso con las puntuaciones clínicas de IHC de cada muestra establecidas con anterioridad enmascaradas.
La concordancia global de todos los casos que se podían evaluar fue del 100% en las tres comparaciones entre sitios en el instrumento, BenchMark ULTRA y el instrumento BenchMark XT. La concordancia global entre el instrumento BenchMark ULTRA y el instrumento BenchMark XT de los casos que se podían evaluar fue del 100% en cada uno de los tres sitios. Los índices de aceptabilidad de fondo y morfología de todos los casos fueron del 100% en ambos instrumentos en los Sitios A y C y > 95% en ambos instrumentos en el Sitio B. Consulte las tablas que aparecen a continuación.
Tabla 32. Concordancia global de la evaluación clínica entre sitios: carcinoma gástrico, todos los casos que se podían evaluar
|
Instrumento BenchMark ULTRA |
Porcentaje de concordancia global (casos positivos y negativos) |
Porcentaje de concordancia global (incluidos los casos equívocos) |
|
Sitio A frente a Sitio B: n/N (%) (CI del 95%) |
30/30 (100%) (88.6 – 100) |
38/42 (90.5%) (77.9 – 96.2) |
|
Sitio A frente a Sitio C: n/N (%) (CI del 95%) |
30/30 (100%) (88.6 – 100) |
35/42 (83.3%) (69.4 – 91.7) |
|
Sitio B frente a Sitio C: n/N (%) (CI del 95%) |
30/30 (100%) (88.6 – 100) |
31/42 (73.8%) (58.9 – 84.7) |
|
Instrumento BenchMark XT |
Porcentaje de concordancia global (casos positivos y negativos) |
Porcentaje de concordancia global (incluidos los casos equívocos) |
|
Sitio A frente a Sitio B: n/N (%) (CI del 95%) |
31/31 (100%) (89.0 – 100.0) |
36/43 (83.7%) (70.0 – 91.9) |
|
Sitio A frente a Sitio C: n/N (%) (CI del 95 %) |
31/31 (100 %) (89.0 – 100.0) |
36/43 (83.7 %) (70.0 – 91.9) |
|
Sitio B frente a Sitio C: n/N (%) (CI del 95%) |
31/31 (100%) (89.0 – 100.0) |
35/43 (81.4%) (67.4 – 90.3) |
Tabla 33. Concordancia global de la evaluación clínica entre plataformas: carcinoma gástrico, todos los casos que se podían evaluar
|
Instrumento BenchMark ULTRA frente al instrumento BenchMark XT |
Porcentaje de concordancia global (casos positivos y negativos) |
Porcentaje de concordancia global (incluidos los casos equívocos) |
|
Sitio A: n/N (%) (CI del 95%) |
40/40 (100%) (91.2 – 100) |
42/44 (95.5%) (84.9 – 98.7) |
|
Sitio B: n/N (%) (CI del 95%) |
34/34 (100%) (89.8 – 100) |
37/42 (88.1%) (75.0 – 94.8) |
|
Sitio C: n/N (%) (CI del 95%) |
32/32 (100%) (89.3 – 100) |
38/44 (86.4%) (73.3 – 93.6) |
Tabla 34. Índices de aceptabilidad de la morfología y la tinción de fondo: carcinoma gástrico, todos los casos
|
Instrumento BenchMark ULTRA |
Sitio A |
Sitio B |
Sitio C |
|
Índices de aceptabilidad de la morfología |
44/44 (100%) |
43/44 (97.7%) |
44/44 (100%) |
|
Índices de aceptabilidad de la tinción de fondo |
44/44 (100%) |
42/44 (95.5%) |
44/44 (100%) |
|
Instrumento BenchMark XT |
Sitio A |
Sitio B |
Sitio C |
|
Índices de aceptabilidad de la morfología |
44/44 (100%) |
43/44 (97.7%) |
44/44 (100%) |
|
Índices de aceptabilidad de la tinción de fondo |
44/44 (100%) |
43/44 (97.7%) |
44/44 (100%) |
Estudio comparativo del instrumento BenchMark y del instrumento BenchMark GX con el instrumento BenchMark XT: Carcinoma gástrico
Se tiñeron los portaobjetos con secciones de tres microarrays de tejido que contenían casos de carcinoma gástrico FFPE [aproximadamente 50 casos por microarray de tejido] en un instrumento BenchMark XT, un instrumento BenchMark y un instrumento BenchMark GX con los respectivos protocolos de tinción recomendados para los kits de detección ultraView Universal DAB Detection Kit y iVIEW DAB. Entre los controles figuraban PATHWAY HER-2 4 in 1 Control Slides y un segundo portaobjetos de cada TMA teñido con el reactivo Ig negativo. Un lector (anatomopatólogo) asignó la puntuación a los portaobjetos con tinción.
Los índices de concordancia global (y los límites inferiores de los intervalos de confianza del 95% bilateral) de la tinción con el anticuerpo del clon 4B5, en función de la evaluación clínica (positiva, negativa), fueron los siguientes: Instrumento BenchMark frente al instrumento BenchMark XT con ultraView Universal DAB Detection Kit 98.0% (94.2-99.3), instrumento BenchMark GX frente al instrumento BenchMark XT con ultraView Universal DAB Detection Kit 97.4% (93.6-99.0), instrumento BenchMark frente al instrumento BenchMark XT con iVIEW DAB Detection Kit 96.6% (92.7-98.4), instrumento BenchMark GX frente a BenchMark XT con iVIEW DAB Detection Kit 95.9% (91.8-98.0).
Los índices de concordancia positiva (y los límites inferiores de los intervalos de confianza del 95% bilateral) de la tinción con el anticuerpo del clon 4B5, en función de la evaluación clínica (positiva, negativa), fueron los siguientes: Instrumento BenchMark frente al instrumento BenchMark XT con ultraView Universal DAB Detection Kit 91.7% (64.4-98.5), instrumento BenchMark GX frente al instrumento BenchMark XT con ultraView Universal DAB Detection Kit 78.6% (52.4-92.4), instrumento BenchMark frente al instrumento BenchMark XT con iVIEW DAB Detection Kit 80.0% (54.8-93.0), frente al instrumento BenchMark GX frente al instrumento BenchMark XT con iVIEW DAB Detection Kit 73.3% (48.0-89.1).
Los índices de concordancia negativos (y los límites inferiores de los intervalos de confianza del 95% bilateral) de la tinción con el clon 4B5, en función de la evaluación clínica (positiva, negativa), fueron los siguientes: Instrumento BenchMark frente al instrumento BenchMark XT con ultraView Universal DAB Detection Kit 98.5% (94.8-99.6), instrumento BenchMark GX frente al instrumento BenchMark XT con ultraView Universal DAB Detection Kit 99.3% (96.1-99.9), instrumento BenchMark frente al instrumento BenchMark XT con iVIEW DAB Detection Kit 98.1% (94.6-99.4), instrumento BenchMark GX frente al instrumento BenchMark XT con iVIEW DAB Detection Kit 98.1% (94.5-99.3). En las tablas de más abajo se muestra la presentación 2x2 de los índices de concordancia para cada comparación basada en la evaluación clínica (positiva, negativa).
Tabla 35. Análisis 2x2 de los índices de concordancia entre plataformas del instrumento BenchMark frente al instrumento BenchMark XT con ultraView Universal DAB Detection Kit: carcinoma gástrico
|
Clon 4B5 con ultraView Universal DAB Detection Kit |
|||
|---|---|---|---|
|
Instrumento BenchMark |
Instrumento BenchMark XT |
||
|
Positiva |
Negativa |
Total |
|
|
Positiva |
11 |
2 |
13 |
|
Negativa |
1 |
133 |
134 |
|
Total |
12 |
135 |
147 |
|
n/N |
% (CI del 95%) |
||
|
Porcentaje de concordancia global |
144/147 |
98.0 % (94.2-99.3) |
|
|
Porcentaje de concordancia positiva |
11/12 |
91.7% (64.6-98.5) |
|
|
Porcentaje de concordancia negativa |
133/135 |
98.5% (94.8-99.6) |
|
Tabla 36. Análisis 2x2 de los índices de concordancia entre plataformas del instrumento BenchMark GX frente al instrumento BenchMark XT con ultraView Universal DAB Detection Kit: carcinoma gástrico
|
Clon 4B5 con ultraView Universal DAB Detection Kit |
|||
|---|---|---|---|
|
Instrumento BenchMark GX |
Instrumento BenchMark XT |
||
|
Positiva |
Negativa |
Total |
|
|
Positiva |
11 |
1 |
12 |
|
Negativa |
3 |
140 |
143 |
|
Total |
14 |
141 |
155 |
|
n/N |
% (CI del 95%) |
||
|
Porcentaje de concordancia global |
151/155 |
97.4% (93.6-99.0) |
|
|
Porcentaje de concordancia positiva |
11/14 |
78.6% (52.4-92.4) |
|
|
Porcentaje de concordancia negativa |
140/141 |
99.3% (96.1-99.9) |
|
Tabla 37. Análisis 2x2 de los índices de concordancia entre plataformas del instrumento BenchMark frente al instrumento BenchMark XT con el kit de detección iVIEW DAB: carcinoma gástrico
|
Clon 4B5 con iVIEW DAB Detection Kit |
|||
|
Instrumento BenchMark |
Instrumento BenchMark XT |
||
|
Positiva |
Negativa |
Total |
|
|
Positiva |
12 |
3 |
15 |
|
Negativa |
3 |
156 |
159 |
|
Total |
15 |
159 |
174 |
|
n/N |
% (CI del 95%) |
||
|
Porcentaje de concordancia global |
168/174 |
96.6% (92.7-98.4) |
|
|
Porcentaje de concordancia positiva |
12/15 |
80.0% (54.8-93.0) |
|
|
Porcentaje de concordancia negativa |
156/159 |
98.1% (94.6-99.4) |
|
Tabla 38. Análisis 2x2 de los índices de concordancia entre plataformas del instrumento BenchMark GX frente al instrumento BenchMark XT con el kit de detección iVIEW DAB: carcinoma gástrico
|
Clon 4B5 con iVIEW DAB Detection Kit |
|||
|
Instrumento BenchMark GX |
Instrumento BenchMark XT |
||
|
Positiva |
Negativa |
Total |
|
|
Positiva |
11 |
3 |
14 |
|
Negativa |
4 |
154 |
158 |
|
Total |
15 |
157 |
172 |
|
n/N |
% (CI del 95%) |
||
|
Porcentaje de concordancia global |
165/172 |
95.9% (91.8-98.0) |
|
|
Porcentaje de concordancia positiva |
11/15 |
73.3% (48.0-89.1) |
|
|
Porcentaje de concordancia negativa |
154/157 |
98.1% (94.5-99.3) |
|
USO PREVISTO:
VENTANA HER2 (4B5) RABBIT MONOCLONAL PRIMARY ANTIBODY RXDX está destinado a su uso para la detección semicuantitativa del antígeno HER2 mediante inmunohistoquímica (IHC) en secciones de tejido de mama y de tejido gástrico fijado con formol y embebido en parafina, teñidas en un instrumento BenchMark IHC/ISH.
Este dispositivo IHC está indicado como pruebas diagnósticas con fines terapéuticos para identificar a pacientes con cáncer de mama que son elegibles para recibir un tratamiento con trastuzumab (IHC 3+ o IHC 2+/ISH amplificado), pertuzumab (IHC 3+ o IHC 2+/ISH amplificado), trastuzumab emtansina (IHC 3+ o IHC 2+/ISH amplificado) o trastuzumab deruxtecan (IHC 1+ o IHC 2+/ISH no amplificado). Además, este dispositivo IHC está indicado como prueba diagnóstica con fines terapéuticos para identificar a pacientes con cáncer gástrico para los cuales se considera el tratamiento con trastuzumab (IHC 3+ o IHC 2+/ISH amplificado).
La interpretación de este producto debe correr a cargo de un anatomopatólogo cualificado junto con un examen histológico, la información clínica pertinente y los controles adecuados.
Este anticuerpo está destinado para uso diagnóstico in vitro (IVD).
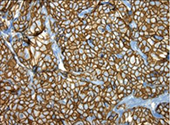
Figura 1. Tinción con VENTANA HER2 (4B5) RABBIT MONOCLONAL PRIMARY ANTIBODY RXDX en carcinoma de mama.

Figura 2. Tinción 3+ con VENTANA HER2 (4B5) RABBIT MONOCLONAL PRIMARY ANTIBODY RXDX en carcinoma gástrico.
LIMITACIONES:
Limitaciones generales:
1 La inmunohistoquímica es un proceso de diagnóstico que comprende varios pasos y requiere una formación especializada en cuanto a la correcta elección de los tejidos y los reactivos, la fijación, el procesamiento, la preparación de portaobjetos de inmunohistoquímica y la interpretación de los resultados de la tinción.
2 La tinción del tejido depende de la manipulación y el procesamiento del tejido antes de la tinción. Una fijación, congelación, descongelación, lavado, secado, calentamiento y seccionado incorrectos o la contaminación con otros tejidos o líquidos puede provocar la aparición de artefactos, el enmascaramiento de anticuerpos o resultados falsos negativos. La existencia de resultados incoherentes puede ser el resultado de la introducción de variaciones en los métodos de fijación e inclusión o puede derivarse de las irregularidades características del tejido.
3 Una contratinción incompleta o excesiva puede comprometer la correcta interpretación de los resultados.
4 La interpretación clínica de cualquier tinción positiva, al igual que su ausencia, es algo que se debe evaluar en función del contexto del historial médico, la morfología y otros criterios histopatológicos. La interpretación clínica de todas las tinciones, o la ausencia de estas, se debe complementar con los estudios morfológicos y los controles correspondientes, así como con otras pruebas diagnósticas. Es responsabilidad del anatomopatólogo cualificado estar familiarizado con los anticuerpos, los reactivos y los métodos que se utilizan para interpretar la preparación de la tinción. La tinción se debe llevar a cabo en un laboratorio certificado y con licencia y bajo la supervisión de un anatomopatólogo, que será el responsable de revisar los portaobjetos teñidos y de garantizar la idoneidad de los controles positivos y negativos.
5 VENTANA proporciona anticuerpos y reactivos con una dilución óptima para su uso siempre que se respeten las instrucciones que se suministran. Cualquier diferencia en la forma de llevar a cabo los procedimientos de prueba recomendados pueden invalidar los resultados previstos. Deben emplearse los controles adecuados y documentarlos. Los usuarios que no sigan los procedimientos de prueba recomendados deberán hacerse responsables de la interpretación de los resultados de la paciente.
6 Este producto no se ha concebido para su uso en citometría de flujo; sus características de rendimiento no se han definido.
7 Los reactivos pueden mostrar reacciones imprevistas en tejidos que no se hayan probado previamente. La posibilidad de encontrarse con reacciones no previstas incluso en los grupos de tejidos probados no se puede eliminar por completo, dada la variabilidad biológica de la expresión del antígeno en neoplasias u otros tejidos anatomopatológicos.38 Póngase en contacto con su representante local de asistencia técnica de Roche si cuenta con reacciones imprevistas documentadas.
8 En los tejidos de pacientes contagiados con el virus de la hepatitis B o que contienen antígeno de superficie de hepatitis B (HBsAg) es posible que se presente una tinción no especifica con la peroxidasa de rábano.39
9 Los resultados falsos positivos se pueden observar por la unión de proteínas no inmunológicas o por los productos de reacción con sustratos. También es posible que aparezcan como consecuencia de la actividad pseudoperoxidasa (eritrocitos), de la actividad peroxidasa endógena (citocromo C) o de la biotina endógena (como en el caso del hígado, del cerebro, de la mama o del riñón) en función del tipo de inmunotinción que se haya utilizado.40
10 Como ocurre en todas las pruebas de inmunohistoquímica, un resultado negativo significa que no se ha detectado el antígeno en concreto, no necesariamente que el antígeno no esté presente en las células o en el tejido que se ha usado para el ensayo.
Limitaciones específicas:
1 Este anticuerpo se ha optimizado, tal y como se indica en la Tabla 1 y la Tabla 2, para los instrumentos BenchMark y las sustancias químicas de detección. No respetar los protocolos de tinción recomendados en la Tabla 1 y la Tabla 2 puede dar lugar a muestras Negative Reagent Control (NRC) inaceptables y a la obtención de muestras teñidas con el anticuerpo VENTANA HER2 (4B5) Assay con una puntuación HER2 modificada. Un aumento del tiempo de incubación del anticuerpo puede producir una tinción inaceptable en el NRC, que podría impedir la evaluación de la muestra con VENTANA HER2 (4B5) Assay. La reducción o ampliación de los tiempos de acondicionamiento celular suelen producir muestras con tinción del anticuerpo VENTANA HER2 (4B5) Assay con puntuaciones de HER2 alteradas, lo que puede dar lugar a la toma de decisiones poco acertadas en el tratamiento de los pacientes. Para obtener más información sobre las variables de fijación, consulte “Immunohistochemistry Principles and Advances”.33
2 El anticuerpo, cuando se utiliza junto con los kit de detección y los accesorios VENTANA, detecta el antígeno que permanece una vez se han llevado a cabo la fijación en formol, el procesamiento del tejido y el corte. Los usuarios que no sigan los procedimientos de prueba recomendados deberán hacerse responsables de la interpretación y validación de los resultados del paciente.
3 Los portaobjetos deberían teñirse inmediatamente, dado que la antigenicidad de las secciones de los cortes de tejido puede disminuir con el tiempo y es posible que se dañen debido a los factores ambientales si se almacenan durante un periodo prolongado. Los portaobjetos que se hayan dejado secar al aire se deberían desecar y conservar a una temperatura de entre 2 y 8°C. Los estudios han demostrado que los portaobjetos sin tinción mantienen la estabilidad del antígeno durante al menos 45 días. Los laboratorios deben validar las fechas de caducidad en su propio entorno si se quiere aplicar una fecha de caducidad superior a los 45 días.
4 No se ha sometido a pruebas la especificidad de la médula ósea. El usuario debe establecer la tinción que considera adecuada en los tejido mencionados anteriormente antes de la interpretación de la información sobre la tinción.
5 La tinción inmunohistoquímica con el clon 4B5 puede dar lugar a tinción citoplasmática y nuclear de la mucosa gástrica normal y, en reducidas ocasiones, de las células neoplásicas del carcinoma gástrico y de la unión gastroesofágica. La naturaleza de esta tinción nuclear y tinción citoplasmática se desconoce actualmente. El patrón de tinción no debe confundirse con una tinción membranosa discontinua que indica la positividad en HER2 en células neoplásicas.
6 Es posible que no todos los ensayos estén registrados en todos los instrumentos. Póngase en contacto con el representante local de servicio Roche para obtener más información.
7 Se ha notificado la presencia de cambios en el estado de HER2 con la progresión metastásica o tras el tratamiento neoadyuvante con quimioterapia. En función de estas observaciones, puede estar justificado obtener una nueva muestra para determinar el estado de HER2 en el momento en el que se vaya a poner el tratamiento, en lugar de basarse en el estado histórico de HER2.41
PREPARACIÓN DE MUESTRAS:
Los tejidos FFPE que se procesan de forma habitual resultan adecuados para su uso con este anticuerpo primario cuando se utilizan con los kits de detección de VENTANA y los instrumentos BenchMark IHC/ISH. El fijador de tejido recomendado es formol tamponado neutro al 10%.29 Los portaobjetos deberían teñirse inmediatamente, ya que la antigenicidad de las secciones de los cortes de tejido puede disminuir con el tiempo.
Se recomienda analizar controles positivos y negativos a la vez que las muestras desconocidas.
Es necesario cortar secciones de tejido de un grosor aproximado de 4 μm y recogerlas en el portaobjetos de vidrio. Los portaobjetos deberán ser del tipo Superfrost Plus o equivalente. Los estudios señalan que las secciones de tejido que se han dejado secar al aire y las secciones de estirpes celulares almacenadas a una temperatura de entre 2 y 8°C se conservan estables durante un mínimo de 45 días. Cada laboratorio debe validar la estabilidad del portaobjetos con el corte para sus propios procedimientos y condiciones de almacenamiento medioambiental.
PRINCIPIO DEL PROCEDIMIENTO:
El ensayo VENTANA HER2 (4B5) Assay contiene un anticuerpo monoclonal de conejo que se une a HER2 en secciones de tejido FFPE. El anticuerpo específico se puede localizar con un conjugado de anticuerpo secundario y HRP (ultraView Universal DAB Detection Kit). El complejo específico de anticuerpo-enzima se puede visualizar a través del precipitado del producto de reacción enzimático. Cada paso se incuba con tiempos y temperaturas específicos. Cuando finaliza cada uno de los pasos de incubación, las secciones se enjuagan en el instrumento con el fin de detener la reacción y eliminar el material que no se ha ligado y que podría impedir la reacción que se quiere lograr en los pasos posteriores. Además, en el instrumento se aplica Liquid Coverslip, que reduce al máximo la evaporación de los reactivos acuosos del portaobjetos que contiene la muestra.
Los casos clínicos se deberían evaluar dentro del contexto del rendimiento de los controles correspondientes. Se recomienda la incorporación de un control de tejido positivo fijado y procesado con el mismo método que la muestra de la paciente (por ejemplo, un carcinoma de mama positivo débil). Además de la tinción con el anticuerpo VENTANA HER2 (4B5) Assay, se debe teñir un segundo portaobjetos con CONFIRM Negative Control Rabbit Ig. Para que la prueba tenga validez, el tejido de control positivo debe presentar tinción de membrana en las células tumorales. Todos estos componentes deben dar resultado negativo en la tinción con CONFIRM Negative Control Rabbit Ig.
Además, se recomienda incluir un portaobjetos de control tisular negativo (como un carcinoma de mama o gástrico negativo en HER2) en cada lote de muestras que se procesen y se analicen en el instrumento BenchMark IHC/ISH. Este control tisular negativo debería teñirse con el anticuerpo VENTANA HER2 (4B5) RxDx para garantizar que la ampliación del antígeno y el resto de los procedimientos previos al tratamiento no generan falsos positivos en las tinciones.
PROCEDIMIENTO DE TINCIÓN:
Los anticuerpos primarios VENTANA se han desarrollado para su uso en los instrumentos BenchMark IHC/ISH junto con los kits de detección de VENTANA y sus accesorios.
Consulte las tablas que aparecen a continuación para ver los protocolos de tinción recomendados.
Este anticuerpo se ha optimizado para tiempos de incubación específicos, pero el usuario debe validar los resultados obtenidos con este reactivo.
Los parámetros de los procedimientos automatizados se pueden visualizar, imprimir y editar según el procedimiento descrito en el Manual del usuario del instrumento. Consulte la hoja de datos de ultraView DAB Detection Kit apropiada para obtener más información sobre los procedimientos de tinción de IHC.
Para obtener más información sobre el uso adecuado de este dispositivo, consulte la hoja de datos del dispensador en línea asociada al N/P 790-7167.
NOTA: Los procedimientos de tinción se resumen en la Tabla 1 (en el caso de evaluación de la positividad de HER2 y evaluación de HER2 bajo en casos de cáncer de mama), Tabla 2 (para evaluación de positividad de HER2 únicamente en casos de cáncer de mama) y Tabla 3 (para evaluación de positividad de HER2 en casos de cáncer gástrico). Los pasos del protocolo de tinción recomendado, incluido el acondicionamiento celular y la incubación del anticuerpo, son los mismos para el cribado de todos los casos de cáncer de mama con HER2; sin embargo, en los procedimientos de tinción se dispone de diferentes niveles de modificación por parte del usuario. Cuando se realiza el cribado de pacientes para una evaluación potencial de HER2 bajo, debe utilizarse el procedimiento de tinción indicado en la Tabla 1. Si la intención de la prueba es notificar el estado de bajo nivel de HER2 (que se define como una puntuación IHC de 1+ o 2+ con un estado de HER2 no amplificado confirmado), se debe emplear el procedimiento de tinción que se recoge en la Tabla 1. Si la intención es notificar el estado de positividad de HER2 (que se define como una puntuación IHC de 3+ o 2+ con un estado de HER2 amplificado confirmado), se debe emplear el procedimiento de tinción que se recoge en la Tabla 1 o en la Tabla 2. Para garantizar la utilización más exhaustiva de los resultados para un estado de HER2 bajo o un estado de HER2 positivo en casos de cáncer de mama, se recomienda encarecidamente usar el procedimiento de tinción indicado en la Tabla 1.
Procedimiento de tinción para todas las evaluaciones de HER2 incluyendo una evaluación potencial de HER2 bajo en muestras de mama:
El protocolo de tinción y el procedimiento que se recogen en esta sección y en la Tabla 1 son adecuados para su uso en todos los casos de cribado de HER2 de carcinomas de mama y se debe emplear durante la evaluación de las muestras del paciente para valorar los posibles tratamientos dirigidos a los niveles bajos de HER2. No respetar los protocolos de tinción recomendados puede dar lugar a muestras teñidas con HER2 inaceptables y a la obtención de una puntuación HER2 distinta, especialmente en casos con una expresión de HER2 baja (IHC 1+). La reducción o ampliación de los tiempos de acondicionamiento celular suelen producir muestras con tinción HER2 con puntuaciones HER2 alteradas, lo que puede dar lugar a la toma de decisiones poco acertadas en el tratamiento de los pacientes. El procedimiento de tinción especificado en la Tabla 1 no permite modificar el acondicionamiento celular ni el tiempo de incubación del anticuerpo para reducir este riesgo, ya que se trata del único protocolo de tinción validado para su uso en la evaluación de muestras de pacientes con HER2 bajo potencial.
Tabla 1. Protocolo de tinción para VENTANA HER2 (4B5) Assay para todas las evaluaciones de HER2 en muestras de mama incluyendo una evaluación potencial de HER2 bajo en un instrumento BenchMark IHC/ISH
|
Tipo de procedimiento |
Método |
||
|
BenchMark GX |
BenchMark XT |
BenchMark ULTRA o BenchMark ULTRA PLUS |
|
|
Procedimiento de tinción |
GX VENTANA HER2 4B5 |
XT VENTANA HER2 4B5 |
ULTRA VENTANA HER2 4B5 |
|
Anticuerpo (primario)* |
VENTANA HER2 4B5 Ab: 16 min o CONFIRM Neg Ctl Rbt Ig:16 min |
VENTANA HER2 4B5 Ab: 16 min o CONFIRM Neg Ctl Rbt Ig:16 min |
VENTANA HER2 4B5 Ab: 12 min o CONFIRM Neg Ctl Rbt Ig:12 min |
|
Contratinción |
Hematoxylin II, 4 minutos |
||
|
Post-contratinción |
Bluing, 4 minutos |
||
* Esta es una condición programada previamente y el usuario no puede seleccionar los pasos.
Procedimientos de tinción alternativos que únicamente se aplican para la evaluación de la posible positividad en HER2 en muestras de mama o gástricas:
Los procedimientos y los protocolos de tinción que se recogen en la Tabla 2 y en la Tabla 3 están indicados para su uso en la evaluación de la positividad en HER2 (definida como sobreexpresión de proteína, puntuación IHC 3+ o 2+ (con un estado de HER2 amplificado confirmado) y no están indicados para la evaluación de niveles bajos de expresión de HER2 (definidos con la puntuación IHC 1+ o 2+ (con un estado de HER2 no amplificado confirmado). Los tiempos de incubación del anticuerpo y de acondicionamiento celular recomendados que se especifican en la Tabla 2 y en la Tabla 3 no están bloqueados y los puede modificar el usuario. Algunas de las posibles razones que existen para querer modificar los parámetros son la variación en la fijación o el procesamiento de los tejidos (al no haberse respetado la fijación ni el procesamiento recomendado de las muestras), diferencias en el instrumento o las condiciones generales del laboratorio y la preferencia del lector. Para obtener más información sobre las variables de fijación, consulte “Immunohistochemistry Principles and Advances”.33 Los usuarios que no sigan los procedimientos de prueba recomendados deberán hacerse responsables de la interpretación y validación de los resultados del paciente.
Tabla 2. Protocolo de tinción recomendado para VENTANA HER2 (4B5) Assay con ultraView DAB Detection Kit en muestras de mama en un instrumento BenchMark IHC/ISH
|
Tipo de procedimiento |
Método |
||
|
BenchMark GX |
BenchMark XT |
BenchMark ULTRA o BenchMark ULTRA PLUS |
|
|
Procedimiento de tinción |
Procedimiento de tinción ultraView DAB |
Procedimiento de tinción ultraView DAB |
Procedimiento de tinción ultraView DAB |
|
Desparafinado |
Seleccionado |
Seleccionado |
Seleccionado |
|
Cell Conditioning (desenmascaramiento del antígeno) |
CC1, Suave |
CC1, Suave |
ULTRA CC1, Suave |
|
Anticuerpo (Primario) |
16 minutos, 37 °C |
16 minutos, 37 °C |
12 minutos, 36 °C |
|
ultraWash |
Seleccionado |
||
|
Contratinción |
Hematoxylin II, 4 minutos |
||
|
Post-contratinción |
Bluing, 4 minutos |
||
Tabla 3. Protocolo de tinción recomendado para VENTANA HER2 (4B5) Assay con ultraView DAB Detection Kit en muestras gástricas en un instrumento BenchMark IHC/ISH
|
Tipo de procedimiento |
Método |
||
|---|---|---|---|
|
BenchMark GX |
Benchmark XT |
BenchMark ULTRA |
|
|
Procedimiento de tinción |
Procedimiento de tinción ultraView DAB |
Procedimiento de tinción ultraView DAB |
Procedimiento de tinción ultraView DAB |
|
Desparafinado |
Seleccionado |
Seleccionado |
Seleccionado |
|
Cell Conditioning (desenmascaramient o del antígeno) |
CC1, Suave |
CC1, Suave |
ULTRA CC1, Suave |
|
Anticuerpo (Primario) |
16 minutos, 37 °C |
16 minutos, 37 °C |
12 minutos, 36 °C |
|
ultraWash |
Seleccionado |
||
|
Contratinción |
Hematoxylin II, 4 minutos |
||
|
Post-contratinción |
Bluing, 4 minutos |
||
IMPORTANCIA CLÍNICA:
El cáncer de mama es el carcinoma diagnosticado con mayor frecuencia en mujeres en el mundo.16 La detección temprana y la selección del tratamiento adecuado tienen una importante repercusión en la supervivencia global.7,17 Aproximadamente entre el 15 y el 30 por ciento de los cánceres ductales invasivos de mama son positivos a HER2.7,10 Casi todos los casos de la enfermedad de Paget en la mama y hasta un 90 por ciento de los casos del carcinoma ductal in situ de tipo comedo son positivos.18,19 El estado positivo en HER2 ha definido un subgrupo de pacientes de cáncer de mama que pueden beneficiarse del tratamiento orientado a HER2 durante más de 20 años.7,17 La población con resultados positivos en HER2 se ha definido históricamente como aquellas pacientes que presentan sobreexpresión de proteína HER2 según la evaluación mediante inmunohistoquímica (IHC) en función de un sistema de puntuación de IHC semicuantitativo (0, 1+, 2+ o 3+) y/o la amplificación del gen según la evaluación con hibridación in situ (ISH).17 La positividad en HER2 se ha relacionado estrechamente con la sobreexpresión de proteína (puntuación de IHC de 3+). En casos con una sobreexpresión límite (puntuación IHC 2+, equívoca) es posible que tenga que realizarse una prueba reflex confirmatoria para evaluar la amplificación del gen según el algoritmo de evaluación de HER2 definido.17
Los fármacos terapéuticos disponibles en el mercado, como Herceptin (trastuzumab), PERJETA (pertuzumab) y KADCYLA (ado-trastuzumab emtansina/trastuzumab emtansina) han demostrado ofrecer un beneficio clínico en los pacientes con cáncer de mama con HER2-positivo deteniendo y, en algunos casos, invirtiendo el crecimiento de su cáncer.20,24 Trastuzumab y pertuzumab son anticuerpos monoclonales humanizados que se unen a la proteína HER2 en la superficie celular e interrumpen la transducción de la señal HER2 mediada.20,24,25 Trastuzumab emtansina es un conjugado de anticuerpo y fármaco compuesto de trastuzumab y el agente citotóxico DM1 conjugado por medio de un enlazador que puede disasociarse.23 Solo los pacientes con cáncer de mama de HER2 positivo (puntuación IHC 3+ o 2+ con un estado amplificado HER2 confirmado) deben beneficiarse del tratamiento con trastuzumab (Herceptin), pertuzumab (PERJETA) o trastuzumab emtansina (KADCYLA).16,18
Aproximadamente el 40-50 por ciento de los pacientes con cáncer de mama tienen tumores que no demuestran amplificación del gen HER2 y no sobreexpresan el receptor; con todo, se detectan niveles bajos de expresión HER2.8,10 Los casos con una expresión de HER2 baja (puntuación IHC 1+ o 2+ (con un estado no amplificado de HER2 confirmado)) suelen considerarse HER2 negativos y se excluyen de las opciones de tratamiento enfocadas en HER2.8 Recientemente, se ha observado un beneficio del tratamiento anti-HER2 con trastuzumab deruxtecan (ENHERTU®) en pacientes con cáncer de mama con bajos niveles de la expresión HER2.6,27 trastuzumab deruxtecan es un conjugado de fármaco anticuerpo que contiene una base de anticuerpo monoclonal (trastuzumab) enfocado en HER2, un enlazar que puede disasociarse y un derivado de exatecan permeable de membrana celular (una carga de inhibidor I de topoisomerasa).10
El diagnóstico in vitro para la evaluación del estado HER2 en pacientes con cáncer de mama es importante como ayuda clínica en la determinación de la terapia con trastuzumab (Herceptin), pertuzumab (PERJETA), trastuzumab emtansina (KADCYLA) o trastuzumab deruxtecan (ENHERTU®).17 La detección basada en IHC de la expresión de proteína HER2 se utiliza como ayuda en la evaluación de los pacientes con cáncer de mama para los cuales se consideran tratamientos enfocados de HER2 con trastuzumab (Herceptin), pertuzumab (PERJETA), trastuzumab emtansina (KADCYLA) o trastuzumab deruxtecan (ENHERTU®).
El cáncer gástrico es el quinto cáncer más común y la causa principal de muerte relacionada con el cáncer en todo el mundo.13 La cirugía es el tratamiento más común para el cáncer gástrico.14,28 Sin embargo, la mayoría de casos de cáncer gástrico se detectan en un estado avanzado y la cirugía suele ser difícil de realizar.14,28 La quimioterapia se utiliza para tratar el cáncer gástrico avanzado incluso si la supervivencia de estos pacientes es baja.14,28 La terapia enfocada de HER2 con trastuzumab es un elemento fundamental en el tratamiento del cáncer de mama invasivo y tiene un valor terapéutico en el tratamiento de pacientes con cáncer gástrico con una sobreexpresión del receptor.12,14 La demostración de la amplificación del gen HER2 y/o la sobreexpresión de proteína es fundamental para seleccionar pacientes para la terapia con trastuzumab.12,14 Los estudios clínicos han demostrado que los pacientes con cáncer de mama o cáncer gástrico con una alta sobreexpresión de proteína HER2 y/o el beneficio de la amplificación del gen se benefician en mayor medida de trastuzumab.12,21 La detección basada en IHC de la expresión de la proteína HER2 se utiliza como una ayuda en la evaluación del cáncer gástrico en pacientes para los cuales se está considerando el tratamiento con trastuzumab (Herceptin).
RESUMEN Y EXPLICACIÓN:
VENTANA HER2 (4B5) RABBIT MONOCLONAL PRIMARY ANTIBODY RXDX (VENTANA HER2 (4B5) Assay) contiene un anticuerpo monoclonal de conejo (clon 4B5) generado contra el dominio interno del receptor 2 del factor de crecimiento epidérmico humano (HER2).
HER2 se clonó y caracterizó.1 El clon 4B5 parece reaccionar con la proteína 185 kDa de los lisados celulares SK-BR-3 a través de la técnica Western blotting. SK-BR-3 es una línea celular de carcinoma de mama, que tiene una sobreexpresión 128 veces superior de mRNA de HER2.2 El tamaño de la banda identificada está bien relacionada con el tamaño notificado para la proteína HER2 (185 kDa).1 Los experimentos de inmunohistoquímica (IHC) con líneas celulares transfectadas (HEK293) han demostrado que el clon 4B5 tiñe células transfectadas con HER2 y células transfectadas con HER4, aunque no se observó tinción de células transfectadas con HER1 o HER3 y los datos de Western blot con la proteína recombinante HER4 también indicaron que el clon 4B5 reconoce un epítopo de HER4.3 A pesar de ello, no se ha observado que HER2 (4B5) tenga una reacción cruzada con HER4 en la tinción IHC de tejido de mama fijado con formol y embebido en parafina (FFPE).4
HER2 es un receptor transmembrana de tirosina quinasa con una estructura parecida a la del receptor del factor de crecimiento epidérmico.5,6 La amplificación del gen y la correspondiente sobreexpresión de HER2 se ha observado en una variedad de tumores, como los carcinomas de mama.5,6,7 La sobreexpresión de proteína, inducida por la amplificación del gen HER2, es la causa principal de la carcinogénesis inducida por HER2.5 La amplificación del gen suele producir un aumento significativo de los receptores HER2 en la membrana celular.5,6 La sobreexpresión de proteína HER2 aumenta la señal de transducción y regula la proliferación y la diferenciación que, en último término, provoca la formación de tumores.5,6
Un espectro de la expresión de la proteína HER2 se ha observado en ausencia de la amplificación del gen.8 Se han propuesto diversos factores para explicar los niveles intermedios de la expresión de la proteína HER2 en casos de cáncer de mama en ausencia de la amplificación del gen, incluyendo la diafonía entre las trayectorias de señalización del receptor de estrógeno y HER2.8,9 La expresión de la proteína HER2 que no se considera una sobreexpresión puede clasificarse como expresión de HER2 baja.8,10,11
La sobreexpresión de proteína HER2 y/o la amplificación del gen ocurre en adenocarcinoma de unión gastroesofágica y gástrica.12,13,14 Se ha notificado un amplio intervalo de frecuencia de sobreexpresión de HER2 en los diversos estudios publicados. Sin embargo, uno de los conjuntos de datos de cribado más grandes que incluía a 3803 pacientes con adenocarcinoma de unión gastroesofágica y gástrica notificó que el 22 porcentaje de los pacientes tuvieron un resultado positivo para la amplificación del gen o la sobreexpresión de proteína HER2.15
REFERENCIAS:
1 Akiyama T, et al. The product of the human c-erbB-2 Gene: A 185-kilodalton glycoprotein with tyrosine kinase activity. Science. 1986;232: 1644-1646.
2 Kraus MH, Popescu NC, Amsbaugh C, King RC. Overexpression of EGF receptor- related proto-oncogene erbB-2 in human mammary tumor cell lines by different molecular mechanisms. EMBO. 1987;6: 605-610.
3 Schrohl AS, Pedersen HC, Jensen SS, Nielsen SL, Brünner N. Human epidermal growth factor receptor 2 (HER2) immunoreactivity: specificity of three pharmacodiagnostic antibodies. Histopathology. 2011;59(5):975-983.
4 Jay JI, Brunhoeber PS, Smith MH, et al. Immunohistochemical analysis of the monoclonal antibody 4B5 in breast tissue expressing human epidermal growth factor receptor 4 (HER4). Histopathology. 2013;62(4):563-577.
5 Moasser MM. The Oncogene HER2: Its Signaling and Transforming Functions and Its Role in Human Cancer Pathogenesis. Oncogene. 2007;26(45):6469-6487.
6 Hsu JL, Hung MC. The Role of HER2, EGFR, and Other Receptor Tyrosine Kinases in Breast Cancer. Cancer Metastasis Rev. 2016;35(4):575-588.
7 Iqbal N, Iqbal N. Human Epidermal Growth Factor Receptor 2 (HER2) in Cancers: Overexpression and Therapeutic Implications. Mol Biol Int. 2014;2014:852748.
8 Tarantino P, Hamilton E, Tolaney SM, et al. HER2-low Breast Cancer: Pathological and Clinical Landscape. J Clin Oncol. 2020;38:1951-1962.
9 Osborne CK, Shou J, Massarweh S, et al: Crosstalk between estrogen receptor and growth factor receptor pathways as a cause for endocrine therapy resistance in breast cancer. Clin Cancer Res 11:865s-870s, 2005.
10 Eiger D, Agostinetto E, Saude-Conde R, de Azambuja E. The Exciting New Field of HER2-low Breast Cancer Treatment. Cancers (Basel). 2021;13.
11 Zhang H, Katerji H, Turner BM, Hicks DG. HER2-low Breast Cancers. Am J Clin Pathol. 2021.
12 Bang YJ, Van Cutsem E, Feyereislova A, et al: ToGA Trial Investigators: Trastuzumab in combination with chemotherapy versus chemotherapy alone for treatment of HER2-positive advanced gastric or gastro-oesophageal junction cancer (ToGA): A phase 3, open-label, randomised controlled trial. Lancet 2010;376: 687- 697.
13 Subasinghe D, Acott N, Kumarasinghe MP. A Survival Guide to HER2 Testing in Gastric/Gastroesophageal Junction Carcinoma. Gastrointest Endosc. 2019;90(1):44-54.
14 Smyth EC, Verheij M, Allum W, et al. Gastric Cancer: ESMO Clinical Practice Guidelines for Diagnosis, Treatment and Follow-Up. Ann Oncol. 2016;27(suppl 5):v38-v49.
15 Van Cutsem E, Bang YJ, Feng-Yi F, et al. HER2 Screening Data from Toga: Targeting HER2 in Gastric and Gastroesophageal Junction Cancer. Gastric Cancer. 2015;18(3):476-484.
16 The Global Cancer Observatory: Cancer Today (GLOBOCAN) 2020. International
17 Wolff AC, Hammond MEH, Allison KH, et al. Human Epidermal Growth Factor Receptor 2 Testing in Breast Cancer: American Society of Clinical Oncology/College of American Pathologists Clinical Practice Guideline Focused Update. Arch Pathol Lab Med. 2018;142:1364-1382.
18 Dickson RB, and Lippman ME. Genes, Oncogenes, and Hormones. Boston: Kluwer Academic Publishers; 1992.
19 Keatings, L. et al. c-erbB-2 oncoprotein expression in mammary and extramammary Paget’s disease: an immunohistochemical study. Histopathology. 1990;17: 234-247.
20 Hudis CA. Trastuzumab--Mechanism of Action and Use in Clinical Practice. N Engl J Med. 2007;357(1):39-51.
21 Slamon DJ, Leyland-Jones B, Shak S, et al. Use of Chemotherapy Plus a Monoclonal Antibody against Her2 for Metastatic Breast Cancer That Overexpresses HER2. N Engl J Med. 2001;344(11):783-792.
22 Herceptin (Trastuzamab) [Package Insert]. EMEA (European Medicines Agency). http://www.ema.europa.eu/docs/en_GB/document_library/EPAR Published 01/03/2010. Updated 04/02/2011. Accessed October 2010.
23 von Minckwitz G, Huang CS, Mano MS, et al. Trastuzumab Emtansine for Residual Invasive HER2-Positive Breast Cancer. N Engl J Med. 2019;380(7):617-628.
24 Swain SM, Kim SB, Cortés J, Ro J, Semiglazov V, Campone M, Ciruelos E, Ferrero JM, Schneeweiss A, Knott A, Clark E, Ross G, Benyunes MC, Baselga J. Pertuzumab, trastuzumab, and docetaxel for HER2-positive metastatic breast cancer (CLEOPATRA study): overall survival results from a randomised, double-blind, placebo-controlled, phase 3 study. Lancet Oncol. 2013 May;14(6):461-71.
25 Moasser MM, Krop IE. The Evolving Landscape of HER2 Targeting in Breast Cancer. JAMA Oncol. 2015;1(8):1154-1161.
26 Modi S, Park H, Murthy RK, et al. Antitumor Activity and Safety of Trastuzumab Deruxtecan in Patients With HER2-low-Expressing Advanced Breast Cancer: Results From a Phase Ib Study. J Clin Oncol. 2020;38:1887-1896.
27 Modi S, Jacot W, Yamashita T, et al. Trastuzumab Deruxtecan in Previously Treated HER2-Low Advanced Breast Cancer [published online ahead of print, 2022 Jun 5]. N Engl J Med. 2022;10.1056/NEJMoa2203690.
28 Cislo M, Filip AA, Arnold Offerhaus GJ, et al. Distinct Molecular Subtypes of Gastric Cancer: From Lauren to Molecular Pathology. Oncotarget. 2018;9(27):19427-19442.
29 Carson FL, Cappellano C. Histotechnology; A Self-Instructional Text, 5th edition. American Society for Clinical Pathology Press; 2020, 2022.
30 Occupational Safety and Health Standards: Occupational exposure to hazardous chemicals in laboratories. (29 CFR Part 1910.1450). Fed. Register.
31 Directive 2000/54/EC of the European Parliament and Council of 24 June 2020 on the protection of workers from risks related to exposure to biological agents at work.
32 Department of Health, Education and Welfare, National Institute of Occupational Safety and Health, Rockville, MD. “Procedures for the decontamination of plumbing systems containing copper and/or lead azides.” DHHS (NIOSH) Publ No. 78-127, Current 13. August 16, 1976.
33 Roche PC, Hsi ED. Immunohistochemistry-Principles and Advances. Manual of Clinical Laboratory Immunology, 6th edition. (NR Rose Ed.) ASM Press, 2002.
34 College of American Pathologists Laboratory Accreditation Program, Anatomic Pathology Checklist, 2017.
35 CLSI. Quality Assurance for Immunocytochemistry: Approved Guideline. CLSI document MM4-A- (ISBN 1-56238-396-5). CLSI, 940 West Valley Road, Suite 1400, Wayne, PA 19087-1898 USA, 1999.
36 Hoffmann M, et al. Histopathology. 2008:52;797-805.
37 Rüschoff J, Dietel M, Baretton G, Arbogast S, Walch A, Monges G, Chenard MP, Penault-Llorca F, Nagelmeier I, Schlake W, Höfler H, Kreipe HH. HER2 diagnostics in gastric cancer-guideline validation and development of standardized immunohistochemical testing. Virchows Arch. 2010;457(3):299-307.
38 Herman GE, Elfont EA. The taming of immunohistochemistry: the new era of quality control. Biotech Histochem. 1991;66(4):194-199.
39 Omata M, Liew CT, Ashcavai M, Peters RL. Nonimmunologic binding of horseradish peroxidase to hepatitis B surface antigen. A possible source of error in immunohistochemistry. Am J Clin Pathol. 1980;73(5):626-32.
40 Nadji M, Morales AR. Immunoperoxidase: part 1. The technique and its pitfalls. Lab Med. 1983;14:767.
41 Ahn S, Woo JW, Lee K, et al. HER2 status in breast cancer: changes in guidelines and complicating factors for interpretation. J Pathol Trans Med.2020:54:34-44.
42 Thomson TA, Hayes MM, Spinelli JJ, Hiland E, Sawrenko C, and Phillip D, et al. HER-2/neu in breast cancer: interobserver variability and performance of immunohistochemistry with 4 antibodies compared with fluorescent in situ hybridization, Mod Pathol. 2001;14:1079-86.
43 Kay EW, Walsh CJ, Cassidy M, Curran B, Leader M. C-erbB-2 immunostaining: problems with interpretation. J Clin Pathol. 1994;47:816-22.
44 Bilous M, Dowsett M, Hanna W, Isola J, Lebeau A, Moreno A, Penault-Llorca F, Rüschoff J, Tomasic G, van de Vijver M. Current Perspectives on HER2 Testing: A Review of National Testing Guidelines. Mod Pathol. 2003;16:173-182.
Nota: En este documento se ha usado el punto como separador decimal para marcar el borde entre la parte entera y la parte fraccionaria de los numerales con decimales. No se han usado separadores para las unidades de millar.
El resumen de los aspectos de seguridad y rendimiento se puede ver a continuación: https://ec.europa.eu/tools/eudamed
Símbolos:
Ventana usa los siguientes símbolos y signos además de los indicados en la norma ISO 15223-1 (para USA: consulte elabdoc.roche.com/symbols para obtener más información).
|
GTIN |
Número mundial de artículo comercial. |
|
Rx only |
Para USA: |
Propiedad intelectual:
VENTANA, BENCHMARK, CONFIRM, INFORM, PATHWAY y ULTRAVIEW son marcas comerciales de Roche. Todos los demás nombres de productos y marcas comerciales pertenecen a sus respectivos propietarios.
© 2024 Ventana Medical Systems, Inc.
For USA: Rx only
Información de contacto:
ROCHE DIAGNOSTICS GMBH
Sandhofer Strasse 116
68305 Mannheim Germany
+800 5505 6606


