
VENCLEXTA - Tabletas recubiertas
Sustancia(s):
- Venetoclax
Presentaciones:
- 1 Caja, 14 Tabletas, 10 Miligramos
- 1 Caja, 7 Tabletas, 50 Miligramos
- 1 Caja, 120 Tabletas, 100 Miligramos
- 1 Caja, 7 Tabletas, 100 Miligramos
- 1 Caja, 14 Tabletas, 100 Miligramos
- 1 Caja, 180 Tabletas, 100 Miligramos
- 1 Caja, 14 Tabletas, 10 Miligramos
FORMA FARMACÉUTICA Y FORMULACIÓN:
Cada TABLETA contiene:
10, 50 o 100 mg de venetoclax.
Excipientes cbp 1 tableta
INDICACIONES TERAPÉUTICAS:
Leucemia Linfocítica Crónica: VENCLEXTA® está indicado en combinación con rituximab o como monoterapia para el tratamiento de pacientes con leucemia linfocítica crónica (LLC), con o sin deleción del 17p, que han recibido al menos una terapia previa.
Leucemia mieloide aguda: VENCLEXTA® en combinación con un agente hipometilante o en combinación con dosis bajas de citarabina está indicado para los pacientes recién diagnosticados con leucemia mieloide aguda (LMA) que no son elegibles para la quimioterapia intensiva.
FARMACOCINÉTICA Y FARMACODINAMIA:
Mecanismo de Acción: Venetoclax es una molécula pequeña biodisponible oralmente que actúa como un potente inhibidor de la proteína anti-apoptótica BCL-2 (Linfoma de Células B-2). La sobrexpresión de BCL-2 ha sido demostrada en varias neoplasias hematológicas y de tumores sólidos, y ha sido implicada como factor de resistencia para algunos agentes terapéuticos. Venetoclax se une directamente al surco de unión BH3 del BCL-2, desplazando a las proteínas pro-apoptóticas que tienen BH3 como motivador, como BIM, para iniciar la permeabilización de la membrana externa mitocondrial (MOMP, por sus siglas en inglés), activación de caspasas y apoptosis. En estudios no clínicos, venetoclax ha demostrado actividad citotóxica en varias neoplasias de células B y otras malignidades hematológicas.
Propiedades farmacodinámicas:
Electrofisiología cardiaca: El efecto de múltiples dosis de venetoclax, hasta de 1200 mg diarios, en el intervalo QTc se evaluó en un brazo de un estudio abierto en 176 pacientes con LLC o Linfoma No Hodgkin previamente tratados. Venetoclax no tuvo efecto en el intervalo QTc y no existió una relación entre la exposición de venetoclax y el cambio en el intervalo QTc.
Propiedades farmacocinéticas:
Absorción: Después de la administración oral, la concentración plasmática máxima de venetoclax se alcanzó entre las 5 y 8 horas después de la administración de la dosis. En el estado estable de venetoclax, el área bajo la curva (AUC, por sus siglas en inglés) aumentó proporcionalmente en el rango de 150 a 800 mg de dosis. Con una dieta baja en grasa, la Cmax de venetoclax fue de 2.1 ± 1.1 μg/mL y la AUC24 fue de 32.8 ± 16.9 μgh/mL a la dosis diaria de 400 mg.
Efecto de los alimentos: La administración de venetoclax con una dieta baja en grasa aumentó la exposición al fármaco aproximadamente 3.4 veces y con una dieta alta en grasas de 5.1 a 5.3 veces en comparación con la administración en ayuno. Venetoclax debe ser administrado con alimentos (Véase Dosis y vía de administración) .
Distribución: Venetoclax se une altamente a proteínas plasmáticas y tiene una fracción libre <0.01 sobre un rango de concentración que va de 1 a 30 μM (0.87-26 μg/mL). La razón plasma-sangre fue de 0.57. De acuerdo a la población estimada el rango del volumen de distribución aparente (Vdss/F) de venetoclax fue de 256 a 321 L en pacientes.
Metabolismo: Los estudios in vitro han demostrado que venetoclax es metabolizado predominantemente por el CYP3A4. M27 fue identificado como el mayor metabolito en plasma, con actividad inhibitoria de BCL-2 que es al menos 58 veces menor que venetoclax in vitro.
Eliminación: De acuerdo a la población estimada, la vida media de eliminación de venetoclax fue de aproximadamente 26 horas. Después de una administración oral de 200 mg de venetoclax radiomarcado ([14C]-venetoclax) en sujetos sanos, > 99.9% de la dosis fue recuperada en heces y < 0.1% fue excretada en orina en 9 días. El 20.8% de la dosis radiomarcada excretada en heces fue de venetoclax sin modificaciones. La farmacocinética de venetoclax no cambia con el tiempo.
Poblaciones específicas:
Edad, raza, sexo y peso: Basado en los análisis de farmacocinética poblacional la edad, raza, sexo y peso no tienen efectos en la eliminación de venetoclax.
Uso pediátrico: No se ha evaluado la farmacocinética de venetoclax en pacientes menores de 18 años de edad.
En un estudio de toxicología de ratones jóvenes fue administrado VENCLEXTA® a una dosis de 10, 30 y 100 mg/kg/día vía oral durante 7 a 60 días de edad. Los signos clínicos encontrados fueron una disminución de la actividad, deshidratación, palidez y una postura encorvada a la dosis de > 30 mg/kg/día. Además se mostró mortalidad y efectos en el peso corporal en la dosis de 100 mg/kg/día. Otros efectos relacionados fueron la disminución de los linfocitos de forma reversible con la dosis de ≥ 10 mg/kg/día, al igual que lo encontrado en ratones adultos, por lo que no se consideró un efecto adverso.
VENCLEXTA® no mostró efectos adversos al nivel NOAEL a la dosis de 10 mg/kg/día en ratones en aproximadamente 0.06 veces la dosis clínica de 400 mg basada en mg/m2 para un niño de 20 kg.
Uso Geriátrico: No se requiere ajuste de dosis para pacientes mayores de 65 años. No existe diferencia significativa en la seguridad y eficacia en pacientes con edad < 65 años en comparación con los de ≥ 65 años en los estudios de combinación con rituximab o monoterapia.
Deterioro Renal: No se han conducido estudios específicos en sujetos con daño renal. No se requiere ajuste de dosis en pacientes con daño renal leve o moderado (CrCl ≥ 30 mL/min), no se ha determinado una dosis recomendada para pacientes con daño renal severo (CrCl < 30 mL/min) o para pacientes en diálisis.
Los pacientes con función renal deteriorada (CrCl < 80 mL/min) pueden requerir un monitoreo y profilaxis de mayor intensidad para reducir el riesgo de SLT al iniciar tratamiento con VENCLEXTA®.
Basado en el análisis farmacocinético de una población que incluyó 211 sujetos con deterioro leve de la función renal (Depuración de creatinina; CrCl ≥ 60 y < 90 mL/min), 83 sujetos con deterioro moderado de la función renal (CrCl ≥ 30 y< 60 mL/min) y 210 sujetos con función renal normal (CrCl ≥ 90 mL/min), la exposición a venetoclax en sujetos con deterioro renal leve o moderado fue similar a la de los sujetos con función renal normal. La farmacocinética de venetoclax no ha sido estudiada en sujetos con deterioro grave (CrCl < 30 mL/min) de la función renal o sujetos en diálisis.
Deterioro Hepático: No se han concluido estudios específicos en sujetos con daño hepático. No se requiere ajuste de dosis en pacientes con daño hepático. No se ha determinado una dosis recomendada para pacientes con daño hepático severo.
Basado en el análisis farmacocinético de una población que incluyó 69 sujetos con deterioro leve de la función hepática, 7 sujetos con deterioro moderado de la función hepática y 429 sujetos con función hepática normal, la exposición a venetoclax fue similar en lo sujetos con deterioro hepático leve y moderado a la de los sujetos con función hepática normal. El deterioro leve de la función hepática fue definido como bilirrubina total normal y aspartato aminotransferasa (AST) > al límite superior normal (ULN, por sus siglas en inglés) o bilirrubina total > 1.0 a 1.5 veces ULN, el deterioro moderado de la función hepática como bilirrubina total > 1.5 a 3.0 veces ULN y el deterioro grave como bilirrubina total > 3.0 veces ULN. La farmacocinética de venetoclax no ha sido estudiada en sujetos con deterioro grave de la función hepática.
Estudios clínicos:
Leucemia Linfocítica Crónica (LLC):
MURANO: Es un estudio clínico de fase 3, abierto, diseñado para evaluar la eficacia y seguridad de la combinación de venetoclax con rituximab, comparándolo con la combinación de bendamustina con rituximab en pacientes con LLC que han recibido al menos una terapia previa. Los pacientes del brazo de venetoclax con rituximab completaron un esquema de ajuste semanal y una dosis de 400 mg de venetoclax diaria del día 1 del ciclo 1 de rituximab por 2 años, siempre y cuando no se presentara progresión de la enfermedad o toxicidad inaceptable. Rituximab se inició con una dosis de 375mg/m2 en el primer ciclo de rituximab y a una dosis de 500 mg/m2 de los ciclos 2 al 6. Cada ciclo fue de 28 días. Los pacientes aleatorizados al grupo de bendamustina recibieron una dosis de 70 mg/m2 en los días 1 y 2, por 6 ciclos, con la aplicación de rituximab previamente descrita. Durante y posterior a completar 24 meses de tratamiento de venetoclax con rituximab los pacientes fueron evaluados de forma continua para la progresión de la enfermedad (PFS) y la supervivencia global (OS).
Un total de 389 pacientes se aleatorizaron; 194 al brazo de venetoclax con rituximab y 195 al brazo de bendamustina con rituximab. Las características demográficas y de la enfermedad de ambos grupos se muestran en la tabla 1.
Tabla 1. Características demográficas y basales del estudio MURANO
|
Características |
Venetoclax + Rituximab (N = 194) |
Bendamustina + Rituximab (N = 195) |
|
Edad, mediana de años (rango) |
64.5 (28.83) |
66 (22-85) |
|
Blancos; % |
96.8 |
96.7 |
|
Hombre;% |
70.1 |
77.4 |
|
Estado físico por ECOG; % |
||
|
0 |
57.2 |
55.7 |
|
1 |
42.3 |
43.3 |
|
2 |
0.5 |
1.0 |
|
Carga tumoral; % |
||
|
Cuenta absoluta de linfocitos ≥ 25 x 109/L |
66.5 |
68.7 |
|
Uno o más nódulos linfáticos ≥ 5 cm |
45.7 |
47.6 |
|
Número de tratamiento previos; % |
||
|
Mediana de número de tratamiento (rango) |
1 (1-5) |
1 (1-4) |
|
1 |
57.2 |
60.0 |
|
2 |
29.4 |
22.1 |
|
≥ 3 |
13.4 |
17.9 |
|
Regímenes de tratamiento previos |
||
|
Mediana de tratamiento en números (rango) |
1 (1-5) |
1 (1-4) |
|
Tratamiento previo con alquilantes, % |
93.3 |
95.4 |
|
Tratamiento previo con análogos de purina, % |
80.5 |
81.4 |
|
Tratamiento previo con anticuerpos monoclonales anti CD20, % |
76.3 |
78.6 |
|
Tratamiento previo con inhibidores de la señalización del receptor de células B, % |
1.5 |
2.6 |
|
Tratados previamente con FCR, % |
54.1 |
55.4 |
|
Refractarios a fludarabina, % |
14.1 |
15.5 |
|
Subtipos de LLC, % |
||
|
Deleción del 17p |
26.6 |
27.2 |
|
Deleción de 11q |
35.3 |
37.9 |
|
Mutación de TP53 |
25.0 |
27.7 |
|
Estado no mutado de IgVH |
68.3 |
68.3 |
|
Tiempo desde el diagnóstico, años; mediana (rango) |
6.44 (0.5-28.4) |
7.11 (0.3-29.5) |
FCR = fludarabina, ciclofosfamida, rituximab.
La mediana de seguimiento al análisis fue de 23.8 meses (rango de 0.0 a 37.4 meses).
El objetivo primario fue la supervivencia libre de progresión (PFS, por sus siglas en inglés) evaluada por los criterios del lnternational Workshop para la LLC (IWCLL, por sus siglas en inglés) y las guías de la actualización del grupo de trabajo del Instituto Nacional de Cáncer de los Estados Unidos del 2008 (NCI-WG, por sus siglas en inglés).
Los resultados de eficacia del estudio MURANO se muestran en la tabla 2. Las curvas de Kaplan-Meier para PFS y OS se muestran en la figura 1 y 2 respectivamente.
Tabla 2. Eficacia del estudio MURANO
|
Evaluación del investigador (INV) |
||
|
Venetoclax + Rituximab (N = 194) |
Bendamustina + Rituximab (N = 195) |
|
|
Supervivencia libre de progresión |
||
|
Número de eventos (%) |
32 (16.5) |
114 (58.5) |
|
Progresión de la enfermedad |
21 |
98 |
|
Eventos de muerte |
11 |
16 |
|
Mediana en meses, (95% CI) |
No alcanzado |
17.0 (15.5, 21.6) |
|
HR (95% CI) |
0.17 (0.11, 0.25) |
|
|
Valor de pa |
p < 0.0001 |
|
|
Estimado a 12 meses, % (95% CI) |
92.7 (89.1, 96.4) |
72.5 (65.9, 79.1) |
|
Estimado a 24 meses, % (95% CI) |
84.9 (79.1, 90.6) |
36.3 (28.5, 44.0) |
|
Tasas de respuesta |
||
|
ORR, % (95% CI) |
93.3 (88.8, 96.4) |
67.7 (60.6, 74.2) |
|
CR+CRi, (%) |
26.8 |
8.2 |
|
nPR, (%) |
3.1 |
6.2 |
|
PR, (%) |
63.4 |
53.3 |
|
Supervivencia global (OS) |
||
|
Número de muertes (%) |
15 (7.7) |
27 (13.8) |
|
Hazard ratio (95% CI) |
0.48 (0.25, 0.90) |
|
|
Tiempo para el siguiente tratamiento de leucemia |
||
|
Número de eventos (%) |
23 (11.9) |
83 (42.6) |
|
Mediana, en meses |
No alcanzado |
26.4 |
|
Hazard ratio (95% CI) |
0.19 (0.12, 0.31) |
|
|
Supervivencia libre de eventos |
||
|
Número de eventos (%) |
33 (17.0) |
118 (60.5) |
|
Mediana en meses |
No alcanzado |
16.4 |
|
Hazard ratio (95% CI) |
0.17 (0.11, 0.25) |
|
CI= intervalo de confianza; CR = remisión completa; CRi = remisión completa con recuperación incompleta de la médula ósea; INV = investigador; MRD = enfermedad mínima residual; NA = no disponible; nPR = remisión parcial nodular; ORR = tasa de respuestas globales (CR + CRi + nPR + PR); PR = remisión parcial; HR = hazard ratio.
a Prueba estratificada de log-rank.
Figura 1. Curva de Kaplan-Meier de PFS evaluada por el investigador con análisis de intención a tratar (ITT) en el estudio MURANO
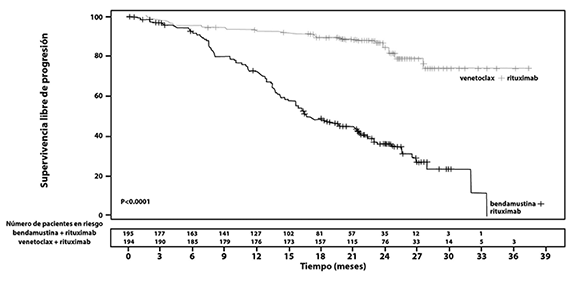
Al momento del análisis primario (datos del 08 de mayo de 2017), 65 pacientes completaron el esquema de 24 meses de venetoclax con rituximab sin progresión y 78 pacientes continuaron recibiendo venetoclax (por más de 18 meses de tratamiento). De los 65 pacientes que continuaron libres de progresión por 24 meses, sólo 2 pacientes progresaron al terminar el tratamiento. 12 pacientes tuvieron una visita de seguimiento a los 3 meses y continuaban libres de progresión, 5 de ellos fueron evaluados a los 6 meses y permanecían sin progresión.
Figura 2. Curva de Kaplan-Meier de supervivencia global con ITT en MURANO
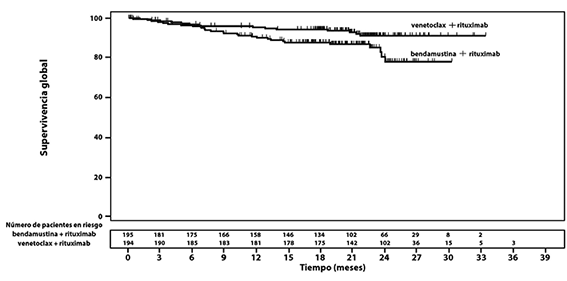
La enfermedad mínima residual (MRD, por sus siglas en inglés) se evaluó por reacción en cadena de la polimerasa alelo específica (ASO-PCR, por sus siglas en inglés) y citometría de flujo. El punto de corte para tener MRD negativa por citometría de flujo en LLC es de 1 por 104 leucocitos. La MRD se mantuvo en sangre periférica en la mayoría de los pacientes (brazo venetoclax + rituximab con 187/194, brazo de bendamustina rituximab 179/195), en un subgrupo de paciente con estudio se presentó MRD en la médula ósea (74/194 en el grupo de venetoclax + rituximab y 41/195 del brazo de bendamustina + rituximab). Las tasas de MRD negativa en sangre periférica, evaluadas en cualquier momento del estudio, fueron de 84% (162/194) para el brazo de venetoclax + rituximab en comparación con 23% en el brazo de bendamustina + rituximab (45/195). La evaluación a los 9 meses de MRD negativa en sangre periférica fue de 62.4% en el brazo de venetoclax + rituximab en comparación con 13.3% en el brazo de bendamustina + rituximab y esta respuesta se ha mantenido por lo menos 9 meses adicionales (59.8% en el brazo de rituximab + venetoclax en comparación con 5.1% en el brazo de bendamustina + rituximab), que son los datos disponibles en el último análisis completado. Las tasas de MRD en médula ósea fueron de 27.3% (53/194) en el brazo de venetoclax + rituximab en comparación con 1.5% (3/195) en el brazo de bendamustina + rituximab.
Existe un beneficio de PFS en el brazo de venetoclax + rituximab en comparación con bendamustina + rituximab en todos los subgrupos analizados, como se muestra en la Figura 3.
Figura 3. Gráfica arbórea del análisis del investigador en diferentes subgrupos en relación al PFS del estudio MURANO

El estado de deleción del 17p se determinó en función de los resultados de la prueba de laboratorio central. La relación de riesgo no estratificada se muestra en el eje X con escala logarítmica. NE = no evaluable.
M13-982: El estudio M13-982 es un estudio clínico de 107 pacientes, multicéntrico, de un solo brazo con pacientes previamente tratados con diagnóstico de LLC con deleción del 17p. El 64.4% de los pacientes fueron hombres y el 97.2% fueron de raza blanca. La mediana de edad fue 67 años (rango de 37-85 años) y la mediana desde el diagnóstico fue de 6.8 años (rango de 0.1 a 32 años; n=106). La mediana de tratamiento previo para LLC fue de 2 (rango de 1-10). Al inicio del estudio, 53.3% de los paciente tenían uno o más nódulos ≥ 5 cm y el 50.5% tenían una cuenta absoluta de linfocitos ≥ 25 x 109/L. El estado físico medido por ECOG al inicio de estudio fue de 39.3% para el valor 0, de 52.3% para el valor 1 y 8.4% para el valor de 2.
El 37.4% (34/91) fueron refractarios a fludarabina, el 81.1% (30/37) presentaban un estado no mutado del gen de IGHV de las inmunoglobulinas y el 23.8% (19/80) presentaron deleción del 11q.
Los pacientes recibieron venetoclax por medio del esquema de ajuste semanal con 20 mg, 50 mg, 100 mg, 200 mg y 400 mg una vez al día. Los pacientes siguieron recibiendo 400 mg de venetoclax una vez al día hasta la progresión o toxicidad inaceptable. La mediana del tiempo para el tratamiento fue de 12.1 meses (rango de 0-21.5 meses).
El objetivo primario fue la tasa de respuesta global (ORR, por sus siglas en inglés) medida por un IRC usando los criterios del IWCLL y las guías del 2008 del NCL-WG. La eficacia se muestra en la tabla 3.
Tabla 3. Resultados de eficacia del estudio M13-982
|
Evaluación del IRC (N=107)ª |
Evaluación del investigador (N=107)ª |
|
|
ORR, % (95% CI) |
79.4 (70.5, 86.6) |
73.8 (64.4, 81.9) |
|
CR + CRi (%) |
7.5 |
15.9 |
|
nPR (%) |
2.8 |
3.7 |
|
PR (%) |
69.2 |
54.2 |
|
DOR, % (95% CI) Estimación a 12 meses |
84.7 (74.5, 91.0) |
89.1 (79.2, 94.4) |
ª Un paciente no presentó la deleción del 17p.
CI = intervalo de confianza; CR = remisión completa; CRi = remisión completa con recuperación incompleta de la médula ósea; DOR = duración de la respuesta; IRC = comité independiente de revisión; nPR = remisión parcial nodal; ORR = tasa de respuestas globales (CR + CRi + nPR + PR); PR = remisión parcial.
La enfermedad mínima residual (MRD, por sus siglas en inglés) se evaluó usando citometría de flujo en 45 de los 107 pacientes que obtuvieron la remisión completa (CR), por sus siglas en inglés), remisión completa con recuperación incompleta de la médula ósea (CRi) o una remisión parcial (PR, por sus siglas en inglés) con enfermedad remante limitada en tratamiento con venetoclax. El punto de corte para obtener un estado negativo de enfermedad mínima residual en paciente con LLC fue de 1 célula LLC por 104 leucocitos en la muestra. Diecisiete por ciento (18/107) de los pacientes fueron MRD negativos en sangre periférica, incluyendo 6 pacientes con MRD negativa en médula ósea.
Se realizó un análisis de estado global de salud (GHS, por sus siglas en inglés) en 73 pacientes así como una evaluación del estado emocional (EF, por sus siglas en inglés), estado funcional (SF, por sus siglas en inglés) y un cuestionario EORTC QLQ-C30 al diagnóstico y durante las 24 semanas. Además se realizó en 74 y 77 pacientes respectivamente evaluación funcional (RF, por sus siglas en inglés) y la escala de fatiga al diagnóstico y las 24 semanas. Posterior al inicio de tratamiento con venetoclax se demostró una mejoría en GHS (16%), EF (10.6%), SF (17.1%), RF (16.2) y en la escala de fatiga (17.5%). La mejoría se observó de manera temprana a la semana 4.
M12-175: El estudio M12-175 es un estudio multicéntrico que incluyó pacientes con diagnóstico de LLC, incluyendo aquellos que tenían la deleción del 17p. La eficacia fue evaluada en 57 pacientes que recibieron una dosis diaria de 400 mg, con previo esquema de ajuste de dosis semanal de 5 semanas. De los 57 pacientes, 75.4 % fueron hombres y el 91.2% de raza blanca. La mediana de edad fue de 66 años (rango de 42-84 años) y la mediana desde el diagnóstico fue de 9 años (rango de 1.1 a 27 años). La mediana de tratamientos previo fue de 3 (rango de 1-11). Al inicio, el 66.7% de los pacientes tenían uno o más nódulos de ≥ 5 cm y el 35.1% de los pacientes presentó una cuenta total de linfocitos superior a ≥ 25 x 109/L. El estado físico medido por ECOG al inicio de estudio fue de 45.5% para el valor 0, de 52.7% para el valor 1 y 1.8% para el valor de 2.
El 75.4% de los pacientes fueron refractarios a fludarabina, el 65,6% (21/23) presentaron un estado no mutado del gen IGHV de la inmunoglobulina, 30.4% (17/56) presentaron deleción del 11q y 21.4% (12/56) presentaron deleción del 17p.
Los pacientes continuaron recibiendo 400 mg de venetoclax como monoterapia hasta la progresión o toxicidad inaceptable. La mediana del tiempo de tratamiento en el momento de la evaluación fue de 11.5 meses (rango 0.5-24.1 meses).
Las respuestas globales y la DOR se evaluaron por el investigador y el IRC, usando los criterios del IWCLL y la actualización de las guías NCI-WG del 2008 . La eficacia se muestra en la tabla 4.
Tabla 4. Resultados de Eficacia del estudio M12-175
|
Evaluación del IRC N=57 |
Evaluación del investigador N=57 |
|
|
ORR, % (95% CI) |
73.7 (60.3, 84.5) |
80.7 (68.1, 90.0) |
|
CR + CRi (%) |
7.0 |
12.3 |
|
nPR (%) |
0 |
3.5 |
|
PR (%) |
66.7 |
64.9 |
|
DOR, % (95% CI) Estimado a 12 meses |
88.8 (67.5, 96.5) |
96.6 (77.9, 99.5) |
CI = intervalo de confianza; CR = remisión completa; CRi = remisión completa con recuperación incompleta de la médula ósea; DOR = duración de la respuesta; IRC = comité independiente de revisión; nPR = remisión parcial nodal; ORR = tasa de respuestas globales (CR + CRi + nPR + PR); PR = remisión parcial.
M14-032: El estudio M14-032 fue un estudio abierto, multicéntrico, que evaluó la eficacia de venetoclax en pacientes con LLC que fueron tratados previamente y progresaron después de ibrutinib (Brazo A) o idelalisib (Brazo B). Los pacientes después de un esquema de ajuste semanal inicial fueron tratados con la dosis de 400 mg al día hasta la progresión o toxicidad inaceptable.
La eficacia fue evaluada por el investigador y un IRC de acuerdo a los criterios del IWCLL y la actualización de las guías del NCL WG del 2008. La evaluación de la respuesta fue realizada a las 8, 12 y 24 semanas para los 64 pacientes en la cohorte general. Para los pacientes de la cohorte de expansión se realizó evaluación a las 12 y 36 semanas.
Un total de 127 pacientes fueron enrolados en el estudio, de los cuales 64 fueron en la cohorte general (43 recibieron antes ibrutinib y 21 idelalisib) y 63 en la cohorte de expansión (48 recibieron antes ibrutinib y 15 idelalisib). La media del tiempo desde el diagnóstico fue de 8.3 años (rango de 0.3-18.5 años). La mediana de tratamiento previo fue de 4 (1-15 tratamientos). La mediana de edad fue de 66 años (rango 28-85 años), 70% fueron hombres y 92% de raza blanca.
Al inicio, el 41% de los pacientes tenían uno o más nódulos de ≥ 5 cm y el 31% de los pacientes presentó una cuenta total de linfocitos superior de ≥ 25 x 109/L.
Los datos de eficacia presentados son con un análisis de los datos del 31 de enero de 2017. El investigador evaluó la eficacia (n=108) incluidos los 64 pacientes de la cohorte general con una evaluación de más de 24 semanas y 7 pacientes en la cohorte de expansión con una evaluación a las 36 semanas. Los resultados de eficacia por el IRC (n=97) incluyeron 64 pacientes de la cohorte general y 33 de la cohorte de expansión.
Los resultados de los 108 pacientes evaluados por el investigador y los 97 evaluados por el IRC se muestran en la tabla 5.
Tabla 5. Resultados de eficacia del estudio M14-032
|
Evaluación del IRC N=97 |
Evaluación del investigador N=108 |
|
|
ORR,% (95% CI) |
73.2 (63.2, 81.7) |
65.7 (56.0, 74.6) |
|
CR + CRi (%) |
1.0 |
9.3 |
|
nPR (%) |
0 |
1.9 |
|
PR (%) |
72.2 |
54.6 |
|
DOR, % (95% CI) Estimado a los 6 meses Estimado a los 12 meses |
N=71 96.8 (87.6, 99.2) NA |
N=71 95.5 (86.8, 98.5) 85.2 (72.0, 92.4) |
|
Tiempo para la primer respuesta, meses (rango) |
2.5 (1.0-8.9) |
2.5 (1.6, 14.9) |
CI = intervalo de confianza; CR = remisión completa; CRi = remisión completa con recuperación incompleta de la médula ósea; DOR = duración de la respuesta; IRC = comité independiente de revisión; nPR = remisión parcial nodal; ORR = tasa de respuestas globales (CR + CRi + nPR + PR); PR = remisión parcial.
La mediana de la duración del tratamiento con venetoclax para los 127 pacientes evaluados por el investigador fue de 10.2 meses (rango de 0.1 a 25.6 meses). La mediana de la duración de tratamiento para los pacientes con venetoclax evaluados por el IRC fue de 12.3 meses (rango de 0.1 a 25.6 meses).
La tasa de MRD negativa en la sangre periférica para los 127 paciente fue de 30% (29/127), incluyendo a 5 pacientes con MRD en médula ósea.
Leucemia mieloide aguda: La eficacia de venetoclax se estudió en dos ensayos no aleatorizados en pacientes con LMA recién diagnosticada que no eran elegibles para quimioterapia intensiva. La eficacia se estableció sobre la base de la tasa de remisión completa (CR)/remisión completa con recuperación hematológica parcial (CRh), la duración de CR/CRh y la tasa de conversión de la dependencia de transfusión a la independencia de la transfusión. La independencia de la transfusión se basó en la ausencia de transfusión de glóbulos rojos o plaquetas durante 56 días consecutivos durante el periodo de tratamiento del estudio y se evaluó en todos los pacientes.
M14-358: La eficacia de venetoclax se estableció en un ensayo clínico no aleatorizado de venetoclax en combinación con azacitidina (n = 84) o decitabina (n = 31) en pacientes recién diagnosticados con LMA que no eran elegibles para quimioterapia intensiva.
Los pacientes recibieron venetoclax a través de un aumento diario hasta una dosis final de 400 mg una vez al día. Durante el aumento, los pacientes recibieron profilaxis SLT y fueron hospitalizados para el seguimiento. La azacitidina a 75 mg/m2 se administró por vía intravenosa o subcutánea en los días 1-7 de cada ciclo de 28 días comenzando en el ciclo 1 día 1. La decitabina a 20 mg/m2 se administró por vía intravenosa los días 1-5 de cada ciclo de 28 días comenzando el Ciclo 1 Día 1. Los pacientes continuaron recibiendo ciclos de tratamiento hasta la progresión de la enfermedad o toxicidad inaceptable. Se implementó la reducción de la dosis de azacitidina en el ensayo clínico para el tratamiento de la toxicidad hematológica; consulte la información completa de prescripción de azacitidina. Las reducciones de dosis para decitabina no se implementaron en el ensayo clínico.
La Tabla 6 resume las características demográficas y de enfermedad iniciales de la población de estudio.
Tabla 6. Características basales del paciente para pacientes con LMA tratados con Venetoclax en combinación con un agente hipometilante
|
Características |
Venetoclax en combinación con Azacitidina N=84 |
Venetoclax en combinación con Decitabina N=1 |
|
Edad en años: Mediana (rango) |
74.5 (61-90) |
72.0 (65-86) |
|
Blancos; % |
91.0 |
87.1 |
|
Hombres; % |
60.7 |
48.4 |
|
Estado funcional de ECOG; % |
||
|
0.1 |
69.0 |
87.1 |
|
2 |
28.6 |
12.9 |
|
3 |
2.4 |
0 |
|
Blastos en médula ósea; % |
||
|
< 30% |
28.6 |
22.6 |
|
≥ 30%-< 50% |
33.3 |
45.2 |
|
≥ 50% |
36.9 |
32.3 |
|
Antecedente de historia de alteraciones hematológicas; % |
25 |
29 |
|
Análisis de mutaciones; % (identificado/probado) |
||
|
TP53 |
27.0 (20/74) |
31.8 (7/22) |
|
IDH1 o 2 |
27.0 (20/74) |
22.7 (5/22) |
|
FLT-3 |
14.9 (11/74) |
13.6 (3/22) |
|
NPM1 |
18.9 (14/74) |
13.6 (3/22) |
|
Riesgo citogenéticoa,b; % |
||
|
Intermedio |
59.5 |
51.6 |
|
Desfavorable |
39.3 |
48.4 |
a Según lo definido por la categorización de riesgo de la Red Nacional del Cáncer (NCCN) v2014.
b Sin mitosis en 1 paciente (excluido el riesgo favorable mediante hibridación fluorescente in situ [FISH]).
La mediana de seguimiento fue de 8,2 meses (rango: 0,4 a 35,5 meses) para venetoclax en combinación con azacitidina y 16,2 meses (rango: 0,7 a 36,7 meses) para venetoclax en combinación con decitabina.
Los resultados de eficacia se muestran en la Tabla 7 y 8 y fueron similares para ambas combinaciones.
Tabla 7. Resultados de eficacia para pacientes recién diagnosticados con LMA tratados con Venetoclax en combinación con un agente hipometilante
|
Venetoclax en combinación con Azacitadina N=84 |
Venetoclax en combinación con Decitabina N=31 |
|
|
Respuesta completa (CR), n (%) 95% CI Mediana de DORª (meses) 95% CI |
34 (40.5) [29.9, 51.7] > 14.6b [14.6, 30.3] |
17 (54.8) [36.0, 72.7] NR [9.7, NR] |
|
CRi, n (%) 95% CI Mediana de DORª (meses) 95% CI |
25 (29.8) [20.3, 40.7] 7.8 [5.6, NR] |
6 (19.4%) [7.5, 37.5] 6.1 [3.0, 16.5] |
|
CR+CRi, n (%) 95%CI Mediana de DORc (meses) 95% CI |
59 (70.2) [59.3, 79.7] > 8.2b [8.2, 30.2] |
23 (74.2) [55.4, 88.1] 15 [5.0, NR] |
|
CRh, n (%) 95%CI Mediana de DORª (meses) 95% CI |
20 (23 .8) [15.2, 34.3] 7.9 [5.8, NR] |
5 (16.1) [5.5, 33.7] 7.2 [2.4, 15.3] |
|
CR+CRh, n (%) 95% CI Mediana DORª (meses) 95% CI |
54 (64.3) [53.1, 74.4] > 8.2b [8.2, 30.3) |
22 (71.0) [52.0, 85.8] 12.7 [7.2, NR] |
|
Independencia trasfucional, n(N (%) Células rojasd Plaquetase |
25/50 (50.0) 15/26 (57.7) |
12/23 (52.2) 3/5 (60.0) |
IC = intervalo de confianza; NR = no alcanzado.
Se definió RC (remisión completa) como recuento absoluto de neutrófilos ≥ 1,000/microlitro, plaquetas ≥ 100,000/microlitro, independencia de transfusión de glóbulos rojos y médula ósea con < 5% de blastos. Ausencia de blastos y explosiones circulantes con varillas Auer; ausencia de enfermedad extramedular.
La CRh (remisión completa con recuperación hematológica parcial) se definió como < 5% de blastos en la médula ósea, sin evidencia de enfermedad y recuperación parcial de los conteos sanguíneos periféricos (plaquetas > 50,000/microlitro y ANC > 500/microlitro).
La CRi (remisión completa con recuperación de sangre incompleta) se definió de la misma manera que todos los criterios para RC excepto neutropenia residual < 1,000/microlitro o trombocitopenia < 100,000/microlitro.
a DOR (duración de la respuesta) se definió como el tiempo transcurrido desde la primera respuesta de CR o CRh a la primera fecha de recaída, progresión clínica de la enfermedad o muerte debido a la progresión de la enfermedad, lo que ocurriera antes.
b Los datos aún no están maduros.
c DOR (duración de la respuesta) se definió como el tiempo transcurrido desde la primera respuesta de CR o CRi a la primera fecha de recaída, progresión clínica de la enfermedad o muerte debido a la progresión de la enfermedad, lo que ocurriera antes.
d Evaluado para pacientes que fueron dependientes al inicio del estudio para la transfusión de glóbulos rojos.
e Evaluado para pacientes que fueron dependientes al inicio del estudio para la transfusión de plaquetas.
Tabla 8. Tiempo hasta la respuesta en pacientes con LMA tratados con [Venetoclax] en combinación con un agente hipometilante
|
Venetoclax en combinación con Azacitadina N=84 |
Venetoclax en combinación con Decitabina N=31 |
|
|
Tiempo medio hasta la MEJOR respuesta de CR (meses) Rango (meses) |
1.9 (0.7-10.9) |
3.6 (1.2-17.6) |
|
Tiempo medio hasta PRIMERA respuesta de CR + CRh (meses) Rango (meses) |
1.0 (0.7-8.9) |
1.8 (0.8-13.8) |
|
Tiempo medio hasta PRIMERA respuesta de CR + CRi (meses) Rango (meses) |
1.2 (0.7-5.5) |
1.9 (0.9-5.4) |
Venetoclax en combinación con azacitidina: Los resultados de eficacia se muestran en la Tabla 7 y 8.
La mediana de la supervivencia general (SG) para los pacientes tratados con venetoclax en combinación con azacitidina fue de 14,9 meses (IC del 95%: 10,2, NR).
Se observaron remisiones (CR o CRh) en los subgrupos con diferentes características basales. Para pacientes con citogenética de riesgo pobre o intermedio, se observaron tasas de remisión similares, la tasa fue de 57.6% o 70.0%, respectivamente. Para los pacientes con las siguientes mutaciones identificadas, las remisiones fueron las siguientes: TP53: 65.0%, IDH1/2: 75.0%, FLT-3: 72.7% y NPM1: 71.4%.
La enfermedad mínima residual (EMR) se evaluó a partir de muestras de aspirado de médula ósea para pacientes que alcanzaron CR o CRh después del tratamiento con venetoclax en combinación con azacitidina. De esos pacientes, el 50% (27/54) lograron EMR con menos de una célula LMA por 103 leucocitos en la médula ósea.
De los pacientes tratados con venetoclax en combinación con azacitidina, el 9,5% (8/84) lograron una CR/CRi y posteriormente recibieron un trasplante de células madre.
Venetoclax en combinación con Decitabine: Los resultados de eficacia se muestran en la Tabla 7 y 8.
La mediana de la supervivencia global (SG) para los pacientes tratados con venetoclax en combinación con decitabina fue de 16,2 meses (IC del 95%: 9,1, NR).
Se observaron remisiones (CR o CRh) en los subgrupos con diferentes características basales. Para pacientes con citogenética de riesgo intermedio o deficiente se observaron tasas de remisión similares, la tasa fue de 73.3% o 68.8%, respectivamente. Para pacientes con las siguientes mutaciones identificadas, las remisiones fueron las siguientes: TP53: 5/7, IDH1/2: 5/5, FLT-3: 1/3 y NPM1: 3/3.
La enfermedad mínima residual (EMR) se evaluó a partir de muestras de aspirado de médula ósea para pacientes que alcanzaron CR o CRh después del tratamiento con [venetoclax] en combinación con decitabina. De esos pacientes, el 36.4% (8/22) lograron EMR con menos de una célula LMA por 103 leucocitos en la médula ósea.
De los pacientes tratados con [venetoclax] en combinación con decitabina, el 9,7% (3/31) lograron una CR/CRi y posteriormente recibieron un trasplante de células madre.
M14-387: La eficacia de venetoclax se estableció en un ensayo clínico no aleatorizado de venetoclax en combinación con dosis bajas de citarabina (n = 82) en pacientes recién diagnosticados con LMA que no eran elegibles para quimioterapia intensiva, incluidos pacientes con exposición previa a agente hipometilante para un trastorno hematológico previo.
Los pacientes iniciaron venetoclax a través de un aumento diario hasta una dosis final de 600 mg una vez al día. Durante el aumento, los pacientes recibieron profilaxis SLT y fueron hospitalizados para el seguimiento. La citarabina en una dosis de 20 mg/m2 se administró por vía subcutánea una vez al día en los días 1-10 de cada ciclo de 28 días que comenzaba en el ciclo 1 día 1. Los pacientes continuaron recibiendo ciclos de tratamiento hasta progresión de la enfermedad o toxicidad inaceptable. La reducción de la dosis de citarabina a dosis bajas no se implementó en los ensayos clínicos.
La Tabla 9 resume las características demográficas y de enfermedad iniciales de la población de estudio.
Tabla 9. Características basales del paciente para pacientes con LMA tratados con Venetoclax en combinación con dosis bajas de citarabina
|
Características |
Venetoclax en combinación con dosis bajas de citarabina N = 82 |
|
Años de edad; mediana (rango) |
74.0 (63-90) |
|
Blanco; % |
94.9 |
|
Hombre; % |
64.6 |
|
Estado física ECOG; % 0-1 2 3 |
70.7 28.0 1.2 |
|
Blastos en médula ósea;% < 30% ≥ 30%-< 50% ≥ 50% |
32.9 22.0 43.9 |
|
Historia de antecedentes de trastornos hematológicos; % |
48.8 |
|
Análisis de mutaciones;% (identificado/probado) |
|
|
TP53 |
14.1 (10/71) |
|
IDH1 o 2 |
25.4 (18/71) |
|
FLT- 3 |
22.5 (16/71) |
|
NPM1 |
12.7 (9/71) |
|
Riesgo citogenético ª; % |
|
|
Intermedio |
59.8 |
|
Mal pronóstico |
31.7 |
|
Sin mitosis |
8.5 |
ª Según lo definido por la categorización de riesgo de la Red Nacional Integral del Cáncer (NCCN).
v2014.
La mediana de seguimiento fue de 7.1 meses (rango: 0.3 a 34.3 meses). Los resultados de eficacia se muestran en la tabla 10 y 11.
Tabla 10. Resultados de eficacia para pacientes recién diagnosticados con LMA tratados con Venetoclax en combinación con dosis bajas de citarabina
|
Venetoclax en combinación con dosis bajas de citarabina N = 82 |
|
|
Respuesta completa (CR), n (%) 95% CI Mediana de DORª (meses) 95% CI |
21 (25.6) [16.6-36.4] NR [10.2, NR] |
|
CRi, n (%) 95% CI Mediana de DORª (meses) 95% CI |
23 (28.0) [18.7, 39.1] 4.7 [2.6, 5.6] |
|
CR+CRi, n (%) 95%CI Mediana de DORc (meses) 95% CI |
44 (53.7) [42.3, 64.7] 8.1 [5.3, 14.9] |
|
CRh, n (%) 95% CI Mediana de DORª (meses) 95% CI |
17 (20.7) [12.6, 31.1] 6.6 [2.8, 11.0] |
|
CR+CRh, n (%) 95% CI Mediana DORª (meses) 95% CI |
38 (46.3) [35.3, 57.7] 11.0 [6.1, NR] |
|
Independencia transfucional, n(N (%) Células rojasd Plaquetase |
23/53 (43.4) 15/23 (65.2) |
IC = intervalo de confianza; NR = no alcanzado.
Se definió RC (remisión completa) como recuento absoluto de neutrófilos ≥ 1,000/microlitro, plaquetas ≥ 100,000/microlitro, independencia de transfusión de glóbulos rojos y médula ósea con < 5% de blastos. Ausencia de blastos y explosiones circulantes con varillas Auer; ausencia de enfermedad extramedular.
La CRh (remisión completa con recuperación hematológica parcial) se definió como < 5% de blastos en la médula ósea, sin evidencia de enfermedad y recuperación parcial de los conteos sanguíneos periféricos (plaquetas > 50,000/microlitro y ANC> 500/microlitro).
La CRi (remisión completa con recuperación de sangre incompleta) se definió de la misma manera que todos los criterios para RC excepto neutropenia residual <1,000/microlitro o trombocitopenia < 100,000/microlitro.
a DOR (duración de la respuesta) se definió como el tiempo transcurrido desde la primera respuesta de CR o CRh a la primera fecha de recaída, progresión clínica de la enfermedad o muerte debido a la progresión de la enfermedad, lo que ocurriera antes.
c DOR (duración de la respuesta) se definió como el tiempo transcurrido desde la primera respuesta de CR o CRi a la primera fecha de recaída, progresión clínica de la enfermedad o muerte debido a la progresión de la enfermedad, lo que ocurriera antes.
d Evaluado para pacientes que fueron dependientes al inicio del estudio para la transfusión de glóbulos rojos.
e Evaluado para pacientes que fueron dependientes al inicio del estudio para la transfusión de plaquetas.
Tabla 11. Tiempo hasta la respuesta en pacientes con LMA tratados con Venetoclax en combinación con dosis bajas de citarabina
|
Venetoclax en combinación con dosis bajas de ciratabina N = 82 |
|
|
Tiempo medio hasta la MEJOR respuesta de CR (meses) Rango (meses) |
3.0 (0.9-22.4) |
|
Tiempo medio hasta PRIMERA respuesta de CR + CRh (meses) Rango (meses) |
1.0 (0.8-9.4) |
|
Tiempo medio hasta PRIMERA respuesta de CR + CRi (meses) Rango (meses) |
1.4 (0.8-14.9) |
La mediana de la supervivencia general (SG) para los pacientes tratados con venetoclax en combinación con una dosis baja de citarabina fue de 10,1 meses (IC del 95%: 5,7 a 14,2).
Se observaron remisiones (CR o CRh) en los subgrupos con diferentes características basales. Para pacientes con citogenética de riesgo deficiente o intermedio, se observaron tasas de remisión similares, la tasa fue de 34.6% o 57.1%, respectivamente.
Para pacientes con las siguientes mutaciones identificadas, las remisiones fueron las siguientes: TP53: 20.0%, IDH1/2: 66.7%, FLT-3: 31.3% y NPM1: 88.9%.
Se evaluó la enfermedad mínima residual (EMR) en la médula ósea para los pacientes que lograron CR o CRh después del tratamiento con venetoclax en combinación con citarabina a dosis bajas. De esos pacientes, el 34.2% (13/38) lograron EMR con menos de una célula LMA por 103 leucocitos en la médula ósea.
De los pacientes tratados con venetoclax en combinación con dosis bajas de citarabina, el 1,2% (1/82) lograron una CR/CRh y posteriormente recibieron un trasplante de células madre.
CONTRAINDICACIONES: En pacientes con Leucemia Linfocítica Crónica el uso concomitante de VENCLEXTA® está contraindicado con inhibidores potentes de CYP3A en el inicio y en la fase de ajuste de la dosis. (Véase Dosis y vía de administración e Interacciones medicamentosas y de otro género).
RESTRICCIONES DE USO DURANTE EL EMBARAZO Y LA LACTANCIA: VENCLEXTA® está contraindicado en mujeres embarazadas.
Datos en animales: En estudios de desarrollo embrio-fetal VENCLEXTA® fue administrado a ratones hembra y conejos hembra embarazadas para evaluar los efectos potenciales post-implantación y el subsecuente desarrollo embrio-fetal durante los periodos respectivos de organogénesis. En los ratones hembra, VENCLEXTA® fue asociado con un aumento de pérdida post-implantación y disminución del peso corporal fetal a una dosis de 150 mg/kg/día (exposición materna 1.2 veces el AUC de la dosis humana recomendada). En los conejos hembra, VENCLEXTA® a una dosis de 300 mg/kg/día produjo toxicidad materna, pero no fetal (exposición materna 0.2 veces el AUC de la dosis humana recomendada). No se observó teratogenicidad en ambos casos.
Reproducción:
Realizar prueba de embarazo.
Mujeres en edad reproductiva deben hacerse la prueba de embarazo antes de iniciar tratamiento con VENCLEXTA®.
Anticoncepción:
Mujeres en edad reproductiva deben utilizar un método anticonceptivo efectivo durante el tratamiento con VENCLEXTA® y por lo menos 30 días después de la última dosis de VENCLEXTA®.
Fertilidad:
Con base en los hallazgos de los estudios en animales, la fertilidad masculina puede ser comprometida con VENCLEXTA®.
Embarazo: VENCLEXTA® no deberá ser utilizado durante el embarazo.
No hay información adecuada y controlada del uso de VENCLEXTA® en mujeres embarazadas. Los estudios en animales han demostrado toxicidad embrio-fetal.
Lactancia:
No se sabe si venetoclax o sus metabolitos se excretan en la leche humana. Los datos disponibles en animales han demostrado la excreción de venetoclax/metabolitos en la leche.
No se puede excluir un riesgo para los recién nacidos/ bebés.
La lactancia debe ser interrumpida durante el tratamiento con VENCLEXTA®.
REACCIONES SECUNDARIAS Y ADVERSAS:
Se han observado las siguientes reacciones adversas:
Experiencia en estudios clínicos en LLC.
MURANO (GO28667): La seguridad de VENCLEXTA® en combinación con rituximab comparado con la combinación de bendamustina + rituximab fue evaluada en un ensayo clínico fase 3 en pacientes con LLC que recibieron al menos una terapia previa (Véase sección Farmacocinética y farmacodinamia, Estudios clínicos). Al momento del análisis de los datos, con una mediana de exposición de 22 meses, se realizó la comparación del brazo de la combinación venetoclax más rituximab con el brazo de bendamustina + rituximab por 6 meses.
Las discontinuaciones de tratamiento por efectos adversos ocurrieron en 16% de los pacientes tratado con la combinación venetoclax más rituximab. Las reducciones de dosis por un evento adverso ocurrieron en el 15% de los pacientes tratados con la combinación de venetoclax más rituximab. Se presentaron interrupciones de dosis en 71% de los pacientes tratados con la combinación de venetoclax más rituximab. El efecto adverso más común que llevó a una modificación de la dosis de venetoclax fue la neutropenia.
La Tabla 12 proporciona las reacciones adversas informadas en MURANO.
Tabla 12. Resumen de los efectos adversos en todos los grados reportados con una incidencia ≥ 10% y ≥ 5% o ≥ 2% en aquellos eventos adversos grado 3 y 4, en pacientes tratados con la combinación de venetoclax más rituximab comparado con bendemustina más rituximab
|
Venetoclax + rituximab (n=194) |
Bendamustina + rituximab (n=188) |
|||
|
Efectos adversos por aparatos y sistemas |
Todos los grados (%) |
Grado 3-4 (%) |
Todos los grados (%) |
Grado 3-4 (%) |
|
Alteraciones en el sistema linfático y sanguíneo |
||||
|
Neutropenia |
61 (muy común) |
58 |
44 |
39 |
|
Alteraciones en el sistema gastrointestinal |
||||
|
Diarrea |
40 (muy común) |
3 |
17 |
1 |
|
Infecciones e infestaciones |
||||
|
Infecciones del tracto respiratorio superior |
22 (muy común) |
2 |
15 |
1 |
|
Alteraciones en la nutrición o metabolismo |
||||
|
Síndrome de lisis tumoral |
3 (común) |
3 |
1 |
1 |
El perfil de seguridad de venetoclax así como otros efectos adversos reportados en el brazo de la combinación de venetoclax más rituximab del estudio MURANO incluyen:
Alteraciones en el sistema linfático y sanguíneo: Anemia (16%), neutropenia febril (4%), linfopenia (0%, considerada una reacción adversa basada en el mecanismo de acción).
Alteraciones en el sistema gastrointestinal: Náusea (21%), estreñimiento 14% y vómito (8%).
Alteraciones generales y sitio de administración: Fatiga (18%).
Infecciones e infestaciones: Neumonía (9%) e infección del tracto urinario (6%).
En el proceso de investigación: Incremento en la creatinina (3%).
Alteraciones en la nutrición o metabolismo: Hipercalemia (6%), hiperfosfatemia (5%), hiperuricemia (4%) e hipocalcemia (2%).
Durante el tratamiento como agente único una vez completado el ajuste de dosis de 5 semanas y después de terminar la combinación de venetoclax más rituximab se reportaron los efectos adversos más comunes (≥ 10% pacientes) y fueron diarrea (19%), neutropenia (14%) e infecciones de tracto respiratorio superior (12%); el efecto más frecuente en el grado 3 y 4 (≥ 2% pacientes) fue la neutropenia (11%).
La seguridad de VENCLEXTA® está basada en la información agrupada de 352 pacientes tratados con VENCLEXTA® en dos estudios fase 2 (M13-982 y M14-032) y en un estudio fase 1 (M12-175) los estudios recluyeron pacientes con LLC previamente tratados, incluyendo 212 pacientes con deleción 17p y 148 pacientes que habían fallado a un inhibidor del receptor de célula B. Los pacientes fueron tratados con 400 mg de VENCLEXTA® como monoterapia, una vez al día, seguido del esquema de ajuste de dosis.
Las reacciones adversas graves más comunes (≥ 2%) no relacionadas a la progresión de la enfermedad, fueron la neumonía y la neutropenia febril.
Las descontinuaciones del tratamiento por efectos no relacionados a la progresión de la enfermedad ocurrieron en el 9% de los pacientes.
Hubo reducción de la dosis en el 13% de los pacientes por reacciones adversas. La interrupción de dosis por efectos adversos fue en 36% de los pacientes. El único efecto adverso que se identificó y que llevó a una reducción de dosis en los eventos adversos más frecuentes (≥ 4%) fue la neutropenia (de un 5 a 4% respectivamente).
Síndrome de Lisis Tumoral: El síndrome de lisis tumoral (SLT) es el riesgo tóxico más importante identificado cuando se inicia VENCLEXTA®. En los estudios fase 1 de búsqueda de dosis, tuvieron un ajuste de dosis más corto, 2 a 3 semanas, y una mayor dosis inicial, en éstos la incidencia de SLT fue de 13% (10/77; 5 SLT por laboratorio y 5 SLT clínico), incluyendo 2 eventos fatales y 3 eventos de falla renal aguda, de los cuales uno requirió diálisis.
El riesgo de SLT se redujo después de revisar el régimen, dosis, medidas de profilaxis y monitoreo (Véase Dosis y vía de administración). En los estudios clínicos de venetoclax, los pacientes con un nódulo linfático medible ≥ 10 cm o aquellos con ambos, ALC ≥ 25 X 109/L y con un nódulo linfático medible ≥ 5 cm fueron hospitalizados para recibir una mayor hidratación y monitoreo durante el primer día de la dosis de 20 mg y 50 mg durante la fase de ajuste de dosis.
En 168 pacientes con LLC que iniciaron con una dosis diaria de 20 mg e incrementaron hasta 400 mg en un periodo de 5 semanas en los estudios M13-982 y M14032, la tasa de SLT fue de 2%. Todos los eventos de SLT fueron por laboratorio (Criterios de laboratorio que cumplieron ≥ 2 de los siguientes criterios dentro de las primeras 24 horas de cada una: Potasio > 6 mmol/L, ácido úrico > 476 μmol/L, calcio < 1.75 mmol/L, o fósforo > 1.5 mmol/L) o eventos de SLT fueron reportados y ocurrieron en pacientes que tuvieron nódulo(s) linfático(s) ≥ 5 cm y/o ALC 25 X 109/L. Todos los eventos se resolvieron en 5 días. No se observó SLT con consecuencias clínicas como falla renal aguda, arritmias cardiacas, muerte súbita y/o convulsiones. Todos los pacientes tuvieron CrCI > 50 mL/min.
Estudio (MURANO): En el estudio de fase 3, abierto, aleatorizado (MURANO), la incidencia de SLT fue de 3% (6/194) en los pacientes de la combinación de venetoclax con rituximab, después de tratar a 77/389 pacientes con las medidas de monitorización y profilaxis del STL realizadas por medio de una enmienda (Véase en Dosis y vías administración). Todos los eventos de SLT ocurrieron en el ajuste de dosis semanal y se resolvieron en 2 días. Los 6 pacientes completaron el esquema de dosis semanal hasta la dosis recomendada diaria de 400 mg de venetoclax. No se observó síndrome de lisis tumoral clínico después de las medidas de profilaxis y monitorización. No se reportaron datos de SLT clínico en pacientes que siguieron el ajuste de dosis de 5 semanas y la profilaxis de SLT (Véase Dosis y vía de administración). No se presentaron datos de SLT clínicos después del ajuste de dosis de 5 semanas y la utilización de las medidas de profilaxis y monitorización (Véase en Dosis y vía de administración). Los rangos de anormalidades de laboratorio ≥ 3 relevantes fueron hipercalemia en 1%, hiperfosfatemia en 1% e hiperuricemia 1%.
Leucemia mieloide aguda:
En combinación con azacitidina o decitabina estudio M14-358:
No se informaron eventos de SLT clínicos o de laboratorio informados con venetoclax en combinación con azacitidina o decitabina con la implementación del programa de aumento de dosis además de las medidas estándar de profilaxis y monitoreo.
En combinación con dosis bajas de citarabina estudio M 14-387: La incidencia de STL fue del 2,4% (2/82) con venetoclax en combinación con dosis bajas de citarabina con la implementación del calendario de aumento de la dosis, además de las medidas estándar de profilaxis y control. Todos los eventos fueron SLT de laboratorio, no hubo informes de SLT clínicos y todos los pacientes pudieron alcanzar la dosis objetivo.
Neutropenia:
Estudio MURANO: La neutropenia es un riesgo identificado en el tratamiento con venetoclax.
En el estudio MURANO en el brazo de VENCLEXTA® con rituximab, la neutropenia se reportó en el 61% de los pacientes (todos los grados). El 43% de los pacientes presentaron una interrupción de la dosis en el brazo de venetoclax con rituximab secundario a neutropenia y el 3% de los pacientes descontinuaron venetoclax por neutropenia. El 32% de los pacientes presentaron neutropenia grado 3 y 26% de los pacientes neutropenia grado 4. La mediana de duración de la neutropenia grado 3 o 4 fue de 8 días (rango de 1-712 días). Las complicaciones clínicas resultado de la neutropenia, así como la neutropenia febril, e infecciones graves grado ≥ 3 sólo se presentaron en tasas bajas en los paciente tratados con venetoclax más rituximab en comparación con el brazo de bendamustina más rituximab; se reportó neutropenia febril del 4% vs 10%, infección grado ≥ 3 en el 18% vs 23%, así como infecciones serias 21% vs 24% respectivamente.
Los efectos adversos están enlistados de acuerdo al sistema, órgano y clase de MedRA y por frecuencia. La frecuencia fue definida como muy común (≥ 1/10), común (≥ 1/100 a < 1/10), poco común (≥ 1/1,000 a <1/100), raro (≥ 1/10,000 a < 1/1,000), muy raro (< 1/10,000), desconocido (no se puede estimar la frecuencia con la información disponible). Dentro de cada grupo de frecuencia los efectos adversos son presentados en orden descendente de acuerdo a su gravedad.
Las reacciones adversas en el estudio fase 3 en pacientes previamente tratados usando monoterapia se presentan en la tabla 13.
Tabla 13. Efectos Adversos reportados en pacientes con LLC tratados con VENCLEXTA® en monoterapia
|
Efecto adverso por sistema |
Frecuencia (Todos los grados) N=352 |
Todos los grados N=352 |
Grado 3-4 N=352 |
|
Alteraciones en el sistema linfático y sanguíneo |
|||
|
Neutropeniaª |
Muy común |
50 |
45 |
|
Anemiab |
Muy común |
33 |
18 |
|
Linfopeniac |
Muy común |
11 |
7 |
|
Neutropenia febril |
Común |
6 |
6 |
|
Alteraciones en el sistema gastrointestinal |
|||
|
Diarrea |
Muy común |
43 |
3 |
|
Náusea |
Muy común |
42 |
1 |
|
Vómito |
Muy común |
16 |
1 |
|
Estreñimiento |
Muy común |
16 |
<1 |
|
Alteraciones generales y sitio de administración |
|||
|
Fatiga |
Muy común |
30 |
3 |
|
Infecciones e infestaciones |
|||
|
Infección del tracto respiratorio superior |
Muy común |
26 |
1 |
|
Neumonía |
Muy común |
12 |
7 |
|
Infección de vías urinarias |
Común |
9 |
1 |
|
Laboratorio |
|||
|
Elevación de la creatinina |
Común |
8 |
< 1 |
|
Alteraciones en la nutrición o metabolismod |
|||
|
N=168 |
N=168 Todos los grados |
N=168 grado ≥ 3 |
|
|
Síndrome de lisis tumorale |
Común |
2 |
2 |
|
Hipercalemiaf |
Muy común |
17 |
1 |
|
Hiperfosfatemiag |
Muy común |
14 |
2 |
|
Hiperuricemiah |
Común |
10 |
< 1 |
|
Hipocalcemiai |
Muy común |
16 |
2 |
a Neutropenia/disminución de la cuenta de neutrófilos.
b Anemia/disminución de la concentración de hemoglobina.
c Linfopenia/disminución de la cuenta de linfocitos.
d Reacción adversa por sistemas que fue reportada en pacientes en el seguimiento de ajuste semanal de dosis de 5 semanas y profilaxis de la lisis tumoral (véase Dosis y vía de administración).
e un evento de lisis tumoral reportado.
f Hiperfosfatemia/incremento del fósforo en sangre.
g Hipercalemia/incremento del potasio en sangre.
h Hiperuricemia/ácido úrico elevado.
i Hipocalcemia/decremento en el calcio sanguíneo.
Experiencia en ensayos clínicos en LMA: La seguridad de venetoclax (dosis diaria de 400 mg) en combinación con agentes hipometilantes (azacitidina [n = 84], o decitabina [n = 31]) y venetoclax (dosis diaria de 600 mg) en combinación con citarabina a dosis bajas (n = 82) se basa en dos ensayos no aleatorizados de pacientes con LMA recién diagnosticada (véase Farmacocinética y farmacodinamia, Estudios clínicos). La duración media de la exposición para los pacientes que toman venetoclax en combinación con azacitidina y decitabina fue de 6,4 meses (rango: 0,1 a 31,9) meses) y 5.7 meses (rango: 0.5 a 35.8 meses), respectivamente. La duración media de la exposición para pacientes que tomaron venetoclax en combinación con dosis bajas de citarabina fue de 4.2 meses (rango: 0.2 a 29.2 meses).
Las tasas de mortalidad a 30 días y 60 días observadas con venetoclax en combinación con azacitidina fueron 2.4% (2/84) y 8.3% (7/84), respectivamente. Las tasas de mortalidad a 30 y 60 días observadas con venetoclax en combinación con decitabina fueron del 6.5% (2/31) y 9.7% (3/31), respectivamente. Las tasas de mortalidad a 30 y 60 días observadas con venetoclax en combinación con citarabina a dosis bajas fueron del 6,1% (5/82) y del 14,6% (12/82), respectivamente.
M14-358:
En combinación con azacitidina: Las reacciones adversas más frecuentes (≥ 30%) de cualquier grado fueron náuseas, diarrea, trombocitopenia, estreñimiento, neutropenia, edema periférico, neutropenia febril, vómitos y fatiga y neumonía.
Se informaron eventos adversos graves en el 73% de los pacientes. Las reacciones adversas graves más frecuentes (≥ 5%) fueron neutropenia febril y neumonía.
Las interrupciones debidas a eventos adversos ocurrieron en el 19% de los pacientes. Las reacciones adversas más frecuentes que condujeron a la interrupción del tratamiento (≥ 2%) fueron la neutropenia febril y la neumonía.
Las interrupciones de la dosis debido a eventos adversos ocurrieron en el 61% de los pacientes. Las reacciones adversas más frecuentes que condujeron a la interrupción de la dosis (≥ 2%) fueron neutropenia febril, disminución del recuento de neutrófilos, neutropenia, neumonía y trombocitopenia.
Las reducciones de dosis debidas a reacciones adversas ocurrieron en 10% de los pacientes. La reacción adversa más frecuente que condujo a la reducción de la dosis (≥ 2%) disminuyó el recuento de neutrófilos.
En combinación con Decitabina: Las reacciones adversas más frecuentes (≥ 30%) de cualquier grado fueron trombocitopenia, neutropenia febril, náuseas, estreñimiento, fatiga, neumonía, diarrea, neutropenia, vómitos y edema periférico.
Se informaron eventos adversos graves en el 77% de los pacientes. Las reacciones adversas graves más frecuentes (≥ 5%) fueron neutropenia febril, neumonía, bacteriemia y sepsis.
Las interrupciones debidas a eventos adversos ocurrieron en el 23% de los pacientes. La reacción adversa más frecuente que condujo a la interrupción del medicamento (≥ 5%) fue la neumonía.
Las interrupciones de la dosis debido a eventos adversos ocurrieron en el 55% de los pacientes. Las reacciones adversas más frecuentes que condujeron a la interrupción de la dosis (≥ 5%) fueron la neutropenia febril, el recuento de neutrófilos disminuido y la neumonía. Las reducciones de dosis debidas a eventos adversos ocurrieron en el 6% de los pacientes. No se informaron eventos para más de un paciente.
Las reacciones adversas notificadas en pacientes con diagnóstico reciente de LMA con venetoclax en combinación con azacitidina o decitabina se presentan en las tablas a continuación (Tabla 14 y Tabla 15).
Tabla 14. Reacciones adversas informadas en ≥ 30% (cualquier grado) o ≥ 5% (grado 3 o 4) de pacientes con LMA tratados con Venetoclax en combinación con azacitidina
|
Reacción adversa por el sistema del cuerpo |
Frecuencia (Cualquier Grado) |
Cualquier grado (%) N=84 |
Grado 3 o 4 (%) N=84 |
|
Trastornos de la sangre y del sistema linfático |
|||
|
Trombocitopeniaª |
Muy común |
50 |
46 |
|
Neutropeniab |
Muy común |
48 |
48 |
|
Neutropenia febril |
Muy común |
37 |
37 |
|
Anemiac |
Muy común |
30 |
30 |
|
Desórdenes gastrointestinales |
|||
|
Náusea |
Muy común |
61 |
1 |
|
Diarrea |
Muy común |
56 |
2 |
|
Constipación |
Muy común |
49 |
2 |
|
Vómito |
Muy común |
36 |
0 |
|
Desórdenes generales y condiciones del sitio de administración |
|||
|
Edema periférico |
Muy común |
38 |
1 |
|
Fatiga |
Muy común |
32 |
6 |
|
Infecciones e infestaciones |
|||
|
Neumonía |
Muy común |
30 |
29 |
|
Bacteriemia |
Común |
4 |
2 |
|
Sepsis |
Común |
4 |
4 |
Reacciones adversas clasificadas utilizando los criterios de terminología común del NCI para eventos adversos versión 4.0.
a Trombocitopenia/recuento de plaquetas disminuido.
b Neutropenia/recuento de neutrófilos disminuido.
c Anemia/la hemoglobina disminuida.
d Neumonía/Neumonía atípica/consolidación pulmonar/Neumocistis jirovecii/neumonía por influenza/neumonía por legionella/neumonía estreptocócica/neumonía fúngica/neumonía por virus respiratorio sincicial/neumonía por Klebsiella/infección pulmonar/neumonía atípica por micobacteriana.
Tabla 15. Reacciones adversas notificadas en ≥ 30% (cualquier grado) o ≥ 5% (grado 3 o 4) de pacientes con LMA tratados con [Venetoclax] en combinación con decitabina
|
Reacción adversa por el sistema del cuerpo |
Frecuencia (Cualquier Grado) |
Cualquier grado (%) N=31 |
Grado 3 o 4 (%) N=31 |
|
Trastornos de la sangre y del sistema linfático |
|||
|
Trombocitopeniaª |
Muy común |
68 |
65 |
|
Neutropenia Febril |
Muy común |
65 |
65 |
|
Neutropeniab |
Muy común |
35 |
35 |
|
Anemiac |
Muy común |
23 |
23 |
|
Desórdenes gastrointestinales |
|||
|
Náusea |
Muy común |
61 |
0 |
|
Constipación |
Muy común |
48 |
0 |
|
Diarrea |
Muy común |
42 |
6 |
|
Vómito |
Muy común |
35 |
0 |
|
Desórdenes generales y condiciones del sitio de administración |
|||
|
Fatiga |
Muy común |
45 |
10 |
|
Edema periférico |
Muy común |
32 |
0 |
|
Infecciones e infestaciones |
|||
|
Neumoníad |
Muy común |
42 |
35 |
|
Bacteriemia |
Común |
23 |
16 |
|
Sepsis |
Común |
10 |
10 |
Reacciones adversas clasificadas utilizando los criterios de terminología común del NCI para eventos adversos versión 4.0.
a Trombocitopenia/recuento de plaquetas disminuido.
b Neutropenia/recuento de neutrófilos disminuido.
c Anemia/la hemoglobina disminuida.
d Neumonía/Neumonía atípica/consolidación pulmonar/Neumonía Neumocistis jirovecii/neumonía por influenza/ neumonía por Legionella/neumonía estreptocócica/neumonía fúngica/neumonía por virus respiratorio sincicial/neumonía por Klebsiella/infección pulmonar/neumonía atípica por micobacteriana.
Anormalidades de laboratorio: La Tabla 16 describe anormalidades comunes de laboratorio informadas durante el tratamiento que fueron nuevas o que empeoraron desde el inicio.
Tabla 16. Anormalidades de laboratorio nuevas o en empeoramiento con Venetoclax informadas en ≥ 40% (cualquier grado) o ≥ 10% (grado 3 o 4) de pacientes con LMA tratados con Venetoclax en combinación con un agente hipometilante
|
[Venetoclax] en combinación con azacitidina |
[Venetoclax] en combinación con Decitabine |
|||
|
Anormalidad de laboratorio |
Todos los Gradosa (%) N=84 |
Grado 3 o 4b (%) N=84 |
Cualquier gradoa (%) N=31 |
Grado3o4 (%) N=31 |
|
Hematología |
||||
|
Disminución absoluta del conteo de neutrófilos |
100 |
98 |
100 |
100 |
|
Disminución absoluta del recuento de glóbulos blancos |
100 |
99 |
97 |
97 |
|
Disminución del conteo de plaquetas |
91 |
81 |
86 |
76 |
|
Disminución absoluta del recuento de linfocitos |
89 |
75 |
100 |
81 |
|
Disminución de la hemoglobina |
56 |
56 |
68 |
65 |
|
Química |
||||
|
Elevación de glucosa |
75 |
12 |
77 |
0 |
|
Calcio bajo |
61 |
8 |
87 |
3 |
|
Albúmina baja |
55 |
5 |
48 |
6 |
|
Potasio bajo |
51 |
7 |
48 |
6 |
|
Sodio bajo |
50 |
8 |
52 |
3 |
|
Fosfato inorgánico bajo |
49 |
19 |
26 |
6 |
|
Elevación de bilirrubina total |
48 |
8 |
32 |
10 |
|
Magnesio bajo |
29 |
0 |
45 |
3 |
a Incluye anormalidades de laboratorio que fueron nuevas o que empeoraron, o que empeoraron desde el inicio del estudio.
M14-387:
En combinación con dosis bajas de citarabina: Las reacciones adversas más frecuentes (≥ 30%) de cualquier grado fueron náuseas, trombocitopenia, diarrea, neutropenia, neutropenia febril, fatiga, estreñimiento y vómitos.
Se informaron eventos adversos graves en el 91% de los pacientes. Las reacciones adversas graves más frecuentes (≥ 5%) fueron neutropenia febril, neumonía y sepsis.
Las interrupciones debidas a eventos adversos ocurrieron en el 29% de los pacientes. Las reacciones adversas más frecuentes que condujeron a la interrupción del fármaco (≥ 2%) fueron trombocitopenia y sepsis.
Las interrupciones de la dosis debido a eventos adversos ocurrieron en el 55% de los pacientes. Las reacciones adversas más frecuentes que condujeron a la interrupción de la dosis (≥ 2%) fueron trombocitopenia, neutropenia, neutropenia febril, vómitos, neumonía y sepsis.
Las reducciones de dosis debidas a eventos adversos ocurrieron en el 7% de los pacientes. La reacción adversa más frecuente que condujo a una reducción de la dosis (≥ 2%) fue la trombocitopenia.
Las reacciones adversas notificadas en pacientes recién diagnosticados con LMA que reciben venetoclax en combinación con citarabina a dosis bajas se presentan en la Tabla 17 .
Tabla 17. Reacciones adversas informadas en ≥ 30% (cualquier grado) o ≥ 5% (grado 3 o 4) de pacientes con LMA tratados con Venetoclax en combinación con dosis bajas de citarabina
|
Reacción adversa por el sistema del cuerpo |
Frecuencia (Cualquier Grado) |
Cualquier grado (%) N=82 |
Grado 3 o 4 (%) N=82 |
|
Trastornos de la sangre y del sistema linfático |
|||
|
Trombocitopeniaª |
Muy común |
60 |
60 |
|
Neutropeniab |
Muy común |
44 |
44 |
|
Neutropenia febril |
Muy común |
43 |
41 |
|
Anemiac |
Muy común |
28 |
28 |
|
Desórdenes gastrointestinales |
|||
|
Náusea |
Muy común |
70 |
2 |
|
Diarrea |
Muy común |
49 |
2 |
|
Constipación |
Muy común |
35 |
0 |
|
Vómito |
Muy común |
30 |
4 |
|
Desórdenes generales y condiciones del sitio de administración |
|||
|
Fatiga |
Muy común |
43 |
7 |
|
Infecciones e infestaciones |
|||
|
Neumoníad |
Muy común |
20 |
18 |
|
Sepsis |
Muy común |
12 |
11 |
Reacciones adversas clasificadas utilizando los criterios de terminología común del NCI para eventos adversos versión 4.0.
a Trombocitopenia/recuento de plaquetas disminuido.
b Neutropenia/recuento de neutrófilos disminuido.
c Anemia/la hemoglobina disminuida.
d Neumonía/Neumonía atípica/consolidación pulmonar/Neumonía Neumocistis jirovecii/neumonía por influenza/ neumonía por Legionella/neumonía estreptocócica/ neumonía fúngica/neumonía por virus respiratorio sincicial/neumonía por Klebsiella/infección pulmonar/neumonía atípica por micobacteriana.
Anormalidades de laboratorio: La Tabla 18 describe anomalías comunes de laboratorio informadas durante el tratamiento que fueron nuevas o que empeoraron desde el inicio.
Tabla 18. Anomalías nuevas o que empeoraron con venetoclax reportadas en > 40% (cualquier grado) o 10% (grado 3 o 4) de pacientes con LMA tratados con venetoclax en combinación con dosis bajas de Citarabina
|
Anormalidad de laboratorio |
Todos los Gradosa (%) N=82 |
Grado 3 o 4b (%) N=82 |
|
Hematología |
||
|
Disminución del conteo de plaquetas |
98 |
95 |
|
Disminución absoluta del conteo de neutrófilos |
97 |
94 |
|
Disminución del conteo de glóbulos blancos |
96 |
95 |
|
Disminución absoluta del recuento de linfocitos |
95 |
65 |
|
Disminución de la hemoglobina |
63 |
62 |
|
Química |
||
|
Elevación de glucosa |
84 |
12 |
|
Calcio bajo |
82 |
15 |
|
Sodio bajo |
63 |
11 |
|
Elevación de bilirrubina total |
63 |
9 |
|
Albúmina baja |
63 |
9 |
|
Potasio bajo |
60 |
20 |
|
Fosfato inorgánico bajo |
55 |
23 |
|
Magnesio bajo |
45 |
1 |
|
Elevación de la fosfatasa alcalina |
41 |
1 |
a Incluye anormalidades de laboratorio que fueron nuevas o que empeoraron, o que empeoraron desde el inicio del estudio.
PRECAUCIONES EN RELACIÓN CON EFECTOS DE CARCINOGÉNESIS, MUTAGÉNESIS, TERATOGÉNESIS Y SOBRE LA FERTILIDAD: No se han realizado estudios de carcinogénesis con venetoclax.
Venetoclax no fue mutagénico en un ensayo bacterial de mutagenicidad in vitro (Ames), no indujo aberraciones numéricas o estructurales en un ensayo de aberraciones cromosómicas in vitro que utilizó linfocitos de sangre periférica humana, y no fue clastogénica en ensayo en el micronúcleo de la medula ósea de ratón in vivo a dosis de hasta 835 mg/kg/día. El metabolito M27 fue negativo para actividad genotóxica in vitro Ames y en un ensayo de aberraciones cromosómicas, en ratones se ha observado un efecto similar a VENCLEXTA® (disminución de la cuenta de linfocitos y de la masa de glóbulos rojos) pero de menor magnitud, consistente con su baja potencia farmacológica in vitro.
Estudios de fertilidad y desarrollo temprano fueron conducidos en ratones machos y hembras. Estos estudios evaluaron apareamiento, fertilización y desarrollo embrionario hasta la implantación. No hubieron efectos de venetoclax en los ciclos de estroma, apareamiento, fertilidad, cuerpo lúteo, implantes uterinos o embriones vivos por camada a dosis de hasta 600 mg/kg/día (exposición de 2.8 y 3.2 veces el AUC de la dosis humana recomendada en ratones machos y hembras, respectivamente) . Sin embargo, un riesgo para la fertilidad masculina humana existe, basado en la toxicidad testicular (pérdida de células germinales) observado en perros en todas las dosis examinadas (exposiciones de 0.5 a 18 veces la exposición en humanos según ABC a la dosis recomendada). La reversibilidad de este hallazgo no ha sido demostrada.
Farmacología Animal y/o Toxicología: En ratas preñadas, las exposiciones maternas sistémicas (AUC) a venetoclax fueron aproximadamente 14 veces mayores que la exposición en humanos a la dosis de 400 mg. Los niveles medibles de radioactividad en los tejidos fetales (hígado, tracto gastrointestinal) fueron > 15 veces menores que los niveles maternos en los mismos tejidos. La radioactividad derivada de Venetoclax no se detectó en sangre, cerebro, ojo, corazón, riñón, pulmón, músculo o médula espinal fetales.
Se administró Venetoclax (dosis única: 150 mg/kg por vía oral) a ratas lactantes durante 8-10 días de parto. Venetoclax en la leche fue 1.6 veces más bajo que en el plasma. El fármaco parental (venetoclax) representaba la mayor parte del material total relacionado con el fármaco en la leche, con trazas de tres metabolitos.
INTERACCIONES MEDICAMENTOSAS Y DE OTRO GÉNERO:
Efectos de otros fármacos sobre VENCLEXTA®.
VENCLEXTA® es metabolizado principalmente por el CYP3A.
lnhibidores de CYP3A: La administración concomitante de ketoconazol aumentó la Cmax de venetoclax en un 130% y la AUC∞ en un 540%.
La administración concomitante de ritonavir aumentó la Cmax de venetoclax en un 140% y el AUC en un 690%.
Comparado con el venetoclax 400 mg administrado solo, la administración concomitante de posaconazol con venetoclax 50 mg y 100 mg resultó en un 61% y 86% más alto que venetoclax Cmax, respectivamente. El AUC24 de venetoclax fue 90% y 144% mayor, respectivamente.
La co-administración de 400 mg una vez al día de Ketokonazol, un inhibidor potente de CYP3A, P-pg y un inhibidor BCRP, por 7 días en 11 pacientes previamente tratados con LNH incrementó la Cmax 130% y el AUC alrededor de 540%.
La coadministración de 50 mg de ritonavir, un inhibidor potente de CYP3A, así como inhibidor de P-gp y OATP1B1/B3 en 6 sujetos sanos por 14 días, incrementó la Cmax 140% y la AUC∞690%.
Comparado con venetoclax 400 mg administrado solo, la administración concomitante de 300 mg de posaconazol, un fuerte inhibidor de CYP3A y P-gp, con venetoclax 50 mg y 100 mg durante 7 días en 12 pacientes recién diagnosticados con LMA resultó en 61% y 86% más venetoclax Cmax, respectivamente. El AUC24 de venetoclax fue 90% y 144% mayor, respectivamente.
Para los pacientes con LLC que requieren el uso concomitante de venetoclax con inhibidores potentes del CYP3A (p. Ej., ltraconazol, ketocona zol, posaconazol, voriconazol, claritromicina, ritonavir) o inhibidores moderados de CYP3A (p. Ej., Ciprofloxacina, diltiazem, eritromicina, dronaderona, fluconazol, verapamilo) administre la dosis de venetoclax de acuerdo con la Tabla 10. Controle a los pacientes más de cerca en busca de signos de toxicidad de venetoclax.
Evite los productos de pomelo (toronja), naranjas de Sevilla y carambola durante el tratamiento con venetoclax, ya que contienen inhibidores de CYP3A.
Reanudar la dosis de VENCLEXTA® utilizada antes de iniciar el inhibidor de CYP3A 2 a 3 días después de la discontinuación del inhibidor (Véase Dosis y vía de administración).
lnhibidores de OATP1B1/1B3 y P-gp: La coadministración de una sola dosis de rifampicina, un inhibidor de OATP1B1/1B3 y P-gp, aumentó la Cmax de VENCLEXTA® un 106% y el AUC∞ un 78% (Véase Propiedades farmacológicas).
Evitar el uso concomitante de venetoclax con inhibidores de la P-gp (por ejemplo, amiodarona, captopril, carvedilol, ciclosporina, felodipina, quercetina, quinidina, ranolazina, ticagrelor) al inicio y durante la fase de rampa; si se debe usar un inhibidor de la P-gp, controle de cerca los signos de toxicidad.
Inductores de CYP3A: La coadministración de una sola dosis de rifampina, un inductor fuerte de CYP3A, disminuyó la Cmax de VENCLEXTA® un 42% y el AUC∞ un 71%. Evitar el uso de VENCLEXTA® con inductores fuertes del CYP3A (carbamazepina, rifampina, fenitoína, hierba de San Juan) o inductores moderados del CYP3A (bosentan, efavirenz, etravirina, modafinil, nafcilina). Considerar tratamientos alternos con menor inducción de CYP3A. (Véase Farmacocinética y farmacodinamia, Propiedades farmacológicas).
Azitromicina: La coadministración de VENCLEXTA® con azitromicina un inhibidor de P-gp disminuye la Cmax un 25% y el AUC∞ un 35%. La coadministración de 500 mg de azitromicina en el primer día seguido de 250 mg por 4 días, en 12 personas sanas, disminuyó la Cmax un 25% y la AUC∞ un 35%. La administración de 500 mg de azitromicina en el día 1 seguido de la administración de 250 mg durante 4 días en pacientes sanos con VENCLEXTA®, disminuyó la Cmax y el AUC∞ de un 25 a 35% respectivamente. No se requieren ajustes de la dosis de venetoclax cuando se administré con azitromicina.
Efectos de VENCLEXTA® sobre otros fármacos:
Warfarina: En un estudio de interacciones fármaco-fármaco en voluntarios sanos, la administración de una sola dosis de VENCLEXTA® con warfarina resultó en un aumento de la Cmax de warfarina del 18 al 28% y el AUC∞ de R-warfarina y S-warfarina. Ya que VENCLEXTA® no fue dosificado en estado estable, es recomendable que el INR (international normalized ratio, por sus siglas en inglés) sea monitorizado de cerca en pacientes recibiendo warfarina.
Digoxina: En un estudio de interacciones fármaco-fármaco en 10 voluntarios sanos, la administración de una dosis única de 100 mg de VENCLEXTA®con 0.5 mg de digoxina, un sustrato de la P-gp, resultó en un aumento de la Cmax de digoxina del 35% y el AUC∞ del 9%.
Substratos de P-gp: La administración de una única dosis de 100 mg de VENCLEXTA® con digoxina terminó en un incremento de la Cmax del 35% de la digoxina y un incremento en la AUC∞ del 9%. Por eso la administración con VENCLEXTA® de sustratos de P-gp con un índice terapéutico estrecho debe evitarse. Un sustrato de la P-gp con un índice terapéutico estrecho puede administrarse si se toma por lo menos 6 horas antes de VENCLEXTA®.
ALTERACIONES EN LOS RESULTADOS DE PRUEBAS DE LABORATORIO: Se han reportado disminución en la cuenta de neutrófilos, eritrocitos y trombocitos en la prueba de biometría hemática completa en los estudios clínicos de pacientes tratados con venetoclax, pudiendo llegar a neutropenia, anemia y trombocitopenia respectivamente. La respuesta a VENCLEXTA® debe monitorearse a través del estudio de biometría hemática completa.
PRECAUCIONES GENERALES:
Síndrome de Lisis Tumoral: El síndrome de lisis tumoral (SLT), incluyendo eventos fatales, ha ocurrido en pacientes con carga tumoral alta cuando han sido tratados con VENCLEXTA® (Véase Reacciones secundarias y adversas).
VENCLEXTA® puede causar reducción rápida del tumor, y por tanto conferir riesgo de SLT en la fase inicial de ajuste de dosis. Puede haber cambios en los electrólitos que requieren rápida intervención tan temprano como a las 6 a 8 horas siguientes de la administración de la primera dosis de VENCLEXTA® y con cada incremento de dosis.
El riesgo de SLT es continuo por muchos factores, incluyendo las comorbilidades. Pacientes con alta carga tumoral (por ejemplo un nódulo linfático con diámetro ≥ 5 cm o ALC -conteo absoluto de linfocitos- ≥ 25 X 109/L) tienen mayor riesgo de presentar SLT con VENCLEXTA®. Función Renal deteriorada aumenta el riesgo. Los pacientes tienen que ser evaluados y recibir profilaxis adecuada para el SLT, incluyendo hidratación y administración de medicamentos para evitar hiperuricemia. El profesional de la salud debe monitorear la química sanguínea y manejar oportunamente las anormalidades. La dosis debe ser interrumpida si es necesario. Si el riesgo incrementa, podrían necesitarse medias intensivas (hidratación intravenosa, monitoreo frecuente, hospitalización) (Véase Dosis y vía de administración).
El uso concomitante de VENCLEXTA® con inhibidores fuertes o moderados de CYP3A aumenta la exposición a VENCLEXTA® y podría aumentar el riesgo de SLT al inicio y en la fase de ajuste de dosis (Véase Dosis y vía de administración e Interacciones medicamentosas y de otro género). Además los inhibidores de la P-gp pueden incrementar la exposición a VENCLEXTA®.
Neutropenia: En pacientes con LLC Grados 3 y 4 de neutropenia han ocurrido en pacientes tratados con VENCLEXTA® como monoterapia y en combinación con rituximab en los diferentes estudios (GO28667/MURANO) (Véase Reacciones secundarias y adversas). En pacientes con LMA, la neutropenia de Grado 3 o 4 es común antes de comenzar el tratamiento. El conteo de neutrófilos puede empeorar con venetoclax en combinación con un agente hipometilante o dosis bajas de citarabina. La neutropenia puede reaparecer con ciclos posteriores de terapia. Monitorear con biometría hemática completa durante el periodo que dure el tratamiento. Se recomienda la reducción o interrupción de las dosis si hay neutropenia grave. Considerar medidas de soporte como terapia antimicrobiana en caso de signos de infección y uso profiláctico de factores de crecimiento (Factor estimulante de crecimiento de granulocitos; G-CSF) (Véase Dosis y vía de administración, Contraindicaciones, Interacciones medicamentosas y de otro género).
Vacunación: La seguridad y eficacia de la vacunación con vacunas de microorganismos vivos atenuados durante o después del tratamiento con VENCLEXTA® no han sido estudiadas. Vacunas de microorganismos vivos no deberán ser administradas durante el tratamiento con VENCLEXTA® y sólo despues de la recuperación de las celulas B.
DOSIS Y VÍA DE ADMINISTRACIÓN: La vía de administración es oral.
Guía de dosificación:
Leucemia Linfocítica Crónica (LLC): VENCLEXTA® en monoterapia debe tomarse oralmente una vez al día hasta la progresión de la enfermedad o que se observe toxicidad. La dosis recomendada de venetoclax es de 400 mg una vez al día una vez que el paciente completó el esquema de ajuste de dosis.
VENCLEXTA® en combinación con rituximab y una vez terminado el ajuste de dosis semanal se administra 24 meses iniciando el conteo en el día 1 del primer ciclo de rituximab. El inicio de rituximab es después de completar el esquema de ajuste de dosis semanal con venetoclax y se haya administrado la dosis de 400 mg por 7 días. Los pacientes deben continuar venetoclax 400 mg una vez al día por 24 meses iniciando desde el día 1 del ciclo 1 de la administración de rituximab.
Régimen de dosificación recomendado: Instruir al paciente a tomar las tabletas de VENCLEXTA® con una comida y agua aproximadamente a la misma hora cada día. Las tabletas de VENCLEXTA® deben ser tragadas no masticadas, aplastadas o rotas antes de tragarlas.
Esquema Semanal de Ajuste de Dosis en monoterapia: La dosis inicial de VENCLEXTA® es de 20 mg al día por 7 días. La dosis de VENCLEXTA® debe ser administrada de acuerdo al esquema semanal de ajuste de dosis hasta los 400 mg al día en un periodo de 5 semanas, como se muestra en la Tabla 19. El esquema semanal de ajuste de dosis de 5 semanas está diseñado para reducir gradualmente la carga de tumor y disminuir el riesgo de SLT.
Tabla 19. Esquema semanal de ajuste de dosis en la fase de inicio en Pacientes con Leucemia Linfocítica Crónica
|
Semana |
Dosis diaria de Venetoclax |
|
1 |
20 mg |
|
2 |
50 mg |
|
3 |
100 mg |
|
4 |
200 mg |
|
5 |
400 mg |
La recomendación después del término del esquema de ajuste de dosis es continuar 400 mg de VENCLEXTA® una vez al día. VENCLEXTA® debe administrarse una vez al día hasta la progresión o al presentar una toxicidad inaceptable.
Esquema Semanal de Ajuste de Dosis en Combinación de VENCLEXTA® con Rituximab: Después de que el paciente ha completado el esquema semanal de ajuste de dosis antes mencionado y ha recibido la dosis de 400 mg por 7 días completos sin evidencia de toxicidad no hematológica de grado 3-4 o toxicidad hematológica grado 4, y no tiene contraindicaciones para el uso de rituximab (Véase IPP-A de rituximab-Mabthera®), el paciente podrá iniciar la terapia combinada de la siguiente forma:
• 6 ciclos de rituximab:
o Las infusiones deben ser administradas en el día 1 de cada ciclo de 28 días; en combinación con la dosis diaria de venetoclax de la siguiente manera:
1. 375 mg/m2 IV en el día 1.
2. 500 mg/m2 IV en el día 1 de los ciclos 2 al 6.
• Se administrarán un total de 6 infusiones de rituximab.
• El paciente debe continuar con 400 mg de VENCLEXTA® una vez al día por 24 meses una vez iniciado el día 1 del ciclo 1 de rituximab.
El paciente debe continuar con 400 mg de VENCLEXTA® una vez al día por 24 meses una vez iniciado el día 1 del ciclo 1 de rituximab.
Leucemia Mieloide Aguda (LMA):
La dosis de VENCLEXTA® depende del agente de combinación: El programa de dosificación de VENCLEXTA® (incluida la fase de rampa) se muestra en la tabla 20. Inicie el agente hipometilante o la dosis baja de citarabina en el día 1.
Tabla 20. Esquema de dosis para la fase de ajuste en pacientes con LMA
|
Día |
VENCLEXTA® Dosis diaria |
|
|
1 |
100 mg |
|
|
2 |
200 mg |
|
|
3 |
400 mg |
|
|
4 y más allá |
400 mg Cuando se dosifica en combinación con un agente hipometilante |
600 mg Cuando se dosifica en combinación con dosis bajas de citarabina |
VENCLEXTA®, en combinación con un agente hipometilante o baja dosis de citarabina, debe continuarse hasta que se observe progresión de la enfermedad o toxicidad inaceptable.
Dosis olvidada: Si el paciente olvida una dosis de VENCLEXTA® dentro de las 8 horas del tiempo que normalmente toma la dosis, el paciente debe tomar la dosis olvidada tan pronto como sea posible y continuar con las dosis diarias programadas. Si el paciente olvida la dosis por más de 8 horas, no deberá tomar la dosis olvidada y continuará con la dosis programada para el día siguiente.
Si el paciente vomita al tomar una dosis, no debe tomar otra dosis ese día. La siguiente dosis la debe tomar en el horario usual.
Evaluación del riesgo de síndrome de lisis tumoral: Los pacientes tratados con [venetoclax] pueden desarrollar el síndrome de lisis tumoral. Consulte la sección apropiada a continuación para obtener detalles específicos sobre la administración.
Leucemia Linfocítica Crónica (LLC): VENCLEXTA® puede causar reducción rápida de la carga tumoral, y por tanto conferir riesgo de SLT en la fase inicial de las 5 semanas de ajuste de dosis. Puede haber cambios en los electrólitos que requieren rápida intervención tan temprano como a las 6 a 8 horas siguientes de la administración de la primera dosis de VENCLEXTA® y con cada incremento de dosis.
El riesgo de SLT es continuo por muchos factores, incluyendo las comorbilidades. Los pacientes con alta carga tumoral (por ejemplo un nódulo linfático con diámetro ≥ 5 cm o ALC ≥ 25 X 109/L) tienen mayor riesgo de presentar SLT con VENCLEXTA®. Función Renal deteriorada (CrCI < 80 mL/min) aumenta el riesgo. El riesgo de presentar SLT puede bajar con la disminución de la carga tumoral producida por el tratamiento con VENCLEXTA® (Véase Precauciones generales).
Realizar evaluación de la carga tumoral, incluyendo valoración radiológica (Tomografía computada). Evaluar la química sanguínea (potasio, ácido úrico, fósforo, calcio y creatinina) en todos los pacientes y corregir las anormalidades pre-existentes antes de iniciar el tratamiento con VENCLEXTA®.
Profilaxis para el síndrome de lisis tumoral:
Leucemia Linfocítica Crónica (LLC):
Seguir las medidas de profilaxis enlistadas a continuación. Utilizar medidas más intensivas (incluyendo hospitalización) si el riesgo incrementa:
• Hidratación: asegurar una hidratación adecuada antes de iniciar la terapia con VENCLEXTA® y durante la fase de ajuste de dosis, especialmente el primer día de cada ajuste de dosis. Administre fluidos intravenosos como sea necesario basado en el riego de SLT o para aquellos pacientes que no pueden mantener hidratación oral adecuada.
• Agentes anti-hiperuricémicos: administrar agentes anti-hiperuricémicos (alopurinol) para pacientes con niveles altos de ácido úrico o en riesgo de SLT. Iniciar 2 a 3 días antes del inicio de VENCLEXTA®; considerar continuar durante la fase de ajuste de dosis.
• Estudios de laboratorio:
o Antes de la dosis:
• Para todos los pacientes, evaluar la química sanguínea antes de iniciar el tratamiento con VENCLEXTA® para evaluar la función renal y corregir anormalidades pre-existentes. Re-evaluar la química sanguínea antes de iniciar cada ajuste de dosis de VENCLEXTA®.
o Después de la dosis:
• Para pacientes en riesgo de SLT, monitorear la química sanguínea a las 6 u 8 horas y a las 24 horas posteriores al inicio de VENCLEXTA®. Corregir las anormalidades electrolíticas pronto. No administrar la siguiente dosis hasta que los resultados de la química sanguínea de las 24 horas hayan sido evaluados. Seguir el mismo esquema de monitorización hasta llegar a la dosis de 50 mg y cuando el paciente continúe con riesgo de SLT en las dosis subsecuentes.
• Hospitalización: basado en la evaluación clínica del médico, algunos pacientes, especialmente aquellos con alto riesgo de SLT, pueden requerir ser hospitalizados en el día de la primer dosis de VENCLEXTA® para recibir una mayor profilaxis y monitoreo durante las primeras 24 horas. Considerar hospitalización para los subsecuentes ajustes de dosis basados en la revaluación del riesgo.
Leucemia mieloide aguda:
Siga las medidas de profilaxis que se enumeran a continuación:
• Todos los pacientes deben tener recuento de glóbulos blancos < 25 x 109/L antes del inicio de [venetoclax] y puede ser necesaria una citorreducción antes del tratamiento.
• Todos los pacientes deben recibir medidas profilácticas que incluyan una hidratación adecuada y agentes antihiperuricémicos antes del inicio de la primera dosis y durante la fase de arranque.
• Evalúe la química sanguínea (potasio, ácido úrico, fósforo, calcio y creatinina) y corrija las anomalías preexistentes antes de iniciar el tratamiento con venetoclax.
• Monitorear la química sanguínea para TLS en la pre-dosis, de 6 a 8 horas después de cada nueva dosis durante la rampa y 24 horas después de alcanzar la dosis final.
• Para pacientes con factores de riesgo para TLS (p. Ej., Blastocitos circulantes, alta carga de leucemia en la médula ósea, niveles elevados de pretratamiento LDH o reducción de la función renal) se deben considerar medidas adicionales, incluido un aumento de la monitorización de laboratorio y la reducción de la dosis inicial de venetoclax.
Modificación de dosis basadas en toxicidad:
Leucemia Linfocitica Crónica (LLC): La interrupción o reducción de dosis pueden ser necesarias. Para los pacientes que han tenido interrupción de dosis por más de una semana durante las primeras 5 semanas de la fase de ajuste de dosis o mayor a 2 semanas completando el esquema semanal de ajuste de dosis, reevaluar el riesgo de SLT para determinar si el reinicio con una dosis menor es necesaria. (Todos o algunos niveles del esquema de ajuste de dosis) (Véase Régimen de dosificación recomendado, Evaluación del riesgo de síndrome de lisis tumoral y Profilaxis para el síndrome de lisis tumoral).
Modificación de dosis para síndrome de lisis tumoral: Si el paciente experimenta cambios en la química sanguínea sugestivos de síndrome de lisis tumoral, retener la siguiente dosis de VENCLEXTA®. Si se resuelve de 24 a 48 horas de la última dosis, reanudar el tratamiento con VENCLEXTA® a la misma dosis.
Para eventos de SLT clínico o cambios en la química sanguínea que requieran más de 48 horas para normalizarse, reanudar el tratamiento con una dosis reducida (véase Tabla 4).
Cuando se reanude el tratamiento con VENCLEXTA® después de la interrupción por SLT, seguir las instrucciones para Profilaxis para el síndrome de lisis tumoral.
Modificación de dosis por otras toxicidades:
Leucemia Linfocitica Crónica (LLC): Retener el tratamiento con VENCLEXTA® por cualquiera de las toxicidades no hematológicas, neutropenia con infección o fiebre grado 3, o toxicidades hematológicas grado 4, excepto linfopenia (Véase Precauciones generales). Para reducir el riesgo de infección asociada con neutropenia, se pueden administrar factores estimulantes de colonia de granulocitos (G-CSF) con VENCLEXTA® si está indicado clínicamente. Una vez que la toxicidad se resuelva a grado 1 o estado basal, la terapia con VENCLEXTA® puede ser reanudada a la misma dosis.
Si la toxicidad es recurrente seguir la guía de reducciones de dosis en la Tabla 10 cuando se reanude el tratamiento con VENCLEXTA® después de la resolución. Una mayor disminución de dosis puede ser indicada por el médico.
Para pacientes que requieren reducción de dosis menor a 100 mg por más de dos semanas, considerar la discontinuación de VENCLEXTA®.
Tabla 21. Reducción de dosis por toxicidad durante el tratamiento con VENCLEXTA®
|
Dosis retenida (mg) |
Dosis reanudada (mg) |
|
400 |
300 |
|
300 |
200 |
|
200 |
100 |
|
100 |
50 |
|
50 |
20 |
|
20 |
10 |
a Continuar con la dosis reducida por una semana antes de incrementar la dosis.
Leucemia mieloide aguda (AML): Realice recuentos sanguíneos con frecuencia mediante la resolución de citopenias. El manejo de algunas reacciones adversas [Véase Precauciones generales] puede requerir interrupciones de la dosis o interrupción permanente de [venetoclax]. La Tabla 22 muestra las pautas de modificación de dosis para toxicidades hematológicas.
Tabla 22. Modificaciones de dosis recomendadas para toxicidadesª
|
Event |
Occurrence |
Action |
|
Toxicidades hematológicas |
||
|
Neutropenia de grado 4 con o sin fiebre o infección; o trombocitopenia de grado 4 (Véase Precauciones generales] |
Ocurrencia antes de lograr la remisión |
Transfundir los productos sanguíneos, administrar profilácticos y tratamientos anti-infecciosos según lo indicado clínicamente. En la mayoría de los casos, el venetoclax y el agente hipometilante o los ciclos de dosis bajas de citarabina no deben interrumpirse debido a las citopenias antes de lograr la remisión. |
|
Primera aparición después de alcanzar la remisión y que dura al menos 7 días |
Retrasar el ciclo de tratamiento posterior de venetoclax y el agente hipometilante o dosis bajas de citarabina y controlar los recuentos sanguíneos. El factor estimulante de colonias de granulocitos (G-CSF) se puede administrar si está clínicamente indicado para la neutropenia. Una vez que la toxicidad se ha resuelto en Grado 1 o 2, se puede reanudar el tratamiento con venetoclax a la misma dosis en combinación con agente hipometilante o dosis bajas de citarabina. |
|
|
Ocurrencias posteriores en ciclos después de lograr la remisión y que duran 7 días o más |
Retrasar el ciclo de tratamiento posterior de venetoclax y el agente hipometilante o dosis bajas de citarabina y controlar los recuentos sanguíneos. El G-CSF se puede administrar si está clínicamente indicado para la neutropenia. Una vez que la toxicidad se ha resuelto en Grado 1 o 2, la terapia con [venetoclax] se puede reanudar a la misma dosis y la duración se puede reducir en 7 días para cada ciclo subsiguiente. |
|
a Las reacciones adversas se clasificaron usando NCI CTCAE versión 4.0.
Modificaciones de dosis para el uso con inhibidores de CYP3A: El uso concomitante de VENCLEXTA® con inhibidores fuertes o moderados del CYP3A aumenta la exposicion (ejemplo la Cmax y la AUC) y el riesgo de SLT al inicio y durante la fase de ajuste de dosis. El uso concomitante de VENCLEXTA® con inhibidores fuertes del CYP3A está contraindicado al inicio y durante la fase de ajuste de dosis (Véase Contraindicaciones).
Evitar el uso concomitante de VENCLEXTA® con inhibidores moderados del CYP3A al inicio y durante la fase de ajuste de dosis. Si un inhibidor moderado del CYP3A debe ser utilizado, siga las recomendaciones para manejar las interacciones medicamentosas resumidas en la Tabla 12. Controle a los pacientes más de cerca en busca de signos de toxicidad [Modificación de dosis basadas en toxicidad].
Reanude la dosis de VENCLEXTA® que fue utilizada antes de la utilización del inhibidor del CYP3A 2 o 3 días después de la descontinuación del inhibidor (Véase Modificación de dosis basadas en toxicidad e Interacciones medicamentosas y de otro género).
Tabla 23. Manejo de interacciones potenciales Venetoclax con inhibidores de CYP3A
|
lnhibidores |
Fase de iniciación y fase de rampa |
Dosis diaria constante (Después de la fase de rampa)ª |
|
|
lnhibidor fuerte de CYP3A |
LLC |
Contraindicado |
Reduzca la dosis de venetoclax a 100 mg o menos. |
|
LMA |
Día 1-10 mg Día 2-20 mg Día 3-50 mg Día 4-100 mg o menos |
||
|
lnhibidor moderado de CYP3A |
Reduzca la dosis de venetoclax en al menos un 50% |
||
a En pacientes con LLC, evite el uso concomitante de venetoclax con inhibidor CYP3A fuerte o moderado. Considere medicamentos alternativos o reduzca la dosis de venetoclax.
MANIFESTACIONES Y MANEJO DE LA SOBREDOSIFICACIÓN O INGESTA ACCIDENTAL: Dosis diarias de hasta 1200 mg de VENCLEXTA® han sido evaluadas en estudios clínicos. No ha habido experiencia de sobredosis en estudios clínicos. Si se sospecha una sobredosis, el tratamiento debe consistir en medidas de soporte general.
PRESENTACIONES:
Para el tratamiento de leucemia linfocítica crónica el paquete de inicio de tratamiento contiene:
Semana 1
Caja con blíster de 14 tabletas de 10 mg.
Semana 2
Caja con blíster de 7 Tabletas de 50 mg.
Semana 3
Caja con blíster de 7 Tabletas de 100 mg.
Semana 4
Caja con blíster de 14 tabletas de 100 mg.
Presentaciones individuales para ajuste de la dosis:
• Caja con blíster de 10 mg con 14 tabletas.
• Caja con blíster de 50 mg con 7 tabletas.
Dosis de mantenimiento:
Caja con Frasco de 120 tabletas de 100 mg.
Para el tratamiento de leucemia mieloide aguda:
Caja con Frasco de 120 tabletas de 100 mg.
o
Caja con Frasco de 180 tabletas de 100 mg.
RECOMENDACIONES SOBRE ALMACENAMIENTO: Consérvese a no más de 30ºC.
LEYENDAS DE PROTECCIÓN:
Léase instructivo anexo. Su venta requiere receta médica. No se use durante el embarazo ni lactancia. No se deje al alcance de los niños. No aceptar si el sello está roto o alterado.
Reporte las sospechas de reacción adversa al correo: farmacovigilancia@cofepris.gob.mx
Titular del registro:
AbbVie lnc.
1 N Waukegan Rd., North Chicago
lllinois (IL) 60064, EUA
Representante legal:
ABBVIE FARMACÉUTICOS, S.A. de C.V.
Autopista México-Querétaro, Km 34.5, Nave-3, lnt. 4-B
Col. San Isidro, C.P. 54740, Cuautitlán lzcalli
México, México
Número de oficio de reconocimiento de medicamento huérfano: 163300EL870091
Fecha de actualización: Junio 29, 2018