VAXOBAN
DESVENLAFAXINA
Tabletas
1 Caja, 14 Tabletas, 50 mg
1 Caja, 28 Tabletas, 50 mg
1 Caja, 30 Tabletas, 50 mg
1 Caja, 14 Tabletas, 100 mg
1 Caja, 28 Tabletas, 100 mg
1 Caja, 30 Tabletas, 100 mg
FORMA FARMACÉUTICA Y FORMULACIÓN:
Cada TABLETA contiene:
Succinato de desvenlafaxina (succinato monohidrato de O-desmetil venlafaxina) equivalente a 50 mg, 100 mg de desvenlafaxina
Excipiente cbp 1 tableta
INDICACIONES TERAPÉUTICAS:
desvenlafaxina es un inhibidor de recaptación de norepinefrina y serotonina (SNRI), está indicado para el tratamiento del trastorno depresivo mayor (MDD). La eficacia de desvenlafaxina ha sido establecida en cuatro estudios de corta duración (8 semanas, controlados con placebo) y dos estudios de mantenimiento en pacientes ambulatorios adultos que cumplieron los criterios DSM-IV para trastorno depresivo mayor.
FARMACOCINÉTICA Y FARMACODINAMIA:
Farmacocinética:
La farmacocinética de dosis única de desvenlafaxina es lineal y proporcional a la dosis en un rango de dosis de 50 a 600 mg/día. Con la administración una vez al día, las concentraciones en plasma en estado estable se alcanzan dentro de aproximadamente 4 a 5 días. En estado estable, la acumulación de dosis múltiples de desvenlafaxina es lineal y predecible a partir del perfil farmacocinético de la dosis única.
Absorción y distribución: La biodisponibilidad oral absoluta de desvenlafaxina después de la administración oral es alrededor de 80%.
Un estudio del efecto de los alimentos que incluyó la administración de desvenlafaxina a participantes sanos en ayunas y después de los alimentos (comida de alto contenido graso, entre 800 y 1,000 calorías) indicó que la Cmáx de desvenlafaxina aumentó alrededor de 16% con alimento, mientras que los ABC fueron similares. No se espera que esta diferencia sea clínicamente significativa; por lo tanto, desvenlafaxina puede tomarse independientemente de las comidas.
La unión a proteínas plasmáticas de desvenlafaxina es baja (30%) y es independiente de la concentración del fármaco. El volumen de distribución de desvenlafaxina en estado estable después de la administración intravenosa es 3.4 L/kg, indicando una distribución en los compartimientos no vasculares.
Metabolismo y eliminación: Desvenlafaxina es metabolizada principalmente mediante la conjugación (mediada por las isoformas UGT) y en menor medida, a través del metabolismo oxidativo CYP3A4 es la isoenzima de citocromo P450 que media el metabolismo oxidativo (N-desmetilación) de desvenlafaxina. La vía metabólica de CYP2D6 no está involucrada y después de la administración de 100 mg, la farmacocinética de desvenlafaxina fue similar en los participantes con fenotipo metabolizador lento y eficiente de CYP2D6. Aproximadamente 45% de desvenlafaxina se excreta inalterada en la orina a las 72 horas después de la administración oral. Aproximadamente 19% de la dosis administrada se excreta como el metabolito glucurónido y < 5% como el metabolito oxidativo (N, O-didesmetilvenlafaxina) en la orina.
Farmacocinética en poblaciones especiales:
Estudios de interacción farmacológica:
Inhibidores de CYP3A4 (ketoconazol): CYP3A4 es una vía menor para el metabolismo de desvenlafaxina. En un estudio clínico, ketoconazol (200 mg dos veces al día) aumentó el área bajo la curva (AUC) de la concentración frente al tiempo de desvenlafaxina (única dosis de 400 mg) en aproximadamente un 43% y la Cmáx en aproximadamente un 8%. El uso concomitante de desvenlafaxina con inhibidores potentes de CYP3A4 podría provocar mayores concentraciones de desvenlafaxina.
Inhibidores de otras enzimas CYP: Teniendo en cuenta los datos obtenidos in vitro, no se espera que los fármacos que inhiben las isoenzimas de CYP 1A1, 1A2, 2A6, 2D6, 2C8, 2C9, 2C19 y 2E1 tengan un efecto significativo sobre el perfil farmacocinético de desvenlafaxina.
Fármacos metabolizados por CYP2D6 (p. ej. desipramina, dextrometorfano, metoprolol, atomoxetina): Estudios in vitro mostraron un mínimo efecto inhibidor de desvenlafaxina sobre CYP2D6. Estudios clínicos han demostrado que desvenlafaxina no ejerce un efecto clínicamente relevante sobre el metabolismo de CYP2D6 a la dosis de 100 mg por día. Cuando se administró desvenlafaxina succinato a una dosis de 100 mg por día junto a una única dosis de 50 mg de desipramina, un sustrato de CYP2D6, la Cmáx y el ABC de desipramina aumentaron en aproximadamente un 25% y 17%, respectivamente. Cuando se administraron 400 mg (8 veces la dosis recomendada de 50 mg), la Cmáx y el ABC de desipramina aumentaron en aproximadamente un 50% y 90%, respectivamente. El uso concomitante de desvenlafaxina con un fármaco metabolizado por CYP2D6 puede provocar mayores concentraciones de dicho fármaco (ver Interacciones farmacológicas: Potencial de desvenlafaxina para afectar otros fármacos).
Fármacos metabolizados por CYP3A4 (midazolam): De forma in vitro, desvenlafaxina no inhibe ni induce la isoenzima CYP3A4. En un estudio clínico, se coadministró desvenlafaxina a 400 mg por día (8 veces la dosis recomendada de 50 mg) con una única dosis de 4 mg de midazolam (un sustrato de CYP3A4). El ABC y la Cmáx de midazolam disminuyeron en aproximadamente un 31% y 16%, respectivamente. El uso concomitante de desvenlafaxina con un fármaco metabolizado por CYP3A4 puede provocar menores exposiciones a ese fármaco.
Fármacos metabolizados por CYP1A2, 2A6, 2C8, 2C9 y 2C19: De forma in vitro, desvenlafaxina no inhibe las isoenzimas CYP1A2, 2A6, 2C8, 2C9 y 2C19 y no se esperaría que afecte la farmacocinética de los fármacos metabolizados por estas isoenzimas de CYP.
In vitro, desvenlafaxina no es un sustrato ni un inhibidor del transportador de la glicoproteína P. Es poco probable que la farmacocinética de desvenlafaxina se vea afectada por fármacos que inhiben el transportador de la glicoproteína P y que desvenlafaxina afecte la farmacocinética de los fármacos que son sustratos del transportador de la glicoproteína P.
Poblaciones especiales:
Edad: En un estudio de participantes sanos que recibieron dosis de hasta 300 mg, hubo un incremento de aproximadamente 32% en la Cmáx y un incremento de 55% en el ABC en participantes mayores de 75 años de edad (n = 17), en comparación con participantes de 18 a 45 años de edad (n = 16). Los participantes de 65 a 75 años de edad (n = 15) no tuvieron un cambio en la Cmáx, sino un incremento aproximado de 32% en el ABC, en comparación con los participantes de 18 a 45 años de edad.
Sexo: En un estudio de participantes sanos que recibieron hasta 300 mg, las mujeres tuvieron una Cmáx aproximadamente 25% mayor y una ABC aproximadamente 10% mayor que los hombres de las mismas edades. No se necesita un ajuste de la dosis con base en el sexo.
Raza: El análisis farmacocinético mostró que la raza (Blanca, n=466, Negra, n=97; Hispánica, n=39; otra n=33) no tuvo un efecto aparente en la farmacocinética de desvenlafaxina. No se necesita un ajuste de la dosis en base a la raza.
Insuficiencia hepática: La disposición de desvenlafaxina succinato después de la administración de 100 mg se estudió en participantes con insuficiencia hepática leve (Child-Pugh A n=8), moderada (Child-Pugh B, n=8) y severa (Child Pugh C, n=8) y a participantes sanos (n=12).
El ABC promedio aumentó en aproximadamente 31% y 35% en pacientes con insuficiencia hepática moderada y severa, respectivamente en comparación con participantes sanos. Los valores de ABC promedio en suero en ayunas fueron similares en participantes con insuficiencia hepática leve y participantes sanos (< 5% diferencia).
La depuración sistémica (CL/F) disminuyó aproximadamente 20% y 36% en pacientes con insuficiencia hepática moderada y severa, respectivamente en comparación con participantes sanos. Los valores de CL/F fueron similares en participantes con insuficiencia hepática leve y participantes sanos (< 5% diferencia).
La t1/2 promedio cambió de aproximadamente 10 horas en participantes sanos y participantes con insuficiencia hepática leve a 13 y 14 horas en insuficiencia hepática moderada y severa, respectivamente. La dosis recomendada en pacientes con insuficiencia hepática es 50 mg/día. El aumento de dosis por encima de 100 mg/día no es recomendado.
Insuficiencia renal: La disposición de desvenlafaxina después de la administración de 100 mg fue estudiada en participantes con nefropatía leve (n=9), moderada (n=8), severa (n=7) y en etapa terminal (ESRD) (n=9) que requería de diálisis y en participantes de control, de edad correspondiente sanos (n=8). La eliminación se correlacionó significativamente con la depuración de creatinina. Se observaron incrementos en ABC de alrededor de 42% en la insuficiencia renal leve (CrCl 24- hr = 50 a 80 mL/min, Cockcroft-Gault [C-G]), alrededor de 56% en insuficiencia renal moderada (CrCl 24-h r =30 a 50 mL/min, C-G), alrededor de 108% en insuficiencia renal severa (CrCl 24-hr ≤ 30 mL/min, C-G), y alrededor de 116% en participantes ESRD, en comparación con participantes de control de edad correspondiente, sanos.
La vida media terminal promedio (t1/2) se prolongó de 11.1 horas en los participantes de control a aproximadamente 13,5, 15,5, 17,6 y 22,8 horas en participantes con insuficiencia renal leve, moderada, severa y ESRD, respectivamente. Menos de 5% del fármaco en el cuerpo fue depurado durante un procedimiento estándar de hemodiálisis de 4 horas.
La dosis máxima recomendada en pacientes con insuficiencia renal moderada es 50 mg al día. Se recomienda el ajuste de dosis de 50 mg interdiarios en pacientes con insuficiencia renal severa o ESRD (ver Dosis y vía de administración: Poblaciones especiales y Uso en poblaciones específicas: Otros factores del paciente).
Farmacodinamia
Mecanismo de acción: Se desconoce el mecanismo exacto de la acción antidepresiva de desvenlafaxina, pero se cree que está relacionado con la potenciación de la serotonina y la norepinefrina en el sistema nervioso central a través de la inhibición de su recaptación. Estudios preclínicos han demostrado que desvenlafaxina es un inhibidor potente y selectivo de la recaptación de serotonina y norepinefrina (IRSN).
Desvenlafaxina carece de afinidad significativa por numerosos receptores, incluyendo muscarínico-colinérgico, H1-histaminérgico o receptores α1-adrenérgicos in vitro. desvenlafaxina también carece de actividad inhibidora de la monoamino oxidasa (MAO).
Cambios en los ECG: Se realizaron electrocardiogramas en los 1,492 pacientes con trastorno depresivo mayor tratados con desvenlafaxina y en los 984 pacientes tratados con placebo en estudios clínicos de hasta 8 semanas de duración. No se observaron diferencias clínicamente relevantes entre los pacientes tratados con desvenlafaxina y los tratados con placebo con respecto a los intervalos QT, QTc, PR y QRS. En un estudio integral sobre el intervalo QTc con criterios determinados de forma prospectiva, desvenlafaxina no provocó la prolongación del intervalo QT. No se observaron diferencias entre los tratamientos con placebo y desvenlafaxina para el intervalo QRS.
CONTRAINDICACIONES:
Desvenlafaxina está contraindicada en:
• Hipersensibilidad a desvenlafaxina succinato, a clorhidrato de venlafaxina o a cualquiera de los excipientes en la formulación de desvenlafaxina. Se ha informado la presencia de angioedema en pacientes tratados con desvenlafaxina (ver Reacciones adversas: Experiencia en estudios clínicos).
• El embarazo, porque se relacionan complicaciones en los neonatos cuando se utiliza desvenlafaxina en el tercer trimestre y durante la lactancia debido a que se excreta en la leche materna. (Ver Restricciones de uso durante el embarazo y la lactancia).
El uso de MAOI destinados a tratar trastornos psiquiátricos con desvenlafaxina o dentro de los 7 días de detener el tratamiento con desvenlafaxina está contraindicado debido un mayor riesgo de sufrir el síndrome de serotonina. El uso de desvenlafaxina dentro de los 14 días de detener un MAOI previsto para tratar trastornos psiquiátricos también está contraindicado (ver Dosis y vía de administración: Pacientes que cambian a o de un Inhibidor de la Monoamino Oxidasa (MAOI) previsto para tratar trastornos psiquiátricos y Advertencias y precauciones: Síndrome de serotonina).
Comenzar la terapia con desvenlafaxina en un paciente que está siendo tratado con MAOI tales como linezolid o azul de metileno por vía intravenosa también está contraindicado debido al mayor riesgo de sufrir el síndrome de serotonina (ver Dosis y vía de administración: Pacientes que cambian a o de un Inhibidor de la Monoamino Oxidasa (MAOI) previsto para tratar trastornos psiquiátricos y Advertencias y precauciones: Síndrome de serotonina).
RESTRICCIONES DE USO DURANTE EL EMBARAZO Y LA LACTANCIA:
Embarazo
Embarazo categoría C
Resumen de los riesgos: No existen estudios adecuados y bien controlados de desvenlafaxina en mujeres embarazadas. En los estudios sobre desarrollo reproductivo que se realizaron en ratas y conejos con desvenlafaxina succinato, no se observó evidencia de teratogenicidad a dosis de hasta 30 veces la dosis humana de 100 mg/día (medida de forma mg/m2) en las ratas y de hasta 15 veces la dosis humana de 100 mg/día (medida de forma mg/m2) en conejos. Se observó un aumento en la mortalidad de las ratas bebé durante los primeros 4 días de lactancia al administrar la dosis durante la gestación y la lactancia, a dosis mayores a 10 veces la dosis humana de 100 mg/día (medidas de forma mg/m2). Debe utilizar desvenlafaxina durante el embarazo sólo si los posibles beneficios justifican los posibles riesgos para el feto. Consideraciones clínicas: Un estudio longitudinal prospectivo con 201 mujeres con antecedentes de depresión mayor y un cuadro de eutimia al comienzo del embarazo demostró que las mujeres que descontinuaron la toma de antidepresivos durante el embarazo eran más propensas a tener una recaída de la depresión mayor que aquellas que continuaron tomando los antidepresivos.
Datos en seres humanos: Los neonatos expuestos a SNRI (Inhibidores de la recaptación de serotonina y norepinefrina) o SSRI (Inhibidores selectivos de la recaptación de serotonina), al final del tercer trimestre han desarrollado complicaciones que requieren de hospitalización prolongada, soporte respiratorio y alimentación por sonda. Dichas complicaciones pueden surgir inmediatamente después del parto. Los hallazgos clínicos informados han incluido alteración respiratoria, cianosis, apnea, convulsiones, inestabilidad de la temperatura, dificultad para alimentarse, vómitos, hipoglucemia, hipotonía, hipertonía, hiperreflexia, temblores, incomodidad nerviosa, irritabilidad y constante llanto. Estas características son consistentes con el efecto tóxico directo de SSRI y SNRI o posiblemente, un síndrome de descontinuación del fármaco. Se debe indicar que, en algunos casos, el cuadro clínico es consistente con el síndrome de serotonina (ver Advertencias y precauciones: Síndrome de serotonina).
Datos en animales: Al administrar desvenlafaxina de forma oral a ratas y conejas preñadas durante el periodo de organogénesis a dosis máximas de 300 mg/kg/día y 75 mg/kg/día, respectivamente, no se observaron efectos teratogénicos. Estas dosis representan 30 veces la dosis de los seres humanos de 100 mg (medida de forma mg/m2) en las ratas y 15 veces la dosis de los seres humanos de 100 mg/día (medida de forma mg/m2) en las conejas. Sin embargo, disminuyeron los pesos fetales y se retrasó la osificación esquelética en las ratas, lo cual se asoció con toxicidad materna a la dosis más elevada, y no se observaron efectos a una dosis de 10 veces la dosis humana de 100 mg/día (medida de forma mg/m2). Al administrar desvenlafaxina succinato de forma oral a ratas preñadas durante toda la gestación y la lactancia, hubo una disminución en el peso de las ratas bebés y un aumento en la mortalidad de éstas durante los primeros cuatro días de lactancia con la dosis más elevada de 300 mg/kg/día. Se desconoce la causa de estas muertes. La dosis a la cual no se observaron muertes en las ratas bebés era de 10 veces la dosis humana de 100 mg/día (medida de forma mg/m2). El crecimiento y desarrollo reproductivo de las crías después del destete no se vieron afectados por el tratamiento de la madre con desvenlafaxina succinato a una dosis de 30 veces la dosis humana de 100 mg/día (medida de forma mg/m2).
Lactancia: desvenlafaxina (O-desmetilvenlafaxina) se excreta en la leche humana. Debido al potencial de reacciones adversas serias en infantes lactantes de desvenlafaxina, se debe tomar una decisión de si descontinuar o no la lactancia o descontinuar el fármaco, tomando en cuenta la importancia del fármaco para la madre.
REACCIONES SECUNDARIAS Y ADVERSAS:
Las siguientes reacciones adversas se discuten en mayor detalle en otras secciones:
• Hipersensibilidad (ver Contraindicaciones).
• Pensamientos y conductas suicidas en adolescentes y adultos jóvenes (ver Advertencias y precauciones: Pensamientos y conductas suicidas en niños, adolescentes y adultos jóvenes).
• Síndrome de serotonina (ver Advertencias y precauciones: Síndrome de serotonina).
• Presión arterial elevada (ver Advertencias y precauciones: Presión arterial elevada).
• Hemorragia anormal (ver Advertencias y precauciones: Sangrado anormal).
• Glaucoma de ángulo estrecho (ver Advertencias y precauciones: Glaucoma de ángulo estrecho).
• Activación de manía/hipomanía (ver Advertencias y precauciones: Activación de la manía e hipomanía).
• Síndrome de descontinuación (ver Advertencias y precauciones: Síndrome de descontinuación).
• Convulsión (ver Advertencias y precauciones: Convulsión).
• Hiponatremia (ver Advertencias y precauciones: Hiponatremia).
• Enfermedad pulmonar intersticial y neumonía eosinofílica (ver Advertencias y precauciones: Enfermedad pulmonar intersticial y neumonía eosinofílica).
Experiencia en estudios clínicos: Debido a que los ensayos clínicos se realizan bajo condiciones muy diferentes, las tasas de reacciones adversas observadas en los ensayos clínicos de un fármaco no pueden compararse de forma directa con las tasas de los estudios clínicos de otro fármaco y podrían no reflejar las tasas observadas en la práctica clínica.
Exposición del paciente: Desvenlafaxina se evaluó respecto a seguridad en 8,394 pacientes diagnosticados con trastorno depresivo mayor que participaron en estudios previos a la comercialización de dosis múltiple, representando 2,784 años-paciente de exposición. De los 8,394 pacientes totales expuestos a por lo menos una dosis de desvenlafaxina, 2,116 fueron expuestos a desvenlafaxina por 6 meses, representando 1,658 años-paciente de exposición y 421 fueron expuestos por un año, representando 416 añospaciente de exposición. Reacciones adversas informadas como motivos de descontinuación del tratamiento: En el conjunto de estudios de 8 semanas controlados con placebo, previos a la comercialización, en pacientes con MDD, 1,834 pacientes recibieron desvenlafaxina (50 a 400 mg). De los 1,834 pacientes, el 12% descontinuó el tratamiento debido a una reacción adversa, en comparación con el 3% de los 1,116 pacientes tratados con placebo. A la dosis recomendada de 50 mg, la tasa de descontinuación debido a una reacción adversa de desvenlafaxina (4,1%) fue similar a la tasa de descontinuación del placebo (3,8%). Para la dosis de 100 mg de desvenlafaxina, la tasa de descontinuación debido a una reacción adversa fue de 8,7%.
Las reacciones adversas más frecuentes que provocaron la descontinuación en al menos un 2% y a una tasa superior a la del placebo en los pacientes tratados con desvenlafaxina en los estudios a corto plazo, de un máximo de 8 semanas, fueron: náuseas (4%); mareos y vómitos (2% cada uno). En un estudio a largo plazo, de un máximo de 9 meses, la reacción adversa más frecuente fue el vómito (2%).
Reacciones adversas comunes en los estudios MDD controlados con placebo: Las reacciones adversas más frecuentes que se observaron en los pacientes con MDD tratados con desvenlafaxina en el conjunto de estudios de 8 semanas, con dosis fija y controlados con placebo, previos a la comercialización (incidencia ≥ 5% y al menos dos veces la tasa del placebo en los grupos de dosis de 50 o 100 mg) fueron: náuseas, mareos, insomnio, hiperhidrosis, estreñimiento, somnolencia, disminución del apetito, ansiedad y trastornos específicos de la función sexual masculina.
La Tabla 2 muestra la incidencia de reacciones adversas comunes que se produjeron en ≥ 2% de pacientes MDD tratados con desvenlafaxina y dos veces la tasa del placebo a cualquier dosis en el conjunto de estudios de 8 semanas, con dosis fija y controlados con placebo, previos a la comercialización.
Tabla 2. Reacciones adversas comunes (≥ 2% en cualquier grupo de dosis fija y dos veces la tasa del placebo) en el conjunto de estudios previos a la comercialización controlados con placebo de 8 semanas de MDD
|
Porcentaje de pacientes que informan una reacción |
|||||
|---|---|---|---|---|---|
|
Sistema órgano clase |
desvenlafaxina |
||||
|
Término preferido |
Placebo (n=636) |
50 mg (n=317) |
100 mg (n=424) |
200 mg (n=307) |
400 mg (n=317) |
|
Trastornos cardiacos |
|||||
|
Presión arterial incrementada |
1 |
1 |
1 |
2 |
2 |
|
Trastornos gastrointestinales |
|||||
|
Náusea |
10 |
22 |
26 |
36 |
41 |
|
Boca seca |
9 |
11 |
17 |
21 |
25 |
|
Estreñimiento |
4 |
9 |
9 |
10 |
14 |
|
Vómitos |
3 |
3 |
4 |
6 |
9 |
|
Trastornos generales y condiciones del sitio de administración |
|||||
|
Fatiga |
4 |
7 |
7 |
10 |
11 |
|
Escalofríos |
1 |
1 |
< 1 |
3 |
4 |
|
Sensación de nerviosismo |
1 |
1 |
2 |
3 |
3 |
|
Trastornos del metabolismo y la nutrición |
|||||
|
Disminución del apetito |
2 |
5 |
8 |
10 |
10 |
|
Trastornos del sistema nervioso |
|||||
|
Mareos |
5 |
13 |
10 |
15 |
16 |
|
Somnolencia |
4 |
4 |
9 |
12 |
12 |
|
Temblores |
2 |
2 |
3 |
9 |
9 |
|
Alteración de la atención |
< 1 |
< 1 |
1 |
2 |
1 |
|
Trastornos psiquiátricos |
|||||
|
Insomnio |
6 |
9 |
12 |
14 |
15 |
|
Ansiedad |
2 |
3 |
5 |
4 |
4 |
|
Nerviosismo |
1 |
< 1 |
1 |
2 |
2 |
|
Sueños anormales |
1 |
2 |
3 |
2 |
4 |
|
Trastornos renal y urinario |
|||||
|
Dificultad urinaria |
0 |
< 1 |
1 |
2 |
2 |
|
Trastornos respiratorio, torácico y del mediastino |
|||||
|
Bostezos |
< 1 |
1 |
1 |
4 |
3 |
|
Trastornos de la piel y del tejido subcutáneo |
|||||
|
Hiperhidrosis |
4 |
10 |
11 |
18 |
21 |
|
Sentidos especiales |
|||||
|
Visión borrosa |
1 |
3 |
4 |
4 |
4 |
|
Midriasis |
< 1 |
2 |
2 |
6 |
6 |
|
Vértigo |
1 |
2 |
1 |
5 |
3 |
|
Tinitus |
1 |
2 |
1 |
1 |
2 |
|
Disgeusia |
1 |
1 |
1 |
1 |
2 |
|
Trastornos vasculares |
|||||
|
Sofocos |
< 1 |
1 |
1 |
2 |
2 |
Reacciones adversas de la función sexual: La tabla 3 muestra la incidencia de reacciones adversas de la función sexual que se produjeron en ≥2% de los pacientes con MDD tratados con desvenlafaxina en cualquier grupo de dosis fija (conjunto de estudios clínicos de 8 semanas controlados con placebo, de dosis fija, previos a la comercialización).
Tabla 3. Reacciones adversas de la función sexual (≥ 2% en hombres o mujeres en cualquier grupo desvenlafaxina) durante el periodo en terapia
|
Desvenlafaxina |
|||||
|
Placebo (n=239) |
50 mg (n=108) |
100 mg (n=157) |
200 mg (n=131) |
400 mg (n=154) |
|
|
Sólo hombres |
|||||
|
Anorgasmia |
0 |
0 |
3 |
5 |
8 |
|
Disminución de la libido |
1 |
4 |
5 |
6 |
3 |
|
Orgasmo anormal |
0 |
0 |
1 |
2 |
3 |
|
Retraso de la eyaculación |
< 1 |
1 |
5 |
7 |
6 |
|
Disfunción eréctil |
1 |
3 |
6 |
8 |
11 |
|
Trastorno de eyaculación |
0 |
0 |
1 |
2 |
5 |
|
Falla en la eyaculación |
0 |
1 |
0 |
2 |
2 |
|
Disfunción sexual |
0 |
1 |
0 |
0 |
2 |
|
Placebo (n=397) |
50 mg (n=209) |
100 mg (n=267) |
200 mg (n=176) |
400 mg (n=163) |
|
|
Sólo mujeres |
|||||
|
Anorgasmia |
0 |
1 |
1 |
0 |
3 |
Otras reacciones adversas observadas en los estudios clínicos previos y posteriores a la comercialización: Otras reacciones adversas poco frecuentes, no descritas en la etiqueta, que se producen a una incidencia de < 2% en los pacientes MDD tratados con desvenlafaxina fueron:
Trastornos cardiacos: Taquicardia.
Trastornos generales y condiciones del sitio de administración: Astenia.
Investigaciones: Aumento de peso, prueba de función hepática anormal, aumento de la prolactina en sangre. Trastornos musculoesqueléticos y del tejido conectivo: Entumecimiento musculoesquelético.
Trastornos del sistema nervioso: Síncope, convulsión, distonia.
Trastornos psiquiátricos: Despersonalización, bruxismo.
Trastornos renales y urinarios: Retención urinaria.
Trastornos de la piel y tejido subcutáneo: Sarpullido, alopecia, reacción de fotosensibilidad, angioedema.
En estudios clínicos, hubo informes poco comunes de reacciones adversas cardiacas isquémicas incluyendo isquemia del miocardio, infarto de miocardio y oclusión coronaria que requiere de revascularización; estos pacientes tuvieron factores de riesgo cardiaco subyacentes múltiples.
Más pacientes experimentaron estos eventos durante el tratamiento con desvenlafaxina en comparación con el placebo.
Cambios en el laboratorio, ECG y signos vitales observados en los estudios clínicos de MDD: Los siguientes cambios se observaron en estudios previos a la comercialización, de corto plazo, controlados con placebo de MDD con desvenlafaxina.
Lípidos: Las elevaciones en el colesterol total, colesterol LDL (lipoproteínas de baja densidad) y triglicéridos en suero, en ayunas se produjeron en los estudios controlados. Algunas de estas anormalidades se consideraron potencialmente significativas.
El porcentaje de pacientes que superaron el valor de umbral predeterminado se muestra en la Tabla 5.
Tabla 4. Incidencia (%) de pacientes con anormalidades de lípidos de significancia clínica potencial*
|
Desvenlafaxina |
|||||
|
Placebo |
50 mg |
100 mg |
200 mg |
400 mg |
|
|
Colesterol total *(Aumento de ≥ 50 mg/dL y valor absoluto de ≥ 261 mg/dL) |
2 |
3 |
4 |
4 |
10 |
|
Colesterol LDL *(Aumento de ≥ 50 mg/dL y valor absoluto de ≥ 190 mg/dL) |
0 |
1 |
0 |
1 |
2 |
|
Triglicéridos en ayunas *(En ayunas: ≥ 327 mg/dL) |
3 |
2 |
1 |
4 |
6 |
Proteinuria: Se observó proteinuria, mayor que o igual a trazas, en los estudios controlados de dosis fija previos a la comercialización (ver la Tabla 5). Esta proteinuria no estuvo asociada con incrementos en BUN o creatinina y fue generalmente temporal.
Tabla 5. Incidencia (%) de pacientes con proteinuria en los estudios clínicos de dosis fija
|
Desvenlafaxina |
|||||
|
Placebo |
50 mg |
100 mg |
200 mg |
400 mg |
|
|
Proteinuria |
4 |
6 |
8 |
5 |
7 |
Cambios en los signos vitales: La Tabla 6 resume los cambios que se observaron en los estudios previos a la comercialización, de corto plazo, controlados con placebo con desvenlafaxina en pacientes con MDD (dosis de 50 a 400 mg).
Tabla 6. Cambios medios en signos vitales al final en la terapia para todos los estudios de corto plazo, controlados, de dosis fija
|
Desvenlafaxina |
|||||
|
Placebo |
50 mg |
100 mg |
200 mg |
400 mg |
|
|
Presión arterial |
|||||
|
Sistólica supina bp (mm Hg) |
-1,4 |
1,2 |
2,0 |
2,5 |
2,1 |
|
Diastólica supina bp (mm Hg) |
-0,6 |
0,7 |
0,8 |
1,8 |
2,3 |
|
Frecuencia de pulso |
|||||
|
Pulso en posición supina (bpm) |
-0,3 |
1,3 |
1,3 |
0,9 |
4,1 |
|
Peso (kg) |
0,0 |
-0,4 |
-0,6 |
-0,9 |
-1,1 |
El tratamiento con desvenlafaxina en todas las dosis desde 50 mg/día hasta 400 mg/día en estudios controlados se asoció con hipertensión sostenida, definida como presión arterial diastólica supina (PADS) emergente del tratamiento con valores ≥ 90 mm Hg y ≥ 10 mm Hg por encima del valor basal durante 3 visitas consecutivas durante la terapia (véase la Tabla 7). Los análisis de los pacientes en los estudios previos a la comercialización controlados de desvenlafaxina a corto plazo que cumplieron con los criterios de hipertensión sostenida revelaron un aumento consistente en la proporción de pacientes que desarrollaron hipertensión sostenida. Esto se observó en todas las dosis con una sugerencia de una tasa superior con la dosis de 400 mg/día.
Tabla 7. Proporción de pacientes con elevación sostenida de la presión arterial diastólica supina
|
Grupo de tratamiento |
Proporción de pacientes con hipertensión sostenida |
|
Placebo |
0,5% |
|
desvenlafaxina 50 mg/día |
1,3% |
|
desvenlafaxina 100 mg/día |
0,7% |
|
desvenlafaxina 200 mg/día |
1,1% |
|
desvenlafaxina 400 mg/día |
2,3% |
Hipotensión ortostática: En los estudios clínicos previos a la comercialización, controlados con placebo, de corto plazo con dosis de 50 a 400 mg, la hipotensión ortostática sistólica (disminución ≥ 30 mmHg de la posición supina a posición de pie) se produjo más frecuentemente en pacientes ≥ 65 años de edad que recibían desvenlafaxina (8%, 7/87) frente al placebo (2,5%, 1/40) en comparación con los pacientes < 65 años de edad que recibían desvenlafaxina (0,9%, 18/1937) frente al placebo (0,7%, 8/1218).
Experiencia posterior a la comercialización: Se ha identificado la siguiente reacción adversa durante el uso posterior a la aprobación de desvenlafaxina. Debido a que estas reacciones se informan voluntariamente de una población de tamaño incierto, no siempre es posible calcular confiablemente su frecuencia o establecer una relación de causa a la exposición del fármaco.
Trastornos de la piel y tejido subcutáneo: Síndrome de Stevens-Johnson.
INTERACCIONES MEDICAMENTOSAS Y DE OTRO GÉNERO:
Inhibidores de la monoamino oxidasa (MAOI): No utilice los IMAO destinado a tratar trastornos psiquiátricos con desvenlafaxina o dentro de 7 días tras la suspensión del tratamiento con desvenlafaxina. No usar desvenlafaxina dentro de los 14 días de la detención de un IMAO destinado a tratar trastornos psiquiátricos. Además, no empiece a desvenlafaxina en un paciente que está siendo tratado con linezolid o azul de metileno intravenoso (ver Dosis y vía de administración: Pacientes que cambian a o de un inhibidor de la monoaminooxidasa (MAOI) previsto para tratar trastornos psiquiátricos, Contraindicaciones y Advertencias y precauciones: Síndrome de serotonina).
Fármacos serotonérgicos: Basado en el mecanismo de acción de la desvenlafaxina y el potencial para el síndrome de la serotonina, se recomienda precaución cuando desvenlafaxina es co-administrado con otras medicinas que pueden afectar a los sistemas de neurotransmisores serotoninérgicos (ver Dosis y vía de administración: Pacientes que cambian a o de un inhibidor de la monoaminooxidasa (MAOI) previsto para tratar trastornos psiquiátricos, Contraindicaciones y Advertencias y precauciones: Síndrome de serotonina).
Fármacos que interfieren con la hemostasis (por ejemplo, AINES, ácido acetilsalicílico y warfarina): La liberación de serotonina por las plaquetas desempeña un papel importante en la hemostasia. Los estudios epidemiológicos de control de caso y diseño de cohorte han demostrado una asociación entre el uso de fármacos psicotrópicos que interfieren con la recaptación de serotonina y la ocurrencia de sangrado gastrointestinal superior. Estos estudios también han demostrado que el uso concurrente de un AINES o ácido acetilsalicílico puede potenciar este riesgo de sangrado. Se han informado efectos anticoagulantes alterados, incluyendo aumento del sangrado, cuando SSRI y SNRI se coadministran con warfarina. Los pacientes que reciben terapia de warfarina deben ser cuidadosamente monitoreados cuando desvenlafaxina se inicia o descontinúa (ver Advertencias y precauciones: Sangrado anormal).
Posibilidad de que otros medicamentos afecten la desvenlafaxina: Basado en los datos in vitro, no se requiere ajuste de dosis en desvenlafaxina cuando se utiliza de forma concomitante con inhibidores de CYP3A4 y CYP1A1, 1A2, 2A6, 2D6, 2C8, 2C9, 2C19, 2E1, y el transportador de la P-glicoproteína. Los estudios clínicos han demostrado clínicamente significativa interacción farmacocinética entre desvenlafaxina y los inhibidores potentes del CYP3A4 (Figura 1).
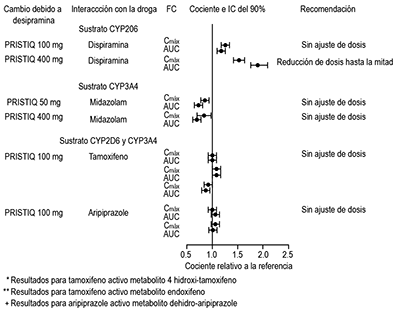
Figura 1. Impacto de otras drogas sobre la farmacocinética de la desvenlafaxina (FC)
 Potencial de desvenlafaxina para afectar otros fármacos:
Potencial de desvenlafaxina para afectar otros fármacos:
Fármacos metabolizados por CYP2D6 (desipramina): Los estudios clínicos han demostrado que desvenlafaxina no tiene un efecto clínicamente relevante en el metabolismo de CYP2D6 a la dosis de 100 mg (figura 2) al día. Los sustratos que se metabolizan principalmente mediante CYP2D6 (p. ej. desipramina, atomoxetina, dextrometorfano, metoprolol, nebivolol, perfenazina, tolterodina) deben administrarse a la dosis original al coadministrarlos con desvenlafaxina de 100 mg o menos o cuando desvenlafaxina es descontinuado. Disminuya hasta a la mitad la dosis de estos sustratos si los coadministra con 400 mg de desvenlafaxina.
No es necesario ajustar la dosis adicionalmente para el uso concomitante de sustratos de las isoezimas CYP3A4, 1A2, 2A6, 2C8, 2C9 y 2C19 y transportador de la P-glicoproteína. Los estudios clínicos han demostrado clínicamente significativa interacción farmacocinética entre sustratos desvenlafaxina y CYP3A4 (Figura 2).
Los estudios clínicos han demostrado que la desvenlafaxina (100 mg diarios) no tiene un efecto clínicamente relevante sobre el tamoxifeno y el aripiprazol, compuestos que son metabolizados por una combinación de ambas enzimas CYP2D6 y CYP3A4 (Figura 2).
Los estudios in vitro mostraron efecto inhibitorio mínima de desvenlafaxina en la isoenzima CYP2D6.
In vitro, la desvenlafaxina no inhibe ni induce la isoenzima CYP3A4.
In vitro, la desvenlafaxina no inhibe las isoenzimas CYP1A2, 2A6, 2C8, 2C9 y 2C19, y Pglicoproteína transportista y no se espera que afecte a la farmacocinética de los fármacos que son sustratos de estas isoenzimas CYP y transportador.
Figura 2. Impacto de la desvenlafaxina la farmacocinética (FC) de desipramina, midazolam, tamoxifeno y aripiprazole
Otros fármacos que contienen desvenlafaxina o venlafaxina: Evite el uso de desvenlafaxina con otros productos que contienen desvenlafaxina o venlafaxina. El uso concomitante de desvenlafaxina con otros productos que contienen desvenlafaxina o venlafaxina aumentará la concentración sanguínea de desvenlafaxina y las reacciones adversas relacionadas con la dosis (ver Reacciones adversas).
Etanol: Un estudio clínico ha demostrado que desvenlafaxina no aumenta el deterioro de las funciones mentales y motoras que provoca el etanol. Sin embargo, al igual que con todos los fármacos que actúan sobre el sistema nervioso central, debe indicarse a los pacientes que deben evitar el consumo de alcohol mientras tomen desvenlafaxina.
Drug-Test Laboratory Interacciones: Se han reportado falso-positivos en las pruebas de inmunoensayo en orina para la detección de fenciclidina (PCP) y las anfetaminas en pacientes que están tomando desvenlafaxina. Esto es debido a la falta de especificidad de las pruebas de detección. Los resultados de pruebas falsos-positivos se puede esperar durante varios días después de suspender el tratamiento con desvenlafaxina. Las pruebas de confirmación, como la cromatografía/espectrometría de masa de gas, distinguirán desvenlafaxina del PCP y las anfetaminas.
ALTERACIONES EN LOS RESULTADOS DE PRUEBAS DE LABORATORIO:
Se han reportado pruebas diagnósticas de inmunoensayos de orina falso-positivas para fenciclidina (PCP) y anfetamina en pacientes que toman desvenlafaxina. Esto se debe a la falta de especificidad de las pruebas diagnósticas. Los resultados falso-positivos de la prueba pueden esperarse durante varios días después de descontinuar la terapia con desvenlafaxina. Pruebas confirmatorias, como cromatografía de gas/espectrometría de masa, distinguirán a desvenlafaxina de PCP y anfetamina.
Puede ocurrir alteración en los tiempos de coagulación y en pruebas de función hepática. Elevación de colesterol y triglicéridos, elevación de prolactina, hiponatremia.
PRECAUCIONES GENERALES:
Pensamientos y conductas suicidas en niños, adolescentes y adultos jóvenes: Los pacientes con trastorno depresivo mayor (MDD), adultos y niños, pueden experimentar empeoramiento de su depresión y/o la emergencia de ideas suicidas y comportamiento suicida (tendencia al suicidio) o cambios inusuales en el comportamiento, ya sea que tomen o no medicaciones antidepresivas y este riesgo puede persistir hasta que se produzca una remisión significativa. El suicidio es un riesgo conocido de depresión y ciertos otros trastornos psiquiátricos, y estos trastornos en sí son los más fuertes pronosticadores de suicidio. Ha habido una preocupación de larga data, sin embargo, que los antidepresivos pueden desempeñar un papel en la inducción del empeoramiento de la depresión y la emergencia de tendencia suicida en ciertos pacientes durante las fases tempranas del tratamiento. Los análisis combinados de los estudios controlados con placebo de corto plazo de los fármacos antidepresivos (SSRI y otros) mostraron que estos fármacos aumentan el riesgo de pensamiento y comportamiento suicida (tendencia al suicidio) en niños, adolescentes y adultos jóvenes (de 18 a 24 años de edad) con trastorno depresivo mayor (MDD) y otros trastornos psiquiátricos. Los estudios de corto plazo no mostraron un incremento en el riesgo de tendencia al suicidio con antidepresivos en comparación con el placebo en adultos mayores de 24 años; hubo una reducción con los antidepresivos en comparación con el placebo en adultos de 65 años y mayores.
Los análisis combinados de los estudios controlados con placebo en niños y adolescentes con MDD, trastorno obsesivo compulsivo (OCD) u otros trastornos psiquiátricos incluyeron un total de 24 estudios de corto plazo de 9 fármacos antidepresivos en más de 4,400 pacientes. Los análisis combinados de los estudios controlados con placebo en adultos con MDD u otros trastornos psiquiátricos incluyeron un total de 295 estudios de corto plazo (duración mediana de 2 meses) de 11 fármacos antidepresivos en más de 77,000 pacientes. Hubo una variación considerable en el riesgo de tendencia al suicidio entre los fármacos, pero una tendencia hacia un incremento en los pacientes más jóvenes para casi todos los fármacos estudiados. Hubo diferencias en el riesgo absoluto de tendencia al suicidio entre las diferentes indicaciones, con la incidencia más alta en MDD. Las diferencias de riesgo (fármaco frente al placebo) sin embargo, fueron relativamente estables dentro del estrato de la edad y entre las indicaciones. Estas diferencias de riesgo (diferencia fármaco-placebo en el número de casos de tendencia al suicidio por 1,000 pacientes tratados) se brindan en la Tabla 1.
Tabla 1
|
Rango de edad |
Diferencia fármaco-placebo en el número de casos de suicidios por 1,000 pacientes tratados |
|
Aumentos en comparación con el placebo |
|
|
< 18 |
14 casos adicionales |
|
18 a 24 |
5 casos adicionales |
|
Disminución en comparación con el placebo |
|
|
25 a 64 |
1 caso menos |
|
≥ 65 |
6 casos menos |
No se produjeron suicidios en ningún estudio de niños. Se produjeron suicidios en estudios de adultos, pero la cantidad no fue suficiente para lograr ninguna conclusión sobre el efecto farmacológico en el suicidio.
Se desconoce si el riesgo de tendencia al suicidio se extienda al uso de largo plazo, es decir, después de varios meses. Sin embargo, existe evidencia sustancial a partir de los estudios de mantenimiento controlados con placebo en adultos con depresión que el uso de antidepresivos puede retrasar la recurrencia de depresión.
Todos los pacientes tratados con antidepresivos para cualquier indicación deben ser monitoreados apropiadamente y observados estrechamente respecto al empeoramiento clínico, tendencia al suicidio y cambios inusuales en el comportamiento, especialmente durante los pocos meses iniciales de un ciclo de terapia farmacológica o en los momentos de cambios de dosis, ya sea aumentos o disminuciones.
Se han informado los siguientes síntomas, ansiedad, agitación, ataques de pánico, insomnio, irritabilidad, hostilidad, agresividad, impulsividad, acatisia (inquietud psicomotriz), hipomanía y manía en pacientes adultos y pediátricos tratados con antidepresivos para el trastorno depresivo mayor así como para otras indicaciones, psiquiátricas y no psiquiátricas. Aunque no se ha establecido una relación de causa entre la emergencia de dichos síntomas y el empeoramiento de la depresión y/o emergencia de impulsos suicidas, existe una preocupación de que dichos síntomas puedan ser precursores del surgimiento de tendencias suicidas.
Se debe considerar cambiar el régimen terapéutico, incluyendo posiblemente descontinuar la medicación, en pacientes cuya depresión empeora persistentemente, o que experimentan tendencia al suicidio emergente o síntomas que podrían ser precursores de empeoramiento de la depresión o tendencia al suicidio, especialmente si estos síntomas son severos, de inicio abrupto o no fueron parte de los síntomas que presentaba el paciente. Si se ha tomado la decisión de descontinuar el tratamiento, la medicación debe ser reducida gradualmente, lo más rápido que sea posible, pero con el conocimiento de que la descontinuación abrupta puede estar asociada con ciertos síntomas (ver Dosis y vía de administración: Descontinuación de desvenlafaxina y Advertencias y precauciones: Síndrome de descontinuación para una descripción de los riesgos de la descontinuación de desvenlafaxina).
Las familias y personas encargadas del cuidado de los pacientes que están siendo tratados con antidepresivos para el trastorno depresivo mayor u otras indicaciones, psiquiátricas y no psiquiátricas, deben ser alertadas sobre la necesidad de monitorear a los pacientes respecto al surgimiento de agitación, irritabilidad, cambios inusuales en el comportamiento y otros síntomas descritos anteriormente, así como la emergencia de tendencia al suicidio e informar acerca de dichos síntomas inmediatamente a los proveedores de atención de la salud. Dicho monitoreo debe incluir la observación diaria por las familias y los encargados de su cuidado. Las prescripciones para desvenlafaxina deben escribirse por la cantidad más pequeña de tabletas consistente con un bueno manejo de los pacientes, con el fin de reducir el riesgo de sobredosis.
Selección de pacientes para trastorno bipolar: Un episodio depresivo mayor puede ser la presentación inicial del trastorno bipolar. Generalmente se cree (aunque no se ha establecido en estudios controlados) que tratar dicho episodio con un antidepresivo, sólo puede aumentar la posibilidad de precipitar un episodio mixto/maniaco en pacientes en riesgo de trastorno bipolar. Se desconoce si alguno de los síntomas descritos anteriormente representa una conversión. Sin embargo, antes de iniciar el tratamiento con un antidepresivo, los pacientes con síntomas depresivos deben ser seleccionados adecuadamente para determinar si están en riesgo de trastorno bipolar, dicha selección debe incluir una historia psiquiátrica detallada, incluyendo historia familiar de suicidio, trastorno bipolar y depresión. Se debe indicar que desvenlafaxina no está aprobado para el uso en el tratamiento de la depresión bipolar.
Síndrome de serotonina: Se ha informado el desarrollo de síndrome de serotonina que potencialmente amenaza la vida con SNRI o SSRI, incluido desvenlafaxina, pero particularmente con el uso concomitante de otros fármacos serotonérgicos (incluyendo triptanos, antidepresivos tricíclicos, fentanilo, litio, tramadol, triptofano, buspirona y hierba de San Juan) y con fármacos con un metabolismo alterado de serotonina (en particular, MAOI, tanto aquellos destinados a tratar trastornos psiquiátricos como también otros, tales como linezolid y azul de metileno por vía intravenosa). Los síntomas del síndrome de serotonina pueden incluir cambios en el estado mental (por ejemplo, agitación, alucinaciones, delirio y coma), inestabilidad autonómica (por ejemplo, taquicardia, presión sanguínea lábil, mareo, diaforesis, enrojecimiento, hipertermia), síntomas neuromusculares (por ejemplo, temblor, rigidez, mioclonia, hiperreflexia, falta de coordinación), ataques convulsivos y/o síntomas gastrointestinales (por ejemplo, náuseas, vómitos, diarrea). Los pacientes deben ser monitoreados por el surgimiento del síndrome de serotonina.
El uso concomitante de desvenlafaxina con MAOI previsto para tratar trastornos psiquiátricos está contraindicado. desvenlafaxina tampoco debe iniciarse en un paciente que está siendo tratado con MAOI tales como linezolid o azul de metileno por vía intravenosa. Todas las notificaciones con azul de metileno que proporcionaron información sobre la vía de administración involucraban la administración intravenosa en el intervalo de dosificación de 1 mg/kg a 8 mg/kg. Ninguna notificación involucró la administración de azul de metileno por otras vías (tales como tabletas orales o inyección local en tejidos) o a dosis menores. Pueden existir circunstancias donde resulta necesario iniciar el tratamiento con un MAOI tal como linezolid o azul de metileno por vía intravenosa en un paciente que toma desvenlafaxina. desvenlafaxina debe descontinuarse antes de iniciar el tratamiento con el MAOI (ver Contraindicaciones y Dosis y vía de administración: Pacientes que cambian a o de un inhibidor de la monoaminooxidasa (MAOI) previsto para tratar trastornos psiquiátricos).
Si el uso concomitante de desvenlafaxina con otros fármacos serotonérgicos, entre ellos, triptanos, antidepresivos tricíclicos, fentanilo, litio, tramadol, buspirona, triptófano y hierba de San Juan es clínicamente necesario, los pacientes deben conocer el mayor riesgo potencial del síndrome de serotonina, particularmente durante el inicio del tratamiento y en los incrementos de la dosis.
El tratamiento con desvenlafaxina y cualquier agente serotonérgico concomitante debe ser descontinuado inmediatamente si se producen los eventos anteriormente mencionados y se debe iniciar el tratamiento sintomático de respaldo.
Presión arterial elevada: Los pacientes que reciben desvenlafaxina deben tener un monitoreo regular de la presión arterial, ya que se observaron incrementos en la presión arterial en los estudios clínicos (ver Reacciones adversas: Experiencia en estudios clínicos). La hipertensión preexistente debe controlarse antes de iniciar el tratamiento con desvenlafaxina. Se debe tener cuidado en tratar a los pacientes con hipertensión preexistente u otra afección subyacente que podría ser comprometida por incrementos en la presión arterial. Casos de presión arterial elevada que requieren de tratamiento inmediato se han informado con desvenlafaxina.
Los incrementos sostenidos de presión arterial podrían tener consecuencias adversas. Para los pacientes que experimentan un incremento sostenido en la presión arterial, mientras reciben desvenlafaxina se debe considerar la reducción o descontinuación de la dosis (ver Reacciones adversas: Experiencia en estudios clínicos). Sangrado anormal: SSRI y SRI incluyendo desvenlafaxina, pueden aumentar el riesgo de eventos de sangrado. El uso concomitante de aspirina, fármacos antiinflamatorios no esteroideos, warfarina y otros anticoagulantes puede incrementar este riesgo. Los informes de caso y estudios epidemiológicos (diseño de cohorte y control de caso) han demostrado una asociación entre el uso de fármacos que interfieren con la recaptación de serotonina y la ocurrencia de sangrado gastrointestinal. Los eventos de sangrado relacionados con SSRI y SNRI han oscilado de equimosis, hematoma, epistaxis y petequia hasta hemorragias que amenazan la vida. Los pacientes deben ser advertidos sobre el riesgo de sangrado asociado con el uso concomitante de desvenlafaxina y AINES, aspirina u otros fármacos que afectan la coagulación o sangrado. Glaucoma de ángulo estrecho: Se ha informado midriasis en asociación con desvenlafaxina, por ende los pacientes con elevación de la presión intraocular o aquellos en riesgo de glaucoma de ángulo estrecho agudo (glaucoma de ángulo cerrado) deben ser monitoreados.
Activación de la manía e hipomanía: Durante todos los estudios de fase 2 y fase 3 de MDD, se informó manía para aproximadamente 0,02% de los pacientes tratados con desvenlafaxina. La activación de la manía/hipomanía ha sido informada en una pequeña proporción de pacientes con trastorno afectivo mayor que fueron tratados con otros antidepresivos comercializados. Así como todos los antidepresivos, desvenlafaxina debe utilizarse con precaución en los pacientes con una historia o historia familiar de manía o hipomanía.
Síndrome de descontinuación: Los síntomas de descontinuación han sido evaluados sistemática y prospectivamente en los pacientes tratados con desvenlafaxina durante los estudios clínicos en Trastorno Depresivo Mayor. La descontinuación o reducción abrupta de la dosis ha estado asociada con la aparición de síntomas nuevos que incluyen mareos, náuseas, cefalea, irritabilidad, insomnio, diarrea, ansiedad, fatiga, sueños anormales e hiperhidrosis. En general, se produjeron eventos de descontinuación más frecuentemente con duración más prolongada de la terapia.
Durante la comercialización de SNRI (Inhibidores de la recaptación de serotonina y norepinefrina) y SSRI (Inhibidores selectivos de recaptación de serotonina), ha habido informes espontáneos de eventos adversos que se produjeron tras la descontinuación de estos fármacos, en particular cuando fue abrupta, incluyendo los siguientes: estado de ánimo disfórico, irritabilidad, agitación, mareos, alteraciones sensoriales (por ejemplo, parestesia, tal como sensaciones de shock eléctrico), ansiedad, confusión, cefalea, letargo, labilidad emocional, insomnio, hipomanía, tinitus y convulsiones. Mientras que estos eventos generalmente son autolimitantes, ha habido informes de síntomas serios de descontinuación.
Los pacientes deben ser monitoreados en relación con estos síntomas cuando descontinúan el tratamiento con desvenlafaxina. Se recomienda una reducción gradual en la dosis en lugar del cese abrupto siempre que sea posible. Si se producen síntomas intolerables después de una disminución en la dosis o tras la descontinuación del tratamiento, se puede considerar reiniciar la dosis prescrita previamente. Posteriormente, el médico puede continuar disminuyendo la dosis, pero a una velocidad más gradual.
Convulsión: Se han informado casos de convulsión en los estudios clínicos previos a la comercialización con desvenlafaxina. Desvenlafaxina no ha sido evaluada sistemáticamente en pacientes con un trastorno de convulsión. Los pacientes con una historia de convulsiones fueron excluidos de los estudios clínicos previos a la comercialización. desvenlafaxina debe prescribirse con precaución en pacientes con trastorno convulsivo. Hiponatremia: Se puede producir hiponatremia como resultado del tratamiento con SSRI y SNRI, incluyendo desvenlafaxina. En muchos casos, esta hiponatremia parece ser el resultado del síndrome de secreción inapropiada de hormona antidiurética (SIADH). Se han informado casos con sodio sérico menor a 110 mmol/L. los pacientes adultos mayores pueden tener un mayor riesgo de desarrollar hiponatremia con SSRI y SNRI. Asimismo, los pacientes que toman diuréticos o que de otro modo tienen un volumen reducido pueden estar en mayor riesgo. La descontinuación de desvenlafaxina debe considerarse en pacientes con hiponatremia sintomática y se debe iniciar la intervención médica apropiada. Los signos y síntomas de hiponatremia incluyen cefalea, dificultad para concentrarse, problemas de memoria, confusión, debilidad e inestabilidad, que puede conducir a caídas. Los signos y síntomas asociados con casos más severos y/o agudos han incluido alucinación, síncope, convulsión, coma, paro respiratorio y muerte.
Enfermedad pulmonar intersticial y neumonía eosinofílica: Enfermedad pulmonar intersticial y neumonía eosinofílica asociada con la terapia de venlafaxina (el fármaco original de desvenlafaxina) se ha informado raramente. La posibilidad de estos eventos adversos debe considerarse en los pacientes tratados con desvenlafaxina que presentan disnea progresiva, tos o malestar en el pecho. Dichos pacientes deben ser sometidos a una evaluación médica inmediata y se debe considerar la descontinuación de desvenlafaxina. Efectos sobre la capacidad para conducir y utilizar máquinas: desvenlafaxina puede producir sedación, mareos. Por consiguiente, se debe indicar a los pacientes que, si experimentan sedación o mareos, deben evitar la realización de tareas potencialmente peligrosas, como conducir o utilizar máquinas.
PRECAUCIONES EN RELACIÓN CON EFECTOS DE CARCINOGÉNESIS, MUTÁGENESIS, TERATOGÉNESIS Y SOBRE LA FERTILIDAD:
Carcinogénesis, mutagénesis, problemas de fertilidad:
Carcinogenicidad: Desvenlafaxina succinato administrado mediante gavaje oral a ratones y ratas por 2 años, no aumentó la incidencia de tumores en ninguno de los estudios.
Los ratones recibieron desvenlafaxina succinato a dosis de hasta 500/300 mg/kg/día (dosificaciones reducidas después de 45 semanas de administración). La dosis de 300 mg/kg/día es 15 veces una dosis humana de 100 mg/día en base mg/m2.
Las ratas recibieron desvenlafaxina succinato a dosis de hasta 300 mg/kg/día (machos) o 500 mg/kg/día (hembras). La dosis más alta es 29 (machos) o 48 (hembras) veces una dosis humana de 100 mg/día en base de mg/m2.
Los ratones, recibieron desvenlafaxina succinado a dosis de hasta 500/300 mg/kg/día (dosificaciones reducidas después de 45 semanas de administración). La dosis de 300 mg/kg/día es 15 veces una dosis humana de 100 mg/día en base mg/m2.
Las ratas, recibieron desvenlafaxina succinato a dosis de hasta 300 mg/día (machos) o 500 mg/kg/día (hembras). La dosis más alta es 29 (machos) o 48 (hembras) veces una dosis humana de 100 mg/día en base mg/kg2.
Mutagenicidad: Desvenlafaxina no fue mutagénica en el ensayo de mutación bacteriana in vitro (Prueba de Ames) y no fue clastogénica en un ensayo de aberración cromosómica in vitro en células CHO, un ensayo de micronúcleos de ratones in vivo o en un ensayo de aberración cromosómica in vivo en ratas. Además, desvenlafaxina no fue genotóxica en el ensayo de mutación directa en célula de mamífero CHO in vitro y fue negativo en el ensayo de transformación de célula embrionaria de ratón BALB/c-3T3 in vitro.
Deterioro de la fertilidad: Cuando se administró desvenlafaxina succinato de forma oral a ratas macho y hembra, hubo una reducción en la fertilidad a la dosis más elevada de 300 mg/kg/día, que representa 30 veces una dosis humana de 100 mg/día (medida en forma de mg/m2). No hubo efecto en la fertilidad a dosis de 100 mg/kg/día, que representa aproximadamente 10 veces una dosis humana de 100 mg/día (medida en forma de mg/m2).
DOSIS Y VÍA DE ADMINISTRACIÓN:
Vía de administración: Oral.
Instrucción general de uso: La dosis recomendada para desvenlafaxina es 50 mg una vez al día, con o sin alimentos.
En estudios clínicos, las dosis de 50 mg a 400 mg por día demostraron ser efectivas, aunque no se demostró un beneficio adicional a dosis superiores a 50 mg por día y las reacciones adversas y descontinuaciones fueron más frecuentes a dosis más altas.
Cuando se descontinúa la terapia, se recomienda la reducción de dosis gradual siempre que sea posible para minimizar los síntomas de descontinuación desvenlafaxina debe tomarse a aproximadamente la misma hora cada día. Las tabletas deben tomarse enteras con líquidos y no divididas, trituradas, masticadas o disueltas.
Poblaciones especiales:
Pacientes con deterioro de la función renal: La dosis máxima recomendada en pacientes con insuficiencia renal moderada (depuración de creatinina [CrCl] a las 24 h = 30 a 50 mL/min, Cockcroft-Gault [C-G]) es 50 mg al día. La dosis máxima recomendada en pacientes con insuficiencia renal severa (CrCl 24-h menos de 30 mL/min, C-G) o nefropatía en etapa terminal (ESRD) es 50 mg interdiario. No se deben administrar dosis suplementarias a pacientes después de la diálisis.
Pacientes con deterioro de la función hepática: La dosis recomendada en pacientes con insuficiencia hepática moderada a grave es 50 mg por día. El aumento de dosis por encima de 100 mg por día no es recomendado. Mantenimiento/continuación/tratamiento ampliado: Es generalmente aceptado que los episodios agudos de trastorno depresivo mayor requieren de varios meses o más tiempo de terapia farmacológica sostenida. Se determinó la eficacia a plazo más largo de desvenlafaxina (50-400 mg) en dos ensayos de mantenimiento. Los pacientes deben ser reevaluados periódicamente para determinar la necesidad del tratamiento continuo. Descontinuación de desvenlafaxina: Se han informado síntomas asociados con la descontinuación de desvenlafaxina, diferentes a SNRI y SSRI. Los pacientes deben ser monitoreados por estos síntomas cuando descontinúan el tratamiento. Se recomienda una reducción gradual de la dosis en lugar de un cese abrupto siempre que sea posible. Si se producen síntomas intolerables después de una disminución en la dosis o tras la descontinuación del tratamiento, entonces se puede considerar reiniciar la dosis prescrita anteriormente. Posteriormente, el médico puede continuar disminuyendo la dosis, pero a una velocidad más gradual.
Pacientes que cambian de otros antidepresivos a desvenlafaxina: Se han informado síntomas de descontinuación cuando se cambia a los pacientes de otros antidepresivos, incluyendo venlafaxina a desvenlafaxina. La reducción gradual del antidepresivo inicial puede ser necesaria para minimizar los síntomas de la descontinuación.
Pacientes que cambian a o de un Inhibidor de la Monoamino Oxidasa (MAOI) previsto para tratar trastornos psiquiátricos: Deben transcurrir al menos 14 días entre la descontinuación de un MAOI previsto para tratar trastornos psiquiátricos y el inicio de la terapia con desvenlafaxina. Inversamente, se debe permitir al menos 7 días después de detener desvenlafaxina antes de comenzar la administración de un MAOI previsto para tratar trastornos psiquiátricos.
Uso de desvenlafaxina con otros MAOI tales como linezolid o azul de metileno: No inicie el tratamiento con desvenlafaxina en un paciente que está siendo tratado con linezolid o azul de metileno por vía intravenosa dado que existe un mayor riesgo de sufrir el síndrome de serotonina. En un paciente que requiere un tratamiento más urgente de una afección psiquiátrica, deben considerarse otras intervenciones, incluida la hospitalización (ver Contraindicaciones). En algunos casos, un paciente que ya está recibiendo terapia con desvenlafaxina puede requerir un tratamiento urgente con linezolid o azul de metileno por vía intravenosa. Si no hay disponibles las alternativas aceptables al tratamiento con linezolid o azul de metileno por vía intravenosa y se considera que los beneficios potenciales del tratamiento con linezolid o azul de metileno por vía intravenosa son mayores que los riesgos del síndrome de serotonina en un paciente en particular, debe detenerse prontamente la terapia con desvenlafaxina y pueden administrarse linezolid o azul de metileno por vía intravenosa. Debe monitorearse el paciente en busca de síntomas del síndrome de la serotonina durante 7 días o hasta 24 horas después de la última dosis de linezolid o azul de metileno por vía intravenosa, lo que ocurra primero. La terapia con desvenlafaxina puede reanudarse 24 horas después de la última dosis de linezolid o azul de metileno por vía intravenosa (ver Advertencias y precauciones: Síndrome de serotonina). El riesgo de administrar azul de metileno por vías no intravenosas (tales como tabletas por vía oral o mediante una inyección local) o en dosis intravenosas mucho más bajas que 1 mg/kg con desvenlafaxina no es claro. No obstante, el médico deberá estar atento a la posibilidad de síntomas emergentes del síndrome de serotonina con tal uso (ver Advertencias y precauciones: Síndrome de serotonina).
Uso en poblaciones específicas:
Uso pediátrico: No se ha establecido la seguridad y eficacia en pacientes pediátricos (ver Advertencias de la caja y Advertencias y precauciones: Pensamientos y conductas suicidas en niños, adolescentes y adultos jóvenes). Cualquier persona que considere el uso de desvenlafaxina en niños o adolescentes debe equilibrar los riesgos potenciales con la necesidad clínica.
Uso geriátrico: De los 4,158 pacientes en los estudios clínicos previos a la comercialización con desvenlafaxina, 6% tenían 65 años o más. No se observaron diferencias generales en seguridad o eficacia entre estos pacientes y pacientes más jóvenes, sin embargo, en los estudios de corto plazo, controlados con placebo, hubo una mayor incidencia de hipotensión ortostática sistólica en pacientes ≥ 65 años de edad en comparación con pacientes < 65 años de edad tratados con desvenlafaxina. Para pacientes adultos mayores, se debe considerar una posible reducción de la depuración renal de desvenlafaxina cuando se determina la dosis. SSRI y SNRI, incluyendo desvenlafaxina han sido asociados con casos de hiponatremia clínicamente significativa en pacientes adultos mayores, que pueden estar en mayor riesgo para este evento adverso. Otros factores del paciente: El efecto de los factores del paciente intrínsecos sobre la farmacocinética de desvenlafaxina se presenta en la Figura 3.
Figura 3: Impacto a los factores intrínsecos (deterioro renal de la función renal y hepática y descripción de la población)
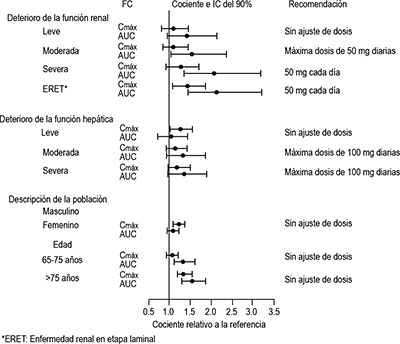
Deterioro de la función renal: En participantes con insuficiencia renal, la depuración de desvenlafaxina disminuyó. En participantes con insuficiencia renal severa CrCl 24 h < 30 mL/min, Coccroft-Gault) y nefropatía en etapa terminal, las vidas medias de eliminación se prolongaron significativamente, aumentando las exposiciones a desvenlafaxina, por lo tanto, se recomienda el ajuste de la dosificación en estos pacientes.
Deterioro de la función hepática: La vida media terminal (t1/2) promedio cambió de aproximadamente 10 horas en participantes sanos con insuficiencia hepática leve a 13 y 14 horas en insuficiencia hepática moderada y severa, respectivamente. La dosis recomendada en pacientes con insuficiencia hepática moderada a grave es 50 mg/día. El aumento de dosis por encima de 100 mg/día no es recomendado.
MANIFESTACIONES Y MANEJO DE LA SOBREDOSIFICACIÓN O INGESTA ACCIDENTAL:
Experiencia humana con sobredosis: Existe experiencia limitada en ensayos clínicos con sobredosis de desvenlafaxina succinato en seres humanos.
desvenlafaxina es el metabolito activo principal de venlafaxina. La experiencia de sobredosis informada con venlafaxina se presenta a continuación; información idéntica puede encontrarse en la sección de Sobredosis del inserto de venlafaxina.
En la experiencia posterior a la comercialización, se ha producido la sobredosis de venlafaxina predominantemente en combinación con alcohol y/u otros fármacos. Los eventos informados más comúnmente en la sobredosis incluyen taquicardia, cambios en el nivel de conciencia (que oscilan de somnolencia a coma), midriasis, convulsiones y vómitos. Se han informado cambios en el electrocardiograma (por ejemplo, prolongación del intervalo QT, bloqueo de la rama cardiaca, prolongación de QRS) taquicardia sinusal y ventricular, bradicardia, hipotensión, rabdomiolisis, vértigo, necrosis hepática, síndrome de serotonina y muerte.
Los estudios retrospectivos publicados informan que la sobredosis de venlafaxina puede estar asociada con un aumento del riesgo de resultados fatales en comparación con aquél observado con productos antidepresivos SSRI pero menor que aquél para los antidepresivos tricíclicos. Los estudios epidemiológicos han demostrado que los pacientes tratados con venlafaxina tienen una carga preexistente más alta de factores de riesgo de suicidio que los pacientes tratados con SSRI.
No es clara la medida en la cual el hallazgo de aumento del riesgo de resultados fatales puede atribuirse a la toxicidad de venlafaxina en la sobredosis, en oposición con ciertas características de los pacientes tratados con venlafaxina.
Manejo de sobredosis: No se conoce antídotos específicos para desvenlafaxina.
En el manejo de la sobredosis, considerar la posibilidad de inclusión de múltiples fármacos.
PRESENTACIONES:
Caja con 14, 28 o 30 tabletas de 100 mg
Caja con 14, 28 o 30 tabletas de 50 mg
RECOMENDACIONES SOBRE ALMACENAMIENTO:
Consérvese a no más de 30 °C.
Consérvese la caja bien cerrada.
LEYENDAS DE PROTECCIÓN:
Su venta requiere receta médica. No se deje al alcance de los niños. No se use durante el embarazo ni lactancia. Prohibida la venta fraccionada del producto.
Reporte las sospechas de reacción adversa al correo: farmacovigilancia@cofepris.gob.mx
Propiedad de:
Sandoz GmbH
Biochemiestraße 10, 6250 Kundl, Tirol
Austria
Representante Legal:
SANDOZ, S.A. de C.V.
La Candelaria No. 186, Col. Atlántida, C.P. 04370
Coyoacán, Ciudad de México, México
Reg. Núm. 116M2023, SSA IV
23330022120515/15Nov2023/IPPA_DRA-Sandoz
®Marca Registrada


