
TEURI
TENOFOVIR
Tabletas
1 Caja, 1 Frasco(s), 30 Tabletas, 300 mg
FORMA FARMACÉUTICA Y FORMULACIÓN:
Cada TABLETA contiene:
Tenofovir Disoproxil Fumarato 300 mg equivalente a 245 mg de Tenofovir Disoproxil
Excipiente cbp 1 tableta
INDICACIONES TERAPÉUTICAS:
Infección por el VIH-1: Tenofovir disoproxil fumarato está indicado en combinación con otros agentes antirretrovirales para el tratamiento de la infección por el VIH-1 en adultos y pacientes pediátricos mayores de 12 años. Al iniciar la administración de Tenofovir disoproxil fumarato para el tratamiento de la infección por el VIH-1, deben tenerse en cuenta los siguientes aspectos:
• Tenofovir disoproxil fumarato no debe ser utilizado en combinación con Emtricitabina/Tenofovir disoproxil fumarato, ni Efavirenz/emticitrabina/tenofovir disoproxil fumarato (véase Precaucione generales).
Hepatitis B crónica: Tenofovir disoproxil fumarato está indicado para el tratamiento de la hepatitis B crónica en adultos. Al iniciar la administración de Tenofovir disoproxil fumarato para el tratamiento de la infección por el VHB, deben tenerse en cuenta los siguientes aspectos:
• Esta indicación se basa principalmente en los datos que se obtuvieron del tratamiento en sujetos que no habían recibido nucleósidos previamente y en una cantidad menor de sujetos que habían recibido previamente lamivudina o dipivoxilo de adefovir. Los sujetos eran adultos que tenían hepatitis B crónica HBeAg positiva y HBeAg negativa con enfermedad hepática compensada (véase Eficacia clínica en pacientes con hepatitis B crónica).
• Tenofovir disoproxil fumarato fue evaluado en una cantidad limitada de sujetos con hepatitis B crónica y enfermedad hepática descompensada (véanse Reacciones secundarias y adversas, Eficacia clínica en pacientes con hepatitis B crónica).
En los ensayos clínicos, el número de sujetos que tenían sustituciones asociadas con la lamivudina o el adefovir al inicio fue demasiado reducido para llegar a conclusiones sobre la eficacia (véanse Farmacocinética y farmacodinamia, Eficacia clínica en pacientes con hepatitis B crónica).
FARMACOCINÉTICA Y FARMACODINAMIA:
Tenofovir disoproxil fumarato (pro fármaco del tenofovir)es una sal de ácido fumárico del éster bis-isopropoxicarboniloximetilo derivado del tenofovir. In vivo, el Tenofovir disoproxil fumarato se convierte en tenofovir, un fosfonato nucleósido acíclico (nucleótido) análogo del 5’ monofosfato de adenosina. El tenofovir muestra actividad contra la transcriptasa reversa del VIH-1.
El nombre químico del Tenofovir disoproxil fumarato es fumarato de 9-[(R)-2-[[bis [[(isopropoxicarbonil) oxi] metoxi] fosfinil] metoxi] propil] adenina (1:1). Su fórmula molecular es C19H30N5O10P • C4H4O4, y su peso molecular es de 635.52. Su fórmula estructural es la siguiente:
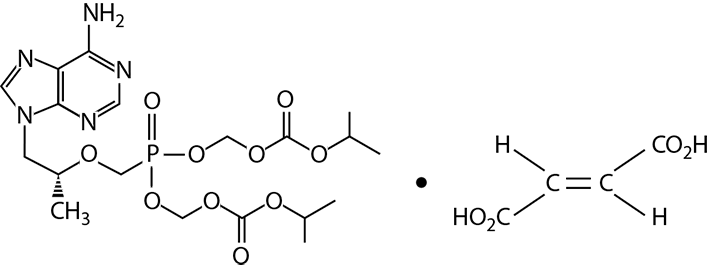
El Tenofovir disoproxil fumarato es un polvo cristalino, entre blanco y blanquecino, escasamente soluble en metanol y etanol.
Estudios clínicos: Se realizaron los siguientes:
Eficacia clínica en pacientes con infección por el VIH-1:
Pacientes adultos sin tratamiento antirretroviral previo:
Estudio 903: Se cuenta con datos que se obtuvieron hasta la semana 144 del estudio 903, un estudio doble ciego, multicéntrico y controlado con principio activo en el que se comparó Fumarato de disoproxilo de tenofovir (300 mg una vez al día) administrado en combinación con lamivudina y efavirenz contra la combinación de estavudina (d4T), lamivudina y efavirenz en 600 sujetos sin tratamiento antirretroviral previo. La media de edad de los sujetos era de 36 años (intervalo de 18 a 64), 74% eran hombres: 64% de raza blanca y 20% de raza negra. La media basal del conteo de células CD4+ era de 279 células/mm3 (intervalo de 3 a 956) y la mediana basal de ARN del VIH-1 plasmático era de 77,600 copias/mi (intervalo de 417 a 5, 130,000). Los sujetos se estratificaron según los valores basales de ARN del VIH-1 y el conteo de células CD4+. El 43% de los sujetos tenía cargas virales basales > 100,000 copias/ml, y 39% tenía conteos de células CD4+ < 200 células/mm3. Los resultados del tratamiento hasta las semanas 48 y 144 se presentan en la tabla 1.
Tabla 1. Resultados del tratamiento asignado aleatoriamente en las semanas 48 y 144 (estudio 903)
|
Resultados |
En la semana 48 |
En la semana 144 |
||
|
Tenofovir disoproxil fumarato + 3TC + EFV (n=299) |
d4T + 3TC + EFV (n=301) |
Tenofovir disoproxil fumarato + 3TC + EFV (n=299) |
d4T + 3TC + EFV (n=301) |
|
|
Sensibles al tratamientoª |
79% |
82% |
68% |
62% |
|
Falla virológicab |
6% |
4% |
10% |
8% |
|
Rebrote |
5% |
3% |
8% |
7% |
|
Sin supresión |
0% |
1% |
0% |
0% |
|
Adición de un antirretroviral |
1% |
1% |
2% |
1% |
|
Muerte |
< 1% |
1% |
< 1% |
2% |
|
Suspensión debido a evento adverso |
6% |
6% |
8% |
13% |
|
Suspensión por otras razonesc |
8% |
7% |
14% |
15% |
a Los sujetos alcanzaron y mantuvieron un valor de ARN-VIH-1 confirmado < 400 copias/ml hasta las semanas 48 y 144.
b Incluye rebrote viral confirmado y falla confirmada para alcanzar < 400 copias/ml hasta las semanas 48 y 144.
c Incluye pérdidas de seguimiento, retiro de sujetos, incumplimiento, violación del protocolo y otras razones.
La proporción de sujetos en los que se logró reducir las concentraciones plasmáticas de ARN-VIH-1 a menos de 400 copias/ml en la semana 144 fue similar entre los dos grupos de tratamiento para la población estratificada según los valores basales de concentración de ARN-VIH-1 (> o = 100,000 copias/ml) y el conteo de células CD4+ (< o ≥ 200 células/mm3). Tras 144 semanas de tratamiento, 62% de los sujetos en el grupo tratado con Tenofovir disoproxil fumarato y 58% de los sujetos en el grupo tratado con estavudina alcanzaron y mantuvieron un valor de ARN-VIH-1 confirmado < 50 copias/ml. El incremento medio en el conteo de células CD4+ con respecto al valor basal fue de 263 células/mm3 en el grupo de Tenofovir disoproxil fumarato y de 283 células/mm3 en el grupo de estavudina.
Hasta la semana 144, 11 sujetos del grupo de Tenofovir disoproxil fumarato y 9 del grupo de estavudina presentaron un nuevo evento de Clase C de acuerdo a la clasificación de los Centros de Control y Prevención de Enfermedades (CDC).
Estudio 934: Se informan los datos obtenidos hasta la semana 144 del estudio 934, un estudio aleatorizado, abierto, controlado con principio activo y multicéntrico en el que se comparó la combinación de emtricitabina + Tenofovir disoproxil fumarato con efavirenz contra la combinación de dosis fijas de zidovudina/lamivudina administradas junto con efavirenz en 511 sujetos sin tratamiento antirretroviral previo.
Desde la semana 96 hasta la semana 144 del estudio, los sujetos recibieron una combinación de dosis fijas de emtricitabina y tenofovir disoproxil fumarato con efavirenz en lugar de emtricitabina + Tenofovir disoproxil fumarato con efavirenz.
La media de edad de los sujetos era de 38 años (intervalo de 18 a 80), 86% eran varones, 59% de raza blanca y 23% de raza negra. La media basal del conteo de células CD4+ era de 245 células/mm3 (intervalo de 2 a 1,191) y la mediana basal de ARN-VIH-1 plasmático era de 5.01 log10 copias/ml (intervalo de 3.56 a 6.54). Los sujetos se estratificaron según el conteo basal de células CD4+ (< o ≥ 200 células/mm3); 41% de los sujetos tenía conteos de células CD4+ < 200 células/mm3 y 51% tenía cargas virales basales > 100,000 copias/ml. En la tabla 2, se presentan los resultados obtenidos al cabo de 48 y 144 semanas en aquellos sujetos que no presentaban resistencia al efavirenz al momento de iniciar el estudio.
Tabla 2. Resultados del tratamiento asignado aleatoriamente en las semanas 48 y 144 (Estudio 934)
|
Resultados |
En la semana 48 |
En la semana 144 |
||
|
FTC + Tenofovir disoproxil fumarato + EFV (n = 244) |
AZT/3TC + EFV (n = 243) |
FTC + Tenofovir disoproxil fumarato + EFV (n = 227)a |
AZT/3TC + EFV (n = 229)a |
|
|
Sensibles al tratamientob |
84% |
73% |
71% |
58% |
|
Fallas virológicasc |
2% |
4% |
3% |
6% |
|
Rebrote |
1% |
3% |
2% |
5% |
|
Sin supresión |
0% |
0% |
0% |
0% |
|
Cambio en el régimen antirretroviral |
1% |
1% |
1% |
1% |
|
Muerte |
< 1% |
1% |
1% |
1% |
|
Suspensión debido a evento adverso |
4% |
9% |
5% |
12% |
|
Suspensión por otras razonesd |
10% |
14% |
20% |
22% |
a Se excluyó del análisis a los sujetos que respondieron al tratamiento en la semana 48 o en la semana 96 (ARN-VIH-1 < 400 copias/ml), pero que no dieron su consentimiento para continuar el estudio después de la semana 48 o la semana 96.
b Los sujetos alcanzaron y mantuvieron un valor de ARN-VIH-1 confirmado < 400 copias/ml hasta las semanas 48 y 144.
c Incluye rebrote viral confirmado y falla confirmada para alcanzar < 400 copias/ml hasta las semanas 48 y 144.
d Incluye pérdidas de seguimiento, retiro de sujetos, incumplimiento, violación del protocolo y otras razones.
Hasta la semana 48, 84% y 73% de los sujetos en el grupo de emtricitabina + Tenofovir disoproxil fumarato y el grupo de zidovudina/lamivudina, respectivamente, alcanzaron y mantuvieron un valor de ARN-VIH-1 < 400 copias/ml (71% y 58% hasta la semana 144).
En este estudio abierto, la diferencia en la proporción de sujetos que alcanzaron y mantuvieron un valor de ARN-VIH-1 < 400 copias/ml hasta la semana 48 se debe en buena medida al número mayor de suspensiones del tratamiento debido a eventos adversos y otras razones en el grupo de zidovudina/lamivudina. Además, 80% de los sujetos en el grupo de emtricitabina + Tenofovir disoproxil fumarato y 70% en el grupo de zidovudina/lamivudina alcanzaron y mantuvieron un valor de ARN del VIH-1 < 50 copias/ml hasta la semana 48 (64% y 56%, respectivamente, hasta la semana 144).
El incremento medio en el conteo de células CD4+ con respecto al valor basal fue de 190 células/mm3 en el grupo de emtricitabina + Tenofovir disoproxil fumarato y de 158 células/mm3 en el grupo de zidovudina/lamivudina en la semana 48 (312 y 271 células/mm3 en la semana 144). Hasta la semana 48, 7 sujetos en el grupo de emtricitabina + Tenofovir disoproxil fumarato y 5 sujetos en el grupo de zidovudina/lamivudina presentaron un nuevo evento de clase C de los CDC (10 y 6 sujetos hasta la semana 144).
Pacientes adultos con tratamiento antirretroviral previo:
Estudio 907: El estudio 907 consistió en un estudio multicéntrico, doble ciego, controlado con placebo, de 24 semanas de duración, en el que se agregó Tenofovir disoproxil fumarato a un régimen de fondo estable con antirretrovirales en 550 sujetos con tratamiento previo. Después de 24 semanas de tratamiento ciego del estudio, a todos los sujetos que continuaban en el estudio se les ofreció Tenofovir disoproxil fumarato de modo abierto durante un periodo adicional de 24 semanas. Los sujetos tenían una media basal del conteo de células CD4+ de 427 células/mm3 (intervalo de 23 a 1,385), una mediana basal de ARN-VIH-1 plasmático de 2,340 (intervalo de 50 a 75,000) copias/ml y una duración media de tratamiento previo contra el VIH-1 de 5.4 años. La media de edad de los sujetos era de 42 años, 85% eran varones, 69% de raza blanca, 17% de raza negra y 12% hispanos.
En la tabla 3, se resumen el porcentaje de sujetos con ARN-VIH-1 < 400 copias/ml y los resultados de los sujetos al cabo de 48 semanas.
Tabla 3. Resultados del tratamiento asignado aleatoriamente (Estudio 907)
|
Resultados |
0-24 semanas |
0-48 semanas |
24-48 semanas |
|
|
Tenofovir disoproxil fumarato (n=368) |
Placebo (n=182) |
Tenofovir disoproxil fumarato (n=368) |
Placebo cruzado a Tenofovir disoproxil fumarato (n=170) |
|
|
ARN-VIH-1 < 400 copias/mla |
40% |
11% |
28% |
30% |
|
Falla virológicab |
53% |
84% |
61% |
64% |
|
Suspensión debido a evento adverso |
3% |
3% |
5% |
5% |
|
Suspensión por otras razonesc |
3% |
3% |
5% |
1% |
a Sujetos con ARN-VIH-1 < 400 copias/ml y sin suspensión previa del fármaco del estudio en las semanas 24 y 48, respectivamente.
b Sujetos con falla de eficacia por valor de ARN-VIH-1 = 400 copias/ml o con valor faltante de ARN del VIH-1 en las semanas 24 y 48, respectivamente.
c Incluye pérdidas de seguimiento, retiro de sujetos, incumplimiento, violación del protocolo y otras razones.
Tras 24 semanas de tratamiento, la proporción de sujetos con ARN-VIH-1 < 50 copias/ml fue mayor en el grupo que recibió Tenofovir disoproxil fumarato que en el que recibió placebo (19% y 1%, respectivamente). El cambio medio en los conteos absolutos de células CD4+ en la semana 24 fue de +11 células/mm3 en el grupo de Tenofovir disoproxil fumarato y de -5 células/mm3 en el grupo de placebo. El cambio medio en los conteos absolutos de células CD4+ en la semana 48 fue de +4 células/mm3 en el grupo Tenofovir disoproxil fumarato.
Hasta la semana 24, un solo sujeto en el grupo de Tenofovir disoproxil fumarato y ninguno de los sujetos en el grupo de placebo presentaron un nuevo evento de clase C de los CDC.
Eficacia clínica en pacientes con hepatitis B crónica:
Hepatitis B crónica HBeAg negativa: El Estudio 102 consistió en un estudio de fase 3, aleatorizado, doble-ciego, controlado con principio activo, en el que se comparó la dosis de 300 mg de Tenofovir disoproxil fumarato con la dosis de 10 mg de Hepsera en 375 sujetos HBeAg- (anti-HBe+), sujetos con función hepática compensada, la mayoría de los cuales no había recibido nucleósidos previamente. La media de edad de los sujetos era de 44 años, 77% eran varones, 25% asiáticos, 65% de raza blanca, 17% había recibido tratamiento previo con interferón alfa y 18% había recibido nucleósidos previamente (16% recibió tratamiento previo con lamivudina). Al inicio, los sujetos tenían una media de 7.8 en la puntuación necroinflamatoria de Knodell; la media del ADN del VHB plasmático era de 6.9 log10 copias/ml; y la media de ALT sérica era de 140 U/I.
Hepatitis B crónica HBeAg positiva: El Estudio 103 consistió en un estudio de fase 3, aleatorizado, doble-ciego, controlado con principio activo, en el que se comparó la dosis de 300 mg de Tenofovir disoproxil fumarato con la dosis de 10 mg de Hepsera en 266 sujetos HBeAg+ con función hepática compensada que no habían recibido nucleósidos previamente. La media de edad de los sujetos era de 34 años, 69% eran varones, 36% asiáticos, 52% de raza blanca, 16% había recibido tratamiento previo con interferón alfa y < 5% había recibido nucleósidos previamente. Al inicio, los sujetos tenían una media de 8.4 en la puntuación necroinflamatoria de Knodell; la media del ADN del VHB plasmático era de 8.7 log10 copias /ml; y la media de ALT sérica era de 147 U/I.
El análisis de los datos primarios se realizó después de que todos los sujetos alcanzaran las 48 semanas de tratamiento; los resultados se resumen a continuación.
El criterio de valoración primario de eficacia en ambos estudios fue la respuesta completa al tratamiento, definida como un valor de ADN del VHB < 400 copias/ml y una mejora de al menos 2 puntos en la puntuación necroinflamatoria de Knodell, sin empeoramiento en la fibrosis de Knodell en la semana 48 (tabla 4).
Tabla 4. Respuesta histológica, virológica, bioquímica y serológica en la semana 48
|
0102 (HBeAg-) |
0103 (HBeAg+) |
|||
|
Tenofovir disoproxil fumarato (n=250) |
Adefovir/dipivoxil (n=125) |
Tenofovir disoproxil fumarato (n=176) |
Adefovir/dipivoxil (n=90) |
|
|
Respuesta completa |
71% |
49% |
67% |
12% |
|
Histología Respuesta histológicaª |
72% |
69% |
74% |
68% |
|
ADN-VHB < 400 copias/ml (< 69 Ul/ml) |
93% |
63% |
76% |
13% |
|
ALT ALT normalizadab |
76% |
77% |
68% |
54% |
|
Serología Pérdida de HBeAg/seroconversión |
NAc |
NAc |
20%/19% |
16%/16% |
|
Pérdida de HBsAg/seroconversión |
0/0 |
0/0 |
3%/1% |
0/0 |
a Mejoría de al menos 2 puntos en la puntuación necroinflamatoria de Knodell, sin empeoramiento en la fibrosis de Knodell.
b La población utilizada en el análisis de la normalización de la ALT incluyó únicamente a sujetos con valores de ALT mayores que el límite superior de lo normal (LSN) al inicio.
c NA = no aplica.
Tratamiento durante 240 semanas: En los Estudios 0102 (HBeAg negativo) y 0103 (HBeAg positivo), los sujetos pasaron a recibir Tenofovir disoproxil fumarato de modo abierto sin interrupción del tratamiento después de recibir el tratamiento doble-ciego durante 48 semanas (ya sea Tenofovir disoproxil fumarato o Hepsera).
La tasa de retención hasta la semana 240 fue de 81% para HBeAg - y de 70% para HBeAg +. En el estudio 102 (HBeAg-), 64% de aquellos que utilizaron tenofovir en forma continua (TDF-TDF) presentaron una carga viral por debajo de las 400 copias/ml, mientras que de los pacientes incluidos en el estudio 103 (HBeAg +), 83% presentaron una carga viral menor a las 400 copias/ml. De aquellos pacientes que cambiaron de ADV a tenofovir en forma abierta a partir de las 48 semanas de tratamiento (ADV-TDF), extensión del estudio, la carga viral permaneció indetectable en 84% y 66% en los estudios 102 y 103, respectivamente.
La respuesta bioquímica (ALT) fue duradera en los sujetos que recibieron tratamiento continuo con TDF por 240 semanas y en aquellos que cambiaron a TDF después de 48 semanas de tratamiento con ADV. 72% de los pacientes que participaron en el estudio 102 (HBeAg-) y 50% de los participantes en el estudio 103 (HBeAg +) alcanzaron la normalización de ALT a la semana 240.
Los análisis de resistencia de la polimerasa/transcriptasa inversa de VHB (pol/RT) aunque se han identificado mutaciones de resistencia después de la administración de otros agentes anti-VHB (lamivudina, adefovir dipivoxil, telbivudina y entecavir), no se han identificado sustituciones de aminoácidos que se asocien a resistencia con tenofovir DF en las regiones pol/RT del VHB durante los primeros 5 años de tratamiento.
Pacientes con hepatitis B crónica y enfermedad hepática descompensada: El Tenofovir disoproxil fumarato fue estudiado en un pequeño ensayo aleatorizado, doble ciego, controlado con principio activo, en el que se evaluó la seguridad del Tenofovir disoproxil fumarato en comparación con otros fármacos antivirales en sujetos con hepatitis B crónica (HBC) y enfermedad hepática descompensada durante 48 semanas.
Se aleatorizaron 45 sujetos adultos (37 varones y 8 mujeres) al grupo de tratamiento con Tenofovir disoproxil fumarato. Al inicio, 69% de los sujetos era HBeAg positivo y 31% era HBeAg negativo. Los sujetos tenían una puntuación media de 7 en la escala de Child-Pugh, una puntuación media de 12 en la escala MELD, un valor medio de ADN-VHB de 5.8 log10 copias/ml y un valor medio de ALT sérica de 61 U/l basal. Los criterios de valoración del estudio consistieron en la suspensión debida a un evento adverso y en el aumento confirmado ≥ 0.5 mg/dl en la creatinina sérica o un nivel confirmado < 2 mg/dl del fósforo sérico (véase Reacciones secundarias y adversas).
Al cabo de 48 semanas, 31/44 (70%) y 12/26 (46%) sujetos tratados con Tenofovir disoproxil fumarato alcanzaron un valor de ADN- VHB < 400 copias/ml y un valor normalizado de ALT, respectivamente. El ensayo no fue diseñado para evaluar el impacto del tratamiento en los criterios de valoración clínicos, como el avance de la enfermedad hepática, la necesidad de un trasplante de hígado o la muerte.
Mecanismo de acción:
El tenofovir disoproxil fumarato es un diéster de fosfonato nucleósido acíclico análogo del monofosfato de adenosina. El tenofovir disoproxil fumarato requiere la hidrólisis inicial del diéster para su conversión a tenofovir y fosforilaciones subsecuentes por enzimas celulares para formar el di fosfato de tenofovir, un finalizador obligado de las cadenas. El di fosfato de tenofovir inhibe la actividad de la transcriptasa reversa del VIH-1 y del VHB al competir con el sustrato natural 5’-trifosfato de desoxiadenosina y, una vez incorporado al ADN, por la terminación de la cadena del ADN. El di fosfato de tenofovir es un inhibidor débil de las ADN polimerasas a, β, y de la ADN-polimerasa mitocondrial g en mamíferos.
Actividad contra el VIH:
Actividad antiviral: La actividad antiviral de tenofovir contra aislados clínicos y de laboratorio del VIH-1 se evaluó en líneas celulares linfoblastoides, en monocitos/macrófagos primarios y en linfocitos de sangre periférica. Los valores de CE50 (50% de la concentración efectiva) del tenofovir estuvieron en el intervalo de 0.04 μM a 8.5 μM. Se observaron efectos entre aditivos y sinérgicos en estudios de combinaciones farmacológicas de tenofovir con inhibidores nucleósidos de la transcriptasa reversa (abacavir, didanosina, lamivudina, estavudina, zalcitabina, zidovudina); inhibidores no-nucleósidos de la transcriptasa reversa (delavirdina, efavirenz, nevirapina); e inhibidores de la proteasa (amprenavir, indinavir, nelfinavir, ritonavir, saquinavir). El tenofovir mostró actividad antiviral en cultivo celular contra los caldos A, B, C, D, E, F, G y O del VIH-1 (los valores de CE50 fluctuaron entre 0.5 μM y 2.2 μM), así como actividad específica de la cepa contra el VIH-2 (CE50 entre 1.6 μM y 5.5 μM).
Resistencia: Se han seleccionado aislados del VIH-1 en cultivo celular con susceptibilidad reducida al tenofovir. Estos virus expresaron una sustitución K65R en la transcriptasa reversa y mostraron una reducción de 2 a 4 veces en la susceptibilidad al tenofovir.
En el estudio 903 realizado en sujetos sin tratamiento previo (Fumarato de disoproxilo de tenofovirt lamivudina + efavirenz frente a estavudina + lamivudina + efavirenz) (véase Estudios clínicos), los análisis genotípicos de los aislados obtenidos de sujetos con falla virológica hasta la semana 144 demostraron que el desarrollo de sustituciones asociadas con la resistencia al efavirenz y a la lamivudina se produce con mayor frecuencia, y sin diferencia entre los grupos de tratamiento. La sustitución K65R se presentó en 8/47 (17%) de los aislados analizados que se obtuvieron de pacientes del grupo de Tenofovir disoproxil fumarato y en 2/49 (4%) de los aislados analizados que se obtuvieron de pacientes del grupo de estavudina. De los 8 sujetos cuyos virus desarrollaron la sustitución K65R en el grupo tratado con Tenofovir disoproxil fumarato hasta la semana 144, 7 de los casos se presentaron durante las primeras 48 semanas de tratamiento y uno en la semana 96. En este estudio, no se identificaron otras sustituciones que provoquen resistencia a Fumarato de disoproxilo de tenofovir.
En el estudio 934 realizado en sujetos sin tratamiento previo (Tenofovir disoproxil fumarato + Emtricitabina + efavirenz frente a zidovudina (AZT)/lamivudina (3TC) + efavirenz) (véase Estudios clínicos), los análisis genotípicos efectuados en los aislados de VIH-1 obtenidos de todos los sujetos que presentaron falla virológica confirmada con > 400 copias/ml de ARN-VIH-1 en la semana 144 o en la suspensión temprana del tratamiento demostraron que las sustituciones asociadas con resistencia al efavirenz se produjeron con mayor frecuencia y fueron similares entre los dos grupos de tratamiento. La sustitución M184V, asociada con resistencia a Emtriva y lamivudina, se observó en 2/19 de los aislados analizados que se obtuvieron de sujetos del grupo de Tenofovir disoproxil fumarato + Emtricitabina y en 10/29 de los aislados analizados que se obtuvieron de sujetos del grupo de zidovudina/lamivudina. Durante las 144 semanas del estudio 934, ninguno de los sujetos presentó una sustitución K65R detectable en su VIH-1, según los análisis genotípicos convencionales.
Resistencia cruzada: Se ha reconocido que existe resistencia cruzada entre ciertos inhibidores de la transcriptasa reversa. La sustitución K65R seleccionada por el tenofovir también es seleccionada en algunos sujetos infectados por el VIH-1 tratados con abacavir, didanosina o zalcitabina. Los aislados de VIH-1 con esta mutación también muestran una susceptibilidad reducida a la emtricitabina y a la lamivudina. Por consiguiente, la resistencia cruzada entre estos fármacos puede ocurrir en pacientes cuyos virus alberguen la sustitución K65R. Los aislados de VIH-1 obtenidos de sujetos (n=20) cuyos VIH-1 expresaron una media de 3 sustituciones de la transcriptasa reversa asociadas con la zidovudina (M41L, D67N, K70R, L210W, T215Y/F o K219Q/E/N) mostraron una disminución de 3.1 veces en la susceptibilidad al tenofovir.
En los estudios 902 y 907 llevados a cabo en sujetos con tratamiento previo (Tenofovir disoproxil fumarato + tratamiento de fondo estándar [TFE] en comparación con placebo+ TFE) (véase Estudios clínicos), 14/304 (5%) de los sujetos tratados con Fumarato de disoproxilo de tenofovir que presentaron falla virológica hasta la semana 96 mostraron una reducción > 1.4 veces (mediana: 2.7 veces) en la susceptibilidad al tenofovir. El análisis genotípico de los aislados obtenidos al inicio del estudio y tras la falla virológica mostró el desarrollo de la sustitución K65R en el gen de la transcriptasa reversa del VIH-1.
La respuesta virológica al tratamiento con Tenofovir disoproxil fumarato se evaluó con respecto al genotipo viral basal (n = 222) en sujetos con tratamiento previo que participaron en los Estudios 902 y 907. En estos estudios clínicos, 94% de los participantes evaluados tenían aislados del VIH-1 basales que expresaban al menos una mutación de inhibidores nucleosídicos de la transcriptasa reversa (INTR). Las respuestas virológicas de los sujetos en el sub estudio genotípico fueron similares a los resultados generales del estudio.
Se realizaron varios análisis exploratorios para evaluar el efecto de sustituciones específicas y patrones de sustitución en los resultados virológicos. Debido a la gran cantidad de posibles comparaciones, no se realizaron pruebas estadísticas. Se observaron diferentes grados de resistencia cruzada de Tenofovir disoproxil fumarato a sustituciones asociadas con la resistencia a la zidovudina preexistentes (M41L, D67N, K70R, L210W, T215Y/F o K219Q/E/N) y parecieron depender del tipo y la cantidad de sustituciones específicas.
Los sujetos tratados con Tenofovir disoproxil fumarato cuyos VIH-1 expresaron 3 o más sustituciones asociadas con la resistencia a la zidovudina que incluían ya sea la sustitución M41L o la L210W de la transcriptasa reversa mostraron respuestas reducidas al tratamiento con Tenofovir disoproxil fumarato; sin embargo, estas respuestas fueron mejores que las obtenidas con placebo.
La presencia de la sustitución D67N, K70R, T215Y/F o K219Q/E/N no pareció afectar a las respuestas al tratamiento con Tenofovir disoproxil fumarato.
Los sujetos cuyos virus expresaban una sustitución L74V sin sustituciones asociadas con la resistencia a la zidovudina (n = 8) tuvieron una respuesta reducida a Tenofovir disoproxil fumarato. Se dispone de datos limitados en el caso de los sujetos cuyos virus expresaban una sustitución Y115F (n = 3), una sustitución Q151M (n = 2) o una inserción T69 (n = 4), todos los cuales tuvieron una respuesta reducida. En los análisis establecidos en el protocolo, la respuesta virológica a Tenofovir disoproxil fumarato no se redujo en sujetos con VIH-1 que expresaron la sustitución M184V asociada con resistencia a abacavir/emtricitabina/lamivudina.
Las respuestas del ARN del VIH-1 en estos sujetos duraron hasta la semana 48.
Análisis fenotípicos de los estudios 902 y 907: El análisis fenotípico basal del VIH-1 obtenido de sujetos con previo (n = 100) demostró una correlación entre la susceptibilidad basal a Tenofovir disoproxil fumarato y la respuesta al tratamiento con este medicamento.
En la tabla 5, se resume la respuesta del ARN del VIH-1 en función de la susceptibilidad basal a Tenofovir disoproxil fumarato.
Tabla 5. Respuesta del ARN- VIH-1 en la semana 24 en función a la susceptibilidad basaI a Tenofovir disoproxil fumarato (intención de tratar)ª
|
Susceptibilidad basal a Tenofovir disoproxil fumaratob |
Cambio en el ARN del VIH-1C (N) |
|
< 1 |
-0.74 (35) |
|
> 1 y ≤ 3 |
-0.56 (49) |
|
> 3 y ≤ 4 |
-0.3 (7) |
|
> 4 |
-0.12 (9) |
a La susceptibilidad al tenofovir se determinó mediante el ensayo recombinante fenotípico Antibiograma (Virco).
b Número de veces en que se incrementó la susceptibilidad en comparación con la cepa silvestre.
c Cambio promedio del ARN del VIH-1 desde el inicio hasta la semana 24 (DAVG24) en log10 copias/ml.
Actividad contra el VHB:
Actividad antiviral: La actividad antiviral del tenofovir contra el VHB se evaluó en la línea celular HepG2 2.2.15. Los valores de CE50 para el tenofovir variaron entre 0.14 y 1.5 μM, con valores de CC50 (50% de la concentración citotóxica) > 100 μM. No se observó actividad antagonista en los estudios de actividad antiviral en combinaciones de cultivos celulares realizados con el tenofovir y los inhibidores nucleósidos de la transcriptasa reversa anti-VHB emtricitabina, entecavir, lamivudina y telbivudina.
Resistencia: Se evaluó anualmente la resistencia genotípica acumulativa a Tenofovir disoproxil fumarato con las secuencias emparejadas de aminoácidos de la transcriptasa reversa del VHB en aislados obtenidos antes y durante el tratamiento de sujetos que recibieron al menos 24 semanas de monoterapia con Tenofovir disoproxil fumarato y que seguían presentando viremia con un valor de ADN del VHB = 400 copias/ml al final de cada año del estudio (o en el momento de la suspensión de la monoterapia con Tenofovir disoproxil fumarato) empleando un análisis según el tratamiento. En cuatro ensayos que se encuentran en curso con Tenofovir disoproxil fumarato (los estudios 102, 103 y 106 realizados en sujetos con enfermedad hepática compensada, y el estudio 108 realizado en sujetos con enfermedad hepática descompensada), 10% (69/660) de los sujetos con enfermedad hepática compensada que recibieron monoterapia con Tenofovir disoproxil fumarato durante un máximo de 240 semanas y 18% (7/39) de los sujetos con enfermedad hepática descompensada que recibieron monoterapia con Tenofovir disoproxil fumarato durante máximo de 48 semanas seguían presentando viremia en el momento de la última medición durante la monoterapia con Tenofovir disoproxil fumarato. En la población de sujetos HBeAg+ sin tratamiento previo con Emtricitabina del estudio 103, 74% (17/23) de los sujetos con un valor de ADN del VHB = 400 copias/ml en el momento de la última medición durante la monoterapia con Tenofovir disoproxil fumarato tenía una carga viral basal > 9 log10 copias/ml.
Se identificaron sustituciones de aminoácidos en la transcriptasa reversa del VHB aparecidas durante el tratamiento en 46% (32/69) de los sujetos de los estudios 102, 103, 106 y 108 con datos genotípicos emparejados evaluables; no se produjeron sustituciones específicas con una frecuencia suficiente para ser asociadas con la resistencia a Tenofovir disoproxil fumarato (análisis genotípicos o fenotípicos).
Resistencia cruzada: Se ha observado resistencia cruzada entre los inhibidores análogos nucleósidos/nucleótidos de la transcriptasa reversa del VHB.
En ensayos basados en células, las cepas del VHB que expresaron las sustituciones rtV173L, rtL180M y rtM204I/V asociadas con la resistencia a la lamivudina y la telbivudina mostraron una susceptibilidad al tenofovir de 0.7 a 3.4 veces mayor en comparación con el virus silvestre. Las sustituciones dobles rtL180M y rtM204I/N redujeron 3.4 veces la susceptibilidad al tenofovir. Las cepas del VHB que expresaron las sustituciones rtL180M, rtT184G, rtS202G/I, rtM204V y rtM250V asociadas con la resistencia al entecavir mostraron una susceptibilidad al tenofovir de 0.6 a 6.9 veces mayor en comparación con el virus silvestre. Una cepa del VHB que expresó a la vez las sustituciones rtL180M, rtT184G, rtS202I y rtM204V presentó una reducción de 6.9 veces en la susceptibilidad al tenofovir.
Las cepas del VHB que expresaron las sustituciones rtA181V y/o rtN236T asociadas con la resistencia al adefovir mostraron unas reducciones de 2.9 a 10 veces en la susceptibilidad al tenofovir en comparación con el virus silvestre. Las cepas que presentaban la sustitución rtA181T mostraron cambios en la susceptibilidad al tenofovir de 0.9 a 1.5 veces mayor en comparación con el virus silvestre.
En los cuatro estudios de tratamiento con Tenofovir disoproxil fumarato, antes del tratamiento con Tenofovir disoproxil fumarato, 14, 15 y 2 sujetos presentaban VHB que albergaba sustituciones asociadas con la resistencia al adefovir (rtA181T/V y/o rtN236T), sustituciones asociadas con la resistencia a la lamivudina (rtM204I/V) o ambas sustituciones, respectivamente. Luego de hasta 144 semanas de tratamiento con Tenofovir disoproxil fumarato, 11 de los 14 sujetos con VHB resistente al adefovir, 12 de los 15 sujetos con VHB resistente a la lamivudina y 1 de los 2 sujetos con VHB resistente tanto al adefovir como a la lamivudina lograron la supresión virológica (ADN del VHB < 400 copias/ml). Dos de los 5 sujetos cuyos virus albergaban las dos sustituciones rtA181T/V y rtN236T y uno de los 5 sujetos cuyos virus albergaban estas sustituciones junto con una sustitución rtM204I seguían presentando viremia luego de hasta 32 semanas de monoterapia con Tenofovir disoproxil fumarato.
Farmacocinética: Se ha evaluado la farmacocinética del tenofovir disoproxil fumarato en voluntarios sanos y en personas infectadas por el VIH-1. La farmacocinética del tenofovir es similar en estas poblaciones.
Absorción: Tenofovir disoproxil fumarato es un pro fármaco diéster hidrosoluble del principio activo tenofovir. En sujetos en ayunas, la biodisponibilidad oral del Tenofovir disoproxil fumarato es de aproximadamente 25%. Después de la administración en ayunas por vía oral de una dosis única de 300 mg de Fumarato de disoproxilo de tenofovir a sujetos infectados por el VIH-1, las concentraciones máximas (Cmáx) en suero se alcanzan en 1.0 ± 0.4 h. Los valores de Cmáx y ABC son de 0.30 ± 0.09 μg/ml y de 2.29 ± 0.69 μg• h/ml, respectivamente.
La farmacocinética del tenofovir es proporcional a la dosis en un intervalo de dosis de 75 a 600 mg de Tenofovir disoproxil fumarato, y no se ve afectada por dosis repetidas.
Distribución: La unión in vitro del tenofovir a las proteínas plasmáticas o séricas humanas es inferior a 0.7 y 7.2%, respectivamente, cuando la concentración del tenofovir está en un intervalo de 0.01 a 25 μg/ml. Tras la administración intravenosa de 1.0 mg/kg y 3.0 mg/kg de tenofovir, el volumen de distribución en estado estable es de 1.3 ± 0.6 l/kg y 1.2 ± 0.4 l/kg, respectivamente.
Metabolismo y eliminación: Los estudios in vitro indican que ni el tenofovir disoproxil ni el tenofovir son sustratos de las enzimas del complejo CYP.
Tras la administración IV de tenofovir, aproximadamente de 70 a 80% de la dosis se recupera sin cambios en la orina dentro de las primeras 72 horas. La semivida de eliminación terminal del tenofovir es de aproximadamente 17 horas tras la administración de una dosis única de Tenofovir disoproxil fumarato por vía oral. Después de dosis orales múltiples de Tenofovir disoproxil fumarato, a razón de 300 mg una vez al día (con alimentos), el 32 ± 10% de la dosis administrada se recupera en la orina en el lapso de 24 horas.
El tenofovir se elimina mediante una combinación de filtración glomerular y secreción tubular activa. Puede haber competencia por la eliminación con otros compuestos que también se eliminan por vía renal.
Efectos de los alimentos en la absorción oral: La administración de Tenofovir disoproxil fumarato después de una comida rica en grasa (~700 a 1000 kcal que contenga de 40 a 50% de grasas) aumenta la biodisponibilidad oral, con un incremento en el ABC0-¥ del tenofovir de aproximadamente 40% y un incremento en la Cmáx de aproximadamente 14%. Sin embargo, la administración de Tenofovir disoproxil fumarato con una comida ligera no tuvo efectos significativos en la farmacocinética del tenofovir si se compara con la administración del fármaco en ayunas.
La comida retrasa el tiempo hasta alcanzar la Cmáx del tenofovir aproximadamente en 1 hora. Cuando el contenido de la dieta no se controló, la Cmáx y el ABC del tenofovir fueron de 0.33 ± 0.12 μg/ml y de 3.32 ± 1.37 μg• h/ml después de dosis múltiples de 300 mg de Tenofovir disoproxil fumarato una vez al día administradas después de consumir alimentos.
Poblaciones especiales:
Raza: No hubo un número suficiente de grupos raciales y étnicos diferentes de la raza blanca para poder determinar adecuadamente las posibles diferencias farmacocinéticas entre estas poblaciones.
Género: La farmacocinética del tenofovir es similar entre los varones y las mujeres.
Pacientes pediátricos de 12 años o más: Se evaluó la farmacocinética del tenofovir en estado estable en 8 sujetos pediátricos infectados por el VIH-1 (12 a < 18 años). Los valores medios (± DE) de Cmáx y ABCtau son de 0.38 ± 0.13 μg/ml y de 3.39 ± 1.22 μg• h/ml, respectivamente. La exposición al tenofovir alcanzada en estos sujetos pediátricos que recibieron dosis diarias de 300 mg de Tenofovir disoproxil fumarato por vía oral fue similar a las exposiciones alcanzadas en adultos que recibieron dosis de 300 mg de Fumarato de disoproxilo de tenofovir una vez al día.
No se han realizado estudios farmacocinéticos en sujetos pediátricos < 12 años de edad.
Pacientes geriátricos: No se han realizado estudios farmacocinéticos en sujetos de edad avanzada (> 65 años).
Pacientes con disfunción renal: La farmacocinética del tenofovir se altera en sujetos con disfunción renal (véase Precauciones generales). En sujetos con depuración de creatinina < 50 ml/min o con enfermedad renal en fase terminal (ERFT) que requieren diálisis, se incrementaron la Cmáx y el ABC0-¥ del tenofovir (tabla 6). Se recomienda que se modifique el intervalo de dosificación de Fumarato de disoproxilo de tenofovir en pacientes con depuración de creatinina < 50 ml/min o en pacientes con ERFT que requieran diálisis (véase Dosis y vía de administración).
Tabla 6. Parámetros farmacocinéticos (media ± DE) del tenofovir en sujetos con diferentes grados de función renal
|
Depuración de creatinina basal (ml/min) |
> 80 (n=3) |
50-80 (n=10) |
30-49 (n=8) |
12-29 (n=11) |
|
Cmáx. (μg/ml) |
0.34 ± 0.03 |
0.33 ± 0.06 |
0.37 ± 0.16 |
0.60 ± 0.19 |
|
ABC0-¥ (μg• h/ml) |
2.18 ± 0.26 |
3.06 ± 0.93 |
6.01 ± 2.50 |
15.98 ± 7.22 |
|
CL/F (ml/min) |
1,043.7 ± 115.4 |
807.7 ± 279.2 |
444.4 ± 209.8 |
177.0 ± 97.1 |
|
CLrenal (ml/min) |
243.5 ± 33.3 |
168.6 ± 27.5 |
100.6 ± 27.5 |
43.0 ± 31.2 |
a. Fumarato de disoproxiI de Tenofovir en dosis única de 300 mg.
El tenofovir se elimina eficientemente mediante hemodiálisis, con un coeficiente de extracción de aproximadamente 54%. Después de una dosis única de 300 mg de fumarato de disoproxilo de tenofovir, una sesión de hemodiálisis de cuatro horas eliminó aproximadamente 10% de la dosis de tenofovir administrada.
Pacientes con disfunción hepática: Se ha estudiado la farmacocinética del tenofovir después de una dosis única de 300 mg de Tenofovir disoproxil fumarato en sujetos no infectados por el VIH y con disfunción hepática de moderada a severa. No hubo alteraciones importantes en la farmacocinética del tenofovir en sujetos con disfunción hepática en comparación con los sujetos con función hepática normal. No se requiere ningún cambio de la dosificación de Tenofovir disoproxil fumarato en los pacientes con disfunción hepática.
CONTRAINDICACIONES:
Fumarato de disoproxilo de tenofovir está contraindicado en pacientes con hipersensibilidad previamente demostrada a cualquiera de los componentes del producto.
Administración concomitante con otros productos: Fumarato de disoproxilo de tenofovir no debe utilizarse en combinación con los productos de dosis fijas combinadas emtricitabina/fumarato de disoproxilo de tenofovir o efavirenz/ emtricitabina/fumarato de disoproxilo, ya que el tenofovir disoproxil fumarato es un componente de estos productos. Fumarato de disoproxilo de tenofovir no debe administrarse en combinación con dipivoxilo de adefovir (véase lnteracciones medicamentosas y de otro género).
RESTRICCIONES DE USO DURANTE EL EMBARAZO Y LA LACTANCIA:
Embarazo: Categoría B para el embarazo. Se realizaron estudios de reproducción en ratas y conejos con dosis hasta 14 y 19 veces mayores que la dosis humana basada en la comparación de la superficie corporal y no se encontró ninguna evidencia de alteración en la fertilidad o daños al feto por el tenofovir. Sin embargo, no hay estudios adecuados y bien controlados con mujeres embarazadas. Como los estudios de reproducción animal no siempre son predictivos de la respuesta humana, Tenofovir disoproxil fumarato sólo debe usarse durante el embarazo si es estrictamente necesario.
Madres en periodo de lactancia: Los centros de control y prevención de enfermedades recomiendan que las madres infectadas por el VIH-1 no amamanten a sus hijos para evitar el riesgo de transmisión postnatal del VIH-1. Los estudios con ratas han demostrado que el tenofovir se secreta en la leche. Se desconoce si el tenofovir se excreta en la leche humana. Debido tanto al riesgo de transmisión del VIH-1 como al potencial de reacciones adversas serias que puede haber en los lactantes, se debe indicar a las madres que no amamanten a sus hijos si están tomando Tenofovir disoproxil fumarato.
REACCIONES SECUNDARIAS Y ADVERSAS:
En otros apartados del prospecto, se tratan las siguientes reacciones adversas:
• Acidosis láctica/hepatomegalia severa con esteatosis (véase Precauciones generales).
• Exacerbación aguda severa de la hepatitis (véase Precauciones generales).
• Nueva aparición o empeoramiento de la disfunción renal (véase Precauciones generales).
• Disminución de la densidad mineral ósea (véase Precauciones generales).
• Síndrome de reconstitución inmunológica (véase Precauciones generales).
Experiencia en ensayos clínicos: Debido a que los ensayos clínicos se realizan en condiciones muy variadas, las tasas de reacciones adversas observadas en los ensayos clínicos de un fármaco no pueden compararse directamente con las tasas en los ensayos clínicos de otro fármaco y puede que no reflejen las tasas observadas en la práctica.
Ensayos clínicos en pacientes adultos con infección por el VIH-1: Más de 12,000 sujetos han recibido tratamiento con Tenofovir disoproxil fumarato solo o en combinación con otros productos farmacéuticos antirretrovirales durante periodos de 28 días a 215 semanas en ensayos clínicos y en estudios de acceso expandido. Un total de 1,544 sujetos han recibido 300 mg una vez al día de Tenofovir disoproxil fumarato en ensayos clínicos; más de 11,000 sujetos han recibido Tenofovir disoproxil fumarato en estudios de acceso expandido.
Las reacciones adversas más frecuentes (incidencia ≥ 10%, grados 2-4) identificadas a partir de cualquiera de los tres grandes ensayos clínicos controlados son erupción cutánea, diarrea, cefalea, dolor, depresión, astenia y náuseas.
Pacientes sin tratamiento antirretroviral previo:
Estudio 903-Reacciones adversas aparecidas durante el tratamiento: Las reacciones adversas más frecuentemente observadas en el estudio controlado comparativo doble ciego en el cual 600 sujetos sin tratamiento previo recibieron Tenofovir disoproxil fumarato (n=299) o estavudina (n=301) en combinación con lamivudina y efavirenz durante 144 semanas (estudio 903) fueron eventos gastrointestinales leves a moderados y mareos.
Las reacciones adversas leves (grado 1) fueron frecuentes, con una incidencia similar en ambos grupos, e incluyeron mareos, diarrea y náuseas. En la tabla 7, se resumen las reacciones adversas específicas de moderadas a severas aparecidas durante el tratamiento.
Tabla 7. Reacciones adversas específicas aparecidas durante el tratamientoa (grados 2-4) notificadas en 5% de cualquier grupo de tratamiento del estudio 903 (0-144 semanas)
|
Tenofovir disoproxil fumarato + 3TC + EFV |
d4T + 3TC + EFV |
|
|
n = 299 |
n = 301 |
|
|
Organismo en general |
||
|
Dolor de cabeza |
14% |
17% |
|
Dolor |
13% |
12% |
|
Fiebre |
8% |
7% |
|
Dolor abdominal |
7% |
12% |
|
Dolor de espalda |
9% |
8% |
|
Astenia |
6% |
7% |
|
Sistema digestivo |
||
|
Diarrea |
11% |
13% |
|
Náuseas |
8% |
9% |
|
Dispepsia |
4% |
5% |
|
Vómitos |
5% |
9% |
|
Trastornos metabólicos |
||
|
Lipodistrofiab |
1% |
8% |
|
Sistema musculoesquelético |
||
|
Artralgia |
5% |
7% |
|
Mialgia |
3% |
5% |
|
Sistema nervioso |
||
|
Depresión |
11% |
10% |
|
Insomnio |
5% |
8% |
|
Mareos |
3% |
6% |
|
Neuropatía periféricac |
1% |
5% |
|
Ansiedad |
6% |
6% |
|
Aparato respiratorio |
||
|
Neumonía |
5% |
5% |
|
Piel y anexos |
||
|
Erupcionesd |
18% |
12% |
a Las frecuencias de las reacciones adversas se basan en todos los eventos adversos aparecidos durante el tratamiento, independientemente de la relación con el fármaco del estudio.
b La lipodistrofia comprende un grupo de eventos adversos descritos por los investigadores, y no es un síndrome definido en el protocolo.
c La neuropatía periférica incluye neuritis y neuropatía periféricas.
d Las erupciones incluyen erupción cutánea, prurito, erupción maculopapular, urticaria, erupción vesiculoampollar y erupción pustular.
Estudio 934-Reacciones adversas aparecidas durante el tratamiento: En el estudio 934, 511 sujetos sin tratamiento antirretroviral previo recibieron Tenofovir disoproxil fumarato/Emtricitabina administrados en combinación con efavirenz (n=257) o zidovudina/lamivudina administradas en combinación con efavirenz (n=254). Las reacciones adversas observadas en este estudio concordaron en general con las reacciones observadas en estudios anteriores tanto en sujetos que no habían recibido tratamiento previo como en aquellos con exposición previa a antirretrovirales (tabla 8).
Tabla 8. Reacciones adversas específicas aparecidas durante el tratamientoa (grados 2-4) notificadas en ≥ 5% de cualquier grupo de tratamiento del estudio 934 (0-144 semanas)
|
Tenofovir disoproxil fumaratob + FTC + EFV |
AZT/3TC + EFV |
|
|
n = 257 |
n = 254 |
|
|
Trastornos gastrointestinales |
||
|
Diarrea |
9% |
5% |
|
Náuseas |
9% |
7% |
|
Vómitos |
2% |
5% |
|
Trastornos generales y alteraciones en el lugar de la administración |
||
|
Fatiga |
9% |
8% |
|
Infecciones e infestaciones |
||
|
Sinusitis |
8% |
4% |
|
Infecciones en las vías respiratorias superiores |
8% |
5% |
|
Nasofaringitis |
5% |
3% |
|
Trastornos del sistema nervioso |
||
|
Dolor de cabeza |
6% |
5% |
|
Mareos |
8% |
7% |
|
Trastornos psiquiátricos |
||
|
Depresión |
9% |
7% |
|
Insomnio |
5% |
7% |
|
Trastornos de la piel y del tejido subcutáneo |
||
|
Erupcionesc |
7% |
9% |
a Las frecuencias de las reacciones adversas se basan en todos los eventos adversos aparecidos durante el tratamiento, independientemente de la relación con el fármaco del estudio.
b Desde la semana 96 hasta la semana 144 del estudio, los sujetos recibieron Emtricitabina + tenofovir disoproxil fumarato con efavirenz, en lugar de Tenofovir disoproxil fumarato + Emtricitabina con efavirenz.
c Las erupciones incluyen erupción cutánea, erupción exfoliativa, erupción generalizada, erupción macular, erupción maculopapular, erupción prurítica y erupción vesicular.
Pacientes con tratamiento antirretroviral previo:
Reacciones adversas aparecidas durante el tratamiento: Las reacciones adversas observadas en sujetos con tratamiento antirretroviral previo concordaron en general con las reacciones observadas en sujetos sin tratamiento previo, e incluyen eventos gastrointestinales leves a moderados, como náuseas, diarrea, vómitos y flatulencia. La proporción de sujetos que suspendieron su participación en los ensayos clínicos debido a reacciones adversas gastrointestinales fue inferior a 1% (estudio 907).
En la tabla 9, se proporciona un resumen de las reacciones adversas moderadas a severas aparecidas durante el tratamiento que se produjeron durante las primeras 48 semanas del estudio 907.
Tabla 9. Reacciones adversas específicas aparecidas durante el tratamientoa (grados 2-4) notificadas en ≥ 3% de cualquier grupo de tratamiento del estudio 907 (0-48 semanas)
|
Tenofovir disoproxil fumarato (n=368) (semanas 0-24) |
Placebo (n=182) (semanas 0-24) |
Tenofovir disoproxil fumarato (n=368) (semanas 0-48) |
Placebo cruzado a Tenofovir disoproxil fumarato (n=170) (semanas 24-48) |
|
|
Organismo en general |
||||
|
Astenia |
7% |
6% |
11% |
1% |
|
Dolor |
7% |
7% |
12% |
4% |
|
Dolor de cabeza |
5% |
5% |
8% |
2% |
|
Dolor abdominal |
4% |
3% |
7% |
6% |
|
Dolor de espalda |
3% |
3% |
4% |
2% |
|
Dolor de pecho |
3% |
1% |
3% |
2% |
|
Fiebre |
2% |
2% |
4% |
2% |
|
Sistema digestivo |
||||
|
Diarrea |
11% |
10% |
16% |
11% |
|
Náuseas |
8% |
5% |
11% |
7% |
|
Vómitos |
4% |
1% |
7% |
5% |
|
Anorexia |
3% |
2% |
4% |
1% |
|
Dispepsia |
3% |
2% |
4% |
2% |
|
Flatulencia |
3% |
1% |
4% |
1% |
|
Aparato respiratorio |
||||
|
Neumonía |
2% |
0% |
3% |
2% |
|
Sistema nervioso |
||||
|
Depresión |
4% |
3% |
8% |
4% |
|
Insomnio |
3% |
2% |
4% |
4% |
|
Neuropatía periféricab |
3% |
3% |
5% |
2% |
|
Mareos |
1% |
3% |
3% |
1% |
|
Piel y anexos |
||||
|
Erupcionesc |
5% |
4% |
7% |
1% |
|
Sudoración |
3% |
2% |
3% |
1% |
|
Sistema musculoesquelético |
||||
|
Mialgia |
3% |
3% |
4% |
1% |
|
Metabolismo |
||||
|
Pérdida de peso |
2% |
1% |
4% |
2% |
a Las frecuencias de las reacciones adversas se basan en todos los eventos adversos aparecidos durante el tratamiento, independientemente de la relación con el fármaco del estudio.
b La neuropatía periférica incluye neuritis y neuropatía periféricas.
c Las erupciones incluyen erupción cutánea, prurito, erupción maculopapular, urticaria, erupción vesiculoampollar y erupción pustular.
Ensayos clínicos en sujetos pediátricos de 12 años o más con infección por el VIH-1: La evaluación de las reacciones adversas se basa en un ensayo aleatorizado (Estudio 321) en el que participaron 87 sujetos pediátricos (12 a < 18 años) infectados por el VIH-1 que recibieron tratamiento con Tenofovir disoproxil fumarato (n = 45) o placebo (n = 42) en combinación con otros agentes antirretrovirales durante 48 semanas. Las reacciones adversas observadas en los sujetos que recibieron tratamiento con Tenofovir disoproxil fumarato concordaron con las reacciones observadas en los ensayos clínicos realizados en adultos.
Los efectos óseos observados en los sujetos pediátricos de 12 años o más concordaron con los efectos observados en los ensayos clínicos realizados en adultos (véase Precauciones generales).
Ensayos clínicos en sujetos adultos con hepatitis B crónica y enfermedad hepática compensada: Reacciones adversas surgidas durante el tratamiento: En los ensayos clínicos controlados en los que participaron sujetos con hepatitis B crónica (0102 y 0103), una mayor cantidad de sujetos tratados con Tenofovir disoproxil fumarato durante el periodo doble-ciego de 48 semanas presentó náuseas: 9% con Tenofovir disoproxil fumarato frente a 2% con adefovir dipodixil. Entre otras reacciones adversas aparecidas durante el tratamiento notificadas en > 5% de los sujetos tratados con Tenofovir disoproxil fumarato, se incluyen: dolor abdominal, diarrea, cefalea, mareos, fatiga, nasofaringitis, dolor de espalda y erupción cutánea.
No se observaron cambios significativos en el perfil de tolerabilidad (frecuencia, naturaleza o severidad de las reacciones adversas) en los sujetos que continuaron el tratamiento con Tenofovir disoproxil fumarato durante un máximo de 96 semanas en estos estudios.
Ensayo clínico en sujetos adultos con hepatitis B crónica y enfermedad hepática descompensada: En un pequeño estudio aleatorizado, doble-ciego, controlado con principio activo (0108), se administró tratamiento con Tenofovir disoproxil fumarato u otros fármacos antivirales durante un máximo de 48 semanas a sujetos con HBC y enfermedad hepática descompensada (véase Estudio clínicos [Eficacia clínica en pacientes con hepatitis B crónica]).
Entre los 45 sujetos que recibieron Tenofovir disoproxil fumarato, las reacciones adversas aparecidas durante el tratamiento, de cualquier grado de severidad, que se informaron con mayor frecuencia fueron dolor abdominal (22%), náuseas (20%), insomnio (18%), prurito (16%), vómitos (13%), mareos (13%) y pirexia (11%). Hasta la semana 48, dos (4%) de los 45 sujetos del estudio murieron debido al avance de la enfermedad hepática. Tres (7%) de los 45 sujetos suspendieron el tratamiento debido a un evento adverso. Cuatro (9%) de los 45 sujetos presentaron un aumento confirmado de 0.5 mg/dl en la creatinina sérica (hasta la semana 48, 1 sujeto también presentó un nivel confirmado de fósforo sérico < 2 mg/dl). Tres de estos sujetos (cada uno de ellos tenía una puntuación = 10 en la escala de Child-Pugh y una puntuación = 14 en el modelo para enfermedad hepática terminal MELD en el momento del ingreso) desarrollaron insuficiencia renal. Debido a que tanto Tenofovir disoproxil fumarato como la enfermedad hepática descompensada pueden afectar la función renal, es difícil de precisar en qué medida.
Tenofovir disoproxil fumarato contribuyó a la disfunción renal en esta población.
Uno de los 45 sujetos presentó una exacerbación hepática mientras estaba en tratamiento durante el estudio de 48 semanas.
Experiencia posterior a la comercialización:
Se han identificado las siguientes reacciones adversas durante el uso posterior a la aprobación de Tenofovir disoproxil fumarato. Debido a que las reacciones posteriores a la comercialización son notificadas voluntariamente por una población de tamaño indeterminado, no siempre es posible calcular de manera fiable su frecuencia ni establecer una relación causal con la exposición al fármaco.
Trastornos del sistema inmunitario:
• Reacción alérgica,
• incluso angioedema.
Trastornos de la nutrición y el metabolismo:
• Acidosis láctica,
• hipopotasemia,
• hipofosfatemia.
Trastornos respiratorios, torácicos y mediastínicos:
• Disnea.
Trastornos gastrointestinales:
• Pancreatitis,
• aumento de la amilasa,
• dolor abdominal.
Trastornos hepatobiliares:
• Esteatosis hepática,
• hepatitis,
• aumento de las enzimas hepáticas (con mayor frecuencia: AST, ALT, gamma GT).
Trastornos de la piel y del tejido subcutáneo:
• Erupción cutánea.
Trastornos musculoesqueléticos y del tejido conjuntivo:
• Rabdomiólisis,
• osteomalacia (se manifiesta como dolor óseo y puede contribuir a las fracturas),
• debilidad muscular,
• miopatía.
Trastornos renales y urinarios:
• Insuficiencia renal aguda,
• insuficiencia renal,
• necrosis tubular aguda,
• síndrome de Fanconi,
• tubulopatía renal proximal,
• nefritis intersticial (incluso casos agudos),
• diabetes insípida nefrogénica,
• disfunción renal,
• aumento de la creatinina,
• proteinuria, poliuria.
Trastornos generales y alteraciones en el lugar de la administración:
• Astenia.
Las siguientes reacciones adversas, enumeradas bajo los encabezados de los sistemas corporales anteriores, pueden producirse a consecuencia de la tubulopatía renal proximal:
• rabdomiólisis,
• osteomalacia,
• hipopotasemia,
• debilidad muscular,
• miopatía,
• hipofosfatemia.
PRECAUCIONES EN RELACIÓN CON EFECTOS DE CARCINOGÉNESIS, MUTAGÉNESIS, TERATOGÉNESIS Y SOBRE LA FERTILIDAD:
Se realizaron estudios a largo plazo sobre la carcinogenicidad del tenofovir disoproxil fumarato oral en ratas y ratones, con exposiciones de hasta aproximadamente 16 veces (ratones) y 5 veces (ratas) mayores que las observadas en seres humanos con la dosis terapéutica para la infección por el VIH-1. En ratones hembras que recibieron la dosis alta, aumentaron los adenomas hepáticos, con exposiciones 16 veces superiores a las de los seres humanos. En ratas sometidas a exposiciones 5 veces mayores que en seres humanos con la dosis terapéutica, el estudio de carcinogenicidad fue negativo.
El tenofovir disoproxil fumarato fue mutagénico en el ensayo de linfoma de ratón in vitro y negativo en la prueba de mutagenicidad bacteriana in vitro (prueba de Ames). En un ensayo in vivo de micronúcleos de ratón, el tenofovir disoproxil fumarato fue negativo cuando se administró a ratones machos.
No hubo efectos en la fertilidad, la capacidad de apareamiento ni el desarrollo embrionario temprano cuando se administró el tenofovir disoproxil fumarato a ratas machos con una dosis equivalente a 10 veces la dosis humana, sobre la base de las comparaciones de la superficie corporal, durante 28 días antes del apareamiento, y a ratas hembras durante 15 días antes del apareamiento hasta el séptimo día de gestación. Sin embargo, hubo una alteración del ciclo estral en las ratas hembras.
INTERACCIONES MEDICAMENTOSAS Y DE OTRO GÉNERO:
En este apartado se describen las interacciones medicamentosas clínicamente relevantes observadas con Tenofovir disoproxil fumarato.
Evaluación de las interacciones medicamentosas: En concentraciones sustancialmente superiores (~300 veces mayores) a aquéllas observadas in vivo, el tenofovir no inhibió el metabolismo del fármaco in vitro mediado por alguna de las siguientes isoformas humanas del CYP: CYP3A4, CYP2D6, CYP2C9 o CYP2E1.
Sin embargo, se observó una disminución pequeña (6%), pero estadísticamente significativa, en el metabolismo del sustrato de CYP1A. En función a los resultados de los experimentos in vitro y la vía de eliminación conocida del tenofovir, la posibilidad de interacciones mediadas por el CYP en las que interviene el tenofovir con otros productos farmacéuticos es baja (véase Farmacocinética y farmacodinamia).
Se ha evaluado Tenofovir disoproxil fumarato en voluntarios sanos en combinación con abacavir, atazanavir, didanosina, efavirenz, emtricitabina, entecavir, indinavir, lamivudina, lopinavir/ritonavir, metadona, nelfinavir, anticonceptivos orales, ribavirina, saquinavir/ritonavir y tacrolimus. En las tablas 10 y 11, se resumen los efectos farmacocinéticos del fármaco coadministrado en la farmacocinética del tenofovir y los efectos de Tenofovir disoproxil fumarato en la farmacocinética del fármaco coadministrado.
Tabla 10. Interacciones medicamentosas: cambios en los parámetros farmacocinéticos de tenofovirª en presencia del fármaco coadministrado
|
Fármaco coadministrado |
Dosis del fármaco coadministrado (mg) |
n |
Cambio % en los parámetros farmacocinéticos del tenofovirb (IC 90%) |
||
|
Cmáx |
ABC |
Cmín |
|||
|
Abacavir |
300 una vez |
8 |
↔ |
↔ |
NC |
|
Atazanavirc |
400 una vez al día x 14 días |
33 |
↑14 (↑8 a ↑20) |
↑24 (↑21 a ↑28) |
↑22 (↑15 a ↑30) |
|
Didanosina (capa entérica) |
400 una vez |
25 |
↔ |
↔ |
↔ |
|
Didanosina (buffer) |
250 o 400 una vez al día x 7 días |
14 |
↔ |
↔ |
↔ |
|
Efavirenz |
600 una vez al día x 14 días |
29 |
↔ |
↔ |
↔ |
|
Emtricitabina |
200 una vez al día x 7 días |
17 |
↔ |
↔ |
↔ |
|
Entecavir |
1 mg una vez al día x 10 días |
28 |
↔ |
↔ |
↔ |
|
Indinavir |
800 tres veces al día x 7 días |
13 |
↑14 (↑3 a ↑33) |
↔ |
↔ |
|
Lamivudina |
150 dos veces al día x 7 días |
15 |
↔ |
↔ |
↔ |
|
Lopinavir/ritonavir |
400/100 dos veces al día x 14 días |
24 |
↔ |
↑32 (↑25 a ↑38) |
↑51 (↑38 a ↑66) |
|
Nelfinavir |
1250 dos veces al día x 14 días |
29 |
↔ |
↔ |
↔ |
|
Saquinavir/ritonavir |
1,000/100 dos veces al día x 14 días |
35 |
↔ |
↔ |
↑23 (↑16 a ↑30) |
|
Tacrolimus |
0.05 mg/kg dos veces al día x 7 días |
21 |
↑13 (↑1 a ↑27) |
↔ |
↔ |
a Los sujetos recibieron 300 mg de Tenofovir disoproxil fumarato una vez al día.
b Aumento = ↑; disminución ↓; sin efecto = ↔; NC = no calculado.
c lnformación para prescribir de Atazanavir.
Después de administrar dosis múltiples a sujetos VIH y VHB negativos que recibieron tratamiento de mantenimiento crónico con metadona o anticonceptivos orales, o dosis únicas de ribavirina, las propiedades farmacocinéticas del tenofovir en estado estable fueron similares a las observadas en estudios anteriores, lo que indica ausencia de interacciones medicamentosas clínicamente significativas entre estos agentes y Tenofovir disoproxil fumarato.
Tabla 11. Interacciones medicamentosas: cambios en los parámetros farmacocinéticos del fármaco coadministrado en presencia de Tenofovir disoproxil fumarato
|
Fármaco coadministrado |
Dosis del fármaco coadministrado (mg) |
n |
Cambio % en los parámetros farmacocinéticos del fármaco coadministradoa (IC 90%) |
||
|
Cmáx |
ABC |
Cmín |
|||
|
Abacavir |
300 una vez |
8 |
↑12 (↓1 a ↑26) |
↔ |
NA |
|
Atazanavirb |
400 una vez al día x 14 días |
34 |
↓21 (↓27 a ↓14) |
↓25 (↓30 a ↓19) |
↓40 (↓48 a ↓32) |
|
Atazanavirb |
Atazanavir/ritonavir 300/100 una vez al día x 42 días |
10 |
↓28 (↓50 a ↑5) |
↓25c (↓42 a ↓3) |
↓23c (↓46 a ↑10) |
|
Efavirenz |
600 una vez al día x 14 días |
30 |
↔ |
↔ |
↔ |
|
Emtricitabina |
200 una vez al día x 7 días |
17 |
↔ |
↔ |
↑20 (↑12 a ↑29) |
|
Entecavir |
1 mg una vez al día x 10 días |
28 |
↔ |
↑13 (↑11 a ↑15) |
↔ |
|
Indinavir |
800 tres veces al día x 7 días |
12 |
↓11 (↓30 a ↑12) |
↔ |
↔ |
|
Lamivudina |
150 dos veces al día x 7 días |
15 |
↓24 (↓34 a ↑12) |
↔ |
↔ |
|
Lopinavir Ritonavir |
Lopinavir/ritonavir 400/100 dos veces al día x 14 días |
24 |
↔ |
↔ |
↔ |
|
↔ |
↔ |
↔ |
|||
|
Metadonad |
40-110 una vez al día x 14 díase |
13 |
↔ |
↔ |
↔ |
|
Nelfinavir Metabolito M8 |
1,250 dos veces al día x 14 días |
29 |
↔ |
↔ |
↔ |
|
↔ |
↔ |
↔ |
|||
|
Anticonceptivos oralesf |
Etinilestradiol/norgestimato (Ortho tricyclen) una vez al día x 7 días |
20 |
↔ |
↔ |
↔ |
|
Ribavirina |
600 una vez |
22 |
↔ |
↔ |
NA |
|
Saquinavir |
Saquinavir/ritonavir 1,000/100 dos veces al día x 14 días |
32 |
↑22 (↑6 a ↑41) |
↑29g (↑12 a ↑48) |
↑47g (↑23 a ↑76) |
|
Ritonavir |
↔ |
↔ |
↑23 (↑3 a ↑46) |
||
|
Tacrolimus |
0.05 mg/kg dos veces al día x 7 días |
21 |
↔ |
↔ |
↔ |
a Aumento = ↑; disminución= ↓; sin efecto= ↔; NA= no se aplica.
b Información para prescribir de Atazanavir.
c En sujetos infectados por el VIH, la adición de tenofovir DF a 300 mg de atazanavir más 100 mg de ritonavir provocó valores de ABC y Cmín del atazanavir que fueron 2.3 y 4 veces más altos que los respectivos valores observados al administrar dosis de 400 mg de atazanavir como monoterapia.
d Las exposiciones a la metadona R (activa), S y total fueron equivalentes cuando se administró sola o con Tenofovir disoproxil fumarato.
e Se mantuvo a cada sujeto en su dosis estable de metadona. No se notificaron alteraciones farmacodinámicas (toxicidad por opiáceos ni síntomas o signos de abstinencia).
f Las exposiciones al etinilestradiol y al 17-desacetil norgestimato (metabolito farmacológicamente activo) fueron equivalentes cuando se administraron solos o con Tenofovir disoproxil fumarato.
g No se prevé que los aumentos en el ABC y la Cmín sean clínicamente relevantes, por lo que no se requieren ajustes de la dosis para la administración concomitante de tenofovir DF y saquinavir reforzado con ritonavir.
La tabla 12 resume las interacciones medicamentosas entre Tenofovir disoproxil fumarato y la didanosina. La administración concomitante de Tenofovir disoproxil fumarato y didanosina debe realizarse con cautela.
Tras la administración de dosis múltiples de Tenofovir disoproxil fumarato junto con 400 mg de didanosina, la Cmáx y el ABC de la didanosina se incrementaron significativamente. Se desconoce el mecanismo de esta interacción.
Cuando se administraron 250 mg de didanosina en cápsulas con capa entérica junto con Tenofovir disoproxil fumarato, las exposiciones sistémicas a la didanosina fueron similares a las observadas tras la administración de las cápsulas de 400 mg con capa entérica solas, en condiciones de ayuno.
Tabla 12. Interacciones medicamentosas: parámetros farmacocinéticos de la didanosina en presencia de Tenofovir disoproxil fumarato
|
Dosis de didanosina (mg)/método de administración |
Método de administración de Tenofovir disoproxil fumaratoa |
n |
Diferencia % (IC 90%) frente a 400 mg de didanosina sola en ayunasb |
|
|
Cmáx |
ABC |
|||
|
Tabletas con buffer |
||||
|
400 una vez al díac x 7 días |
En ayunas 1 hora después de la didanosina |
14 |
↑28 (↑11 a ↑48) |
↑44 (↑31 a ↑59) |
|
Cápsulas con capa entérica |
||||
|
400 una vez, en ayunas |
Con alimentos, 2 h después de la didanosina |
26 |
↑48 (↑25 a ↑76) |
↑48 (↑31 a ↑67) |
|
400 una vez, con alimentos |
Simultáneamente con didanosina |
26 |
↑64 (↑41 a ↑89) |
↑60 (↑44 a ↑79) |
|
250 una vez, en ayunas |
Con alimentos, 2 h después de la didanosina |
28 |
↓10 (↓22 a ↑3) |
↔ |
|
250 una vez, en ayunas |
Simultáneamente con didanosina |
28 |
↔ |
↑14 (0 a ↑31) |
|
250 una vez, con alimentos |
Simultáneamente con didanosina |
28 |
↓29 (↓39 a ↓18) |
↓11 (↓23 a ↑2) |
a Administración junto con una comida ligera (~373 kcal, 20% de grasas).
b Aumento = ↑; disminución = ↓; sin efecto= ↔.
c Incluye a 4 sujetos con peso < 60 kg que recibieron 250 mg de ddl.
Didanosina: La administración concomitante de Tenofovir disoproxil fumarato y didanosina debe realizarse con precaución, y se debe monitorear rigurosamente a los pacientes que reciben esta combinación, a fin de detectar reacciones adversas asociadas a la didanosina. Se debe interrumpir la administración de didanosina en los pacientes que presenten reacciones adversas asociadas a este fármaco.
Cuando se administra junto con Tenofovir disoproxil fumarato, la Cmáx y el ABC de la didanosina (administrada en forma de tabletas con buffer o con capa entérica) aumentaron significativamente (véase la tabla 12). Se desconoce el mecanismo de esta interacción. En concentraciones más altas, la didanosina podría potenciar las reacciones adversas relacionadas con este medicamento, que incluyen pancreatitis y neuropatía.
Se ha observado la supresión de los conteos de células CD4+ en los pacientes que recibieron tenofovir disoproxil fumarato (tenofovir DF) con didanosina en dosis de 400 mg diarios.
En los pacientes que pesan > 60 kg, se debe reducir la dosis de didanosina a 250 mg cuando se administra concomitantemente con Tenofovir disoproxil fumarato. No hay datos disponibles que permitan recomendar un ajuste de la dosis de la didanosina en los pacientes adultos o pediátricos que pesan < 60 kg. Cuando se administran concomitantemente, Tenofovir disoproxil fumarato y la didanosina EC pueden tomarse en ayunas o con una comida ligera (< 400 kcal, 20% de grasas). La administración concomitante de la formulación de tabletas con buffer de didanosina junto con Tenofovir disoproxil fumarato se debe realizar en ayunas.
Atazanavir: Se ha demostrado que el atazanavir aumenta las concentraciones del tenofovir (véase la tabla 10). Se desconoce el mecanismo de esta interacción. Se debe monitorear a los pacientes que reciben atazanavir y Tenofovir disoproxil fumarato para detectar reacciones adversas asociadas a tenofovir disoproxil fumarato. Se debe interrumpir la administración de Tenofovir disoproxil fumarato en los pacientes que presenten reacciones adversas asociadas a este fármaco.
Tenofovir disoproxil fumarato disminuye el ABC y la Cmín. de atazanavir (véase la tabla 11). Cuando se administra concomitantemente con Tenofovir disoproxil fumarato, se recomienda administrar atazanavir en dosis de 300 mg con ritonavir en dosis de 100 mg. No debe administrarse atazanavir sin ritonavir de modo concomitante junto con Tenofovir disoproxil fumarato.
Lopinavir/ritonavir: Se ha demostrado que la combinación de lopinavir/ritonavir aumenta las concentraciones del tenofovir (véase la tabla 10). Se desconoce el mecanismo de esta interacción. Se debe monitorear a los pacientes que reciben lopinavir/ritonavir y Tenofovir disoproxil fumarato para detectar reacciones adversas asociadas a Tenofovir disoproxil fumarato. Se debe interrumpir la administración de Tenofovir disoproxil fumarato en los pacientes que presenten reacciones adversas asociadas a este fármaco.
Fármacos que afectan la función renal: Dado que el tenofovir se elimina principalmente por los riñones, la coadministración de Tenofovir disoproxil fumarato con fármacos que reducen la función renal o que compiten en el proceso de secreción tubular activa puede incrementar las concentraciones séricas del tenofovir y/o de otros fármacos que se eliminan por vía renal. Ejemplos de estos medicamentos, que no constituyen una lista exhaustiva, serían el cidofovir, el aciclovir, el valaciclovir, el ganciclovir y el valganciclovir. Los fármaco que disminuyen la función renal también pueden aumentar las concentraciones séricas del tenofovir.
En el tratamiento de la hepatitis B crónica, Tenofovir disoproxil fumarato no debe administrarse en combinación con dipivoxilo de adefovir.
ALTERACIONES EN LOS RESULTADOS DE PRUEBAS DE LABORATORIO:
Ensayos clínicos en pacientes adultos con infección por el VIH-1:
Pacientes sin tratamiento antirretroviral previo:
Estudio 903: Con excepción de las elevaciones en el colesterol y los triglicéridos en ayunas que fueron más frecuentes en el grupo de estavudina (40% y 9%) que en el de Tenofovir disoproxil fumarato (19% y 1%) respectivamente, las anomalías de laboratorio observadas en este estudio se produjeron con una frecuencia similar en los grupos tratados con Tenofovir disoproxil fumarato y con estavudina. En la tabla 13, se presenta un resumen de las anomalías de laboratorio de grados 3 y 4.
Tabla 13. Anomalías de laboratorio de grado 3/4 notificadas en ≥ 1% de los sujetos tratados con Tenofovir disoproxil fumarato en el estudio 903 (0-144 semanas)
|
Tenofovir disoproxil fumarato + 3TC + EFV |
d4T + 3TC + EFV |
|
|
n=299 |
n=301 |
|
|
Cualquier anomalía de laboratorio ≥ grado 3 |
36% |
42% |
|
Colesterol en ayunas (> 240 mg/dl) |
19% |
40% |
|
Creatina cinasa (V: > 990 U/I; M: > 845 U/I) |
12% |
12% |
|
Amilasa sérica (> 175 U/I) |
9% |
8% |
|
AST (V: > 180 U/I; M: > 170 U/I) |
5% |
7% |
|
ALT (V: > 215 U/I; M: > 170 U/I) |
4% |
5% |
|
Hematuria (> 100 hematíes/CGA) |
7% |
7% |
|
Neutrófilos (< 750/mm3) |
3% |
1% |
|
Triglicéricos en ayunas (> 750 mg/dl |
1% |
9% |
Estudio 934: Las anomalías de laboratorio observadas en este estudio concordaron en general con las anomalías observadas en estudios anteriores (tabla 14).
Tabla 14. Anomalías de laboratorio significativas notificadas en ≥ 1% de los sujetos de cualquier grupo de tratamiento del estudio 934 (0-144 semanas)
|
Tenofovir disoproxil fumaratoa + FTC + EFV |
AZT/3TC + EFV |
|
|
n=257 |
n=254 |
|
|
Cualquier anomalía de laboratorio ≥ grado 3 |
30% |
26% |
|
Colesterol en ayunas (> 240 mg/dl) |
22% |
24% |
|
Creatina cinasa (V: > 990 U/I; M: > 845 U/I) |
9% |
7% |
|
Amilasa sérica (> 175 U/I) |
8% |
4% |
|
Fosfatasa alcalina (> 550 U/I) |
1% |
0% |
|
AST (V: > 180 U/I; M: > 170 U/I) |
3% |
3% |
|
ALT (V: > 215 U/I; M: > 170 U/I) |
2% |
3% |
|
Hemoglobina (< 8.0 mg/dl) |
0% |
4% |
|
Hiperglucemia (> 250 mg/dl) |
2% |
1% |
|
Hematuria (> 75 hematíes/CGA) |
3% |
2% |
|
Glucosuria (≥ 3+) |
< 1% |
1% |
|
Neutrófilos (< 750/mm3) |
3% |
5% |
|
Triglicéridos en ayunas (> 750 mg/dl |
4% |
2% |
a Desde la semana 96 hasta la semana 144 del estudio, los sujetos recibieron emtricitabina + tenofovir disoproxiI fumarato con efavirenz, en lugar de Tenofovir disoproxil fumarato + Emtricitabina con efavirenz.
Pacientes con tratamiento antirretroviral previo:
Estudio 907: Las anomalías de laboratorio observadas en este estudio se produjeron con una frecuencia similar en los grupos tratados con Tenofovir disoproxil fumarato y con placebo. En la tabla 15, se presenta un resumen de las anomalías de laboratorio de grados 3 y 4.
Tabla 15. Anomalías de laboratorio de grado 3/4 notificadas en ≥ 1% de los sujetos tratados con Tenofovir disoproxil fumarato en el estudio 907 (0-48 semanas)
|
Tenofovir disoproxil fumarato (n=368) (semanas 0-24) |
Placebo (n=182) (semanas 0-24) |
Tenofovir disoproxil fumarato (n=368) (semanas 0-48) |
Placebo cruzado a Tenofovir disoproxil fumarato (n=170) (semanas 24-48) |
|
|
Cualquier anomalía de laboratorio ≥ grado 3 |
25% |
38% |
35% |
34% |
|
Triglicéridos (> 750 mg/dl) |
8% |
13% |
11% |
9% |
|
Creatina cinasa (V: > 990 U/I; M: > 845 U/I) |
7% |
14% |
12% |
12% |
|
Amilasa sérica (> 175 U/I) |
6% |
7% |
7% |
6% |
|
Glucosuria (=3+) |
3% |
3% |
3% |
2% |
|
AST (V: > 180 U/I; M: > 170 U/I) |
3% |
3% |
4% |
5% |
|
ALT (V: > 215 U/I; M: > 170 U/I) |
2% |
2% |
4% |
5% |
|
Glucosa sérica (> 250 U/I) |
2% |
4% |
3% |
3% |
|
Neutrófilos (< 750/mm3) |
1% |
1% |
2% |
1% |
Ensayos clínicos en sujetos adultos con hepatitis B crónica y enfermedad hepática compensada:
Estudio 0102: En la tabla 16, se presenta un resumen de las anomalías de laboratorio de grados 3 y 4 hasta la semana 48. Las anomalías de laboratorio de grado 3/4 fueron similares en los sujetos que continuaron el tratamiento con Tenofovir disoproxil fumarato durante un máximo de 144 semanas en estos estudios.
Tabla 16. Anomalías de laboratorio de grado 3/4 notificadas en= 1% de los sujetos tratados con Tenofovir disoproxil fumarato en los estudios 0102 y 0103 (0-48 semanas)
|
Tenofovir disoproxil fumarato (n=426) |
Adefovir dipodixil (n=215) |
|
|
Cualquier anomalía de laboratorio ≥ grado 3 |
19% |
13% |
|
Creatina cinasa (V: > 990 U/I; M: > 845 U/I) |
2% |
3% |
|
Amilasa sérica (> 175 U/I) |
4% |
1% |
|
Glucosuria (= 3+) |
3% |
< 1% |
|
AST (V: > 180 U/I; M: > 170 U/I) |
4% |
4% |
|
ALT (V: > 215 U/I; M: > 170 U/I) |
10% |
6% |
La incidencia general de exacerbaciones de la ALT (definidas como ALT sérica > 2 x valor basal y > 10 x LSN, con o sin síntomas asociados) durante el tratamiento fue similar entre Tenofovir disoproxil fumarato (2.6%) y Hepsera (2%). En general, las exacerbaciones de la ALT se produjeron dentro de las primeras 4 a 8 semanas de tratamiento y estuvieron acompañadas de disminuciones en los niveles de ADN del VHB. Ninguno de los sujetos presentó indicios de descompensación. Las exacerbaciones de la ALT se resolvieron habitualmente en el plazo de 4 a 8 semanas sin modificar la medicación del estudio.
PRECAUCIONES GENERALES:
Acidosis láctica/hepatomegalia severa con esteatosis: Se han reportado casos de acidosis láctica y hepatomegalia severa con esteatosis, incluso casos mortales, en pacientes que recibieron análogos nucleósidos, incluyendo Fumarato de disoproxilo de tenofovir, en combinación con otros antirretrovirales.
La mayoría de estos casos se produjeron en mujeres. La obesidad y la exposición prolongada a nucleósidos pueden ser factores de riesgo. Debe ejercerse particular precaución al prescribir análogos nucleósidos a pacientes con factores de riesgo identificados de hepatopatía, sin dejar de tomar en cuenta que se han reportado casos aun en personas sin ningún factor de riesgo identificado. Debe suspenderse el tratamiento con Tenofovir disoproxil fumarato en cualquier paciente que presente resultados clínicos o de laboratorio que sugieran acidosis láctica o hepatotoxicidad significativa (que puede incluir hepatomegalia y esteatosis aun sin elevaciones marcadas de las transaminasas).
Exacerbación de la hepatitis después de la suspensión del tratamiento: La suspensión del tratamiento anti-VHB, incluido Tenofovir disoproxil fumarato, puede estar asociada con exacerbaciones agudas severas de la hepatitis. Se debe realizar un monitoreo estricto de los pacientes infectados por el VHB que suspendan la administración de Tenofovir disoproxil fumarato con seguimiento clínico y de laboratorio durante al menos varios meses después de interrumpir el tratamiento. Si corresponde, puede estar justificada la reanudación del tratamiento contra la hepatitis B.
Nueva aparición o empeoramiento de la disfunción renal: El tenofovir se elimina principalmente por los riñones. Se han notificado casos de disfunción renal, entre los que se incluyen insuficiencia renal aguda y síndrome de Fanconi (lesión tubular renal con hipofosfatemia severa), asociados con el uso de Fumarato de disoproxilo de tenofovir (véase Reacciones secundarias y adversas). Se recomienda calcular la depuración de creatinina en todos los pacientes antes de iniciar la terapia y según se requiera clínicamente durante el tratamiento con Tenofovir disoproxil fumarato. Debe realizarse una vigilancia sistemática de la depuración de creatinina calculada y del fósforo sérico en los pacientes con riesgo de disfunción renal, incluidos los pacientes que han sufrido anteriormente eventos renales mientras recibían Adefovir dipidoxil.
Se recomienda el ajuste del intervalo de dosificación de Tenofovir disoproxil fumarato y la vigilancia estricta de la función renal en todos los pacientes con una depuración de creatinina <50 ml/min (véase Dosis y vía de administración). No hay información sobre la seguridad ni la eficacia en los pacientes con disfunción renal que hayan recibido Tenofovir disoproxil fumarato siguiendo estas pautas de dosificación, por lo que el beneficio potencial del tratamiento con Tenofovir disoproxil fumarato debe evaluarse en relación con el posible riesgo de toxicidad renal.
Se debe evitar administrar Tenofovir disoproxil fumarato con el uso reciente o concomitante de un medicamento nefrotóxico.
Pacientes con infección concomitante por el VIH-1 y el VHB:
Debido al riesgo de desarrollar resistencia al VIH-1, Tenofovir disoproxil fumarato debe utilizarse en pacientes con infección concomitante por el VIH-1 y el VHB únicamente como parte de un régimen combinado con antirretrovirales adecuado.
Antes de iniciar el tratamiento con Tenofovir disoproxil fumarato, deben ofrecerse pruebas de anticuerpos contra el VIH-1 a todos los pacientes infectados por el VHB. También antes de iniciar el tratamiento con Tenofovir disoproxil fumarato, se recomienda analizar la presencia de hepatitis B crónica en todos los pacientes infectados por el VIH-1.
Disminución de la densidad mineral ósea: Debe considerarse la evaluación de la densidad mineral ósea (DMO) en los adultos y los pacientes pediátricos de 12 años o más que tengan antecedentes de fracturas óseas patológicas u otros factores de riesgo de osteoporosis o pérdida de masa ósea. Si bien no se ha estudiado el efecto de los suplementos de calcio y vitamina D, dichos suplementos pueden ser beneficiosos para todos los pacientes. Se debe obtener asesoramiento adecuado si se sospecha de la presencia de anomalías óseas.
En los sujetos adultos infectados por el VIH-1 y tratados con Tenofovir disoproxil fumarato en el estudio 903 durante 144 semanas, se observó una disminución de la DMO con respecto al valor basal en la columna lumbar y en la cadera en ambos grupos del estudio. En la semana 144, hubo una disminución en el porcentaje medio significativamente mayor con respecto al valor basal de la DMO en la columna lumbar en los sujetos que recibieron Tenofovir disoproxil fumarato + lamivudina + efavirenz (-2.2% ± 3.9) en comparación con los sujetos que recibieron estavudina + lamivudina + efavirenz (-1.0% ± 4.6). Los cambios en la DMO a nivel de la cadera fueron similares en ambos grupos de tratamiento (-2.8% ± 3.5 en el grupo de Tenofovir disoproxil fumarato frente a -2.4% ± 4.5 en el grupo de estavudina). En ambos grupos, la mayor parte de la reducción de la DMO se produjo durante las primeras 24 a 48 semanas del estudio y se mantuvo hasta la semana 144. El porcentaje de sujetos que sufrieron una reducción de al menos 5% de la DMO en la columna o 7% de la DMO en la cadera fue de 28% en el grupo de Tenofovir disoproxil fumarato y de 21% en el grupo de estavudina.
Se reportaron fracturas clínicamente relevantes (excluidos los dedos de las manos y de los pies) en 4 sujetos del grupo de Tenofovir disoproxil fumarato y en 6 sujetos del grupo de estavudina. Además, hubo aumentos significativos en los niveles de marcadores bioquímicos del metabolismo óseo (fosfatasa alcalina ósea sérica, osteocalcina sérica, telopéptido C sérico y telopéptido N urinario) en el grupo de Tenofovir disoproxil fumarato con relación al grupo de estavudina, lo que sugiere mayor recambio óseo. Los niveles séricos de hormona paratiroidea y de vitamina D 1,25 también fueron más altos en el grupo que recibió Tenofovir disoproxil fumarato. Excepto por la fosfatasa alcalina ósea, estos cambios en los valores se mantuvieron dentro del intervalo normal.
En un estudio clínico en el que participaron sujetos pediátricos de 12 años o más que tenían infección por el VIH-1 (Estudio 321), los efectos óseos fueron similares a los de los sujetos adultos. En circunstancias normales, la DMO aumenta rápidamente en este grupo etario. En este estudio, la tasa media de ganancia ósea fue menor en el grupo tratado con Tenofovir disoproxil fumarato que en el grupo que recibió placebo. Seis sujetos tratados con Tenofovir disoproxil fumarato y un sujeto tratado con placebo presentaron una pérdida significativa (> 4%) de la DMO en la columna lumbar al cabo de 48 semanas.
Entre los 28 sujetos que recibieron Tenofovir disoproxil fumarato durante 96 semanas, las puntuaciones Z disminuyeron en -0.341 para la columna lumbar y -0.458 para el cuerpo completo. El crecimiento del esqueleto (estatura) no se vio afectado. Los marcadores de recambio óseo en sujetos pediátricos de 12 años o más que recibieron tratamiento con Tenofovir disoproxil fumarato indican un aumento del recambio óseo, lo cual concuerda con los efectos observados en adultos.
Se desconocen los efectos sobre la salud ósea a largo plazo y el riesgo futuro de fracturas que tienen los cambios en la DMO y en los marcadores bioquímicos asociados con el uso de Tenofovir disoproxil fumarato.
Se han notificado casos de osteomalacia (asociada a tubulopatía renal proximal y que puede contribuir a las fracturas) en relación con el uso de Tenofovir disoproxil fumarato (véase Experiencia posterior a la comercialización).
No se han estudiado los efectos óseos de Tenofovir disoproxil fumarato en pacientes con infección crónica por el VHB.
Redistribución de la grasa corporal:
En los pacientes infectados por el VIH, se han descrito eventos de redistribución/acumulación de grasa corporal, incluyendo obesidad central; acumulación de grasa dorsocervical (Joroba de búfalo), emaciación periférica o facial, agrandamiento de mamas y “aspecto cushingoide" en pacientes que recibieron tratamiento antirretroviral combinado. El mecanismo y las consecuencias a largo plazo de estos eventos se desconocen actualmente. No se ha establecido una relación causal.
Síndrome de reconstitución inmunológica:
Se han reportado casos de este síndrome en pacientes infectados por el VIH que reciben tratamiento antirretroviral combinado, incluido Tenofovir disoproxil fumarato. Durante la fase inicial del tratamiento antirretroviral combinado, los pacientes cuyos sistemas inmunitarios responden al tratamiento pueden desarrollar una respuesta inflamatoria a infecciones oportunistas residuales o indolentes (tales como la infección causada por Mycobacterium avium, el citomegalovirus, la neumonía por Pneumocystis jirovecii (PCP) o la tuberculosis), que pueden requerir evaluación y tratamiento adicionales. Falla virológica temprana: Los estudios clínicos en sujetos infectados por el VIH han demostrado que algunos regímenes que contienen solamente tres inhibidores nucleósidos de la transcriptasa reversa (ITRAN) son generalmente menos eficaces que los regímenes de tres fármacos que contienen dos ITRAN en combinación con un inhibidor no nucleósido de la transcriptasa reversa o un inhibidor de la proteasa del VIH-1. En particular, se han notificado falla virológica temprana y tasas altas de sustituciones de resistencia. Por lo tanto, los regímenes de tres nucleósidos deben emplearse con precaución. Se debe monitorear atentamente a los pacientes que reciban un régimen únicamente con tres nucleósidos y debe considerarse la modificación de su tratamiento.
Toxicología y/o farmacología en animales: El tenofovir y el tenofovir disoproxil fumarato administrados a ratas, perros y monos en estudios toxicológicos con exposiciones (según los valores de ABC) mayores o iguales a 6 veces las observadas en los seres humanos ocasionaron toxicidad ósea. En los monos, la toxicidad ósea se diagnosticó como osteomalacia. La osteomalacia que se observó en los monos mostró ser reversible al reducir la dosis o al suspender el uso de tenofovir. En ratas y perros, la toxicidad ósea se manifestó como densidad mineral ósea reducida. Se desconocen los mecanismos subyacentes en la toxicidad ósea.
En 4 especies animales se observó evidencia de toxicidad renal. En diferentes grados, se observaron incrementos de creatinina sérica, BUN (nitrógeno ureico en sangre), glucosuria, proteinuria, fosfaturia y/o calciuria, además de disminuciones del fosfato sérico en estos animales. Estas toxicidades se observaron en exposiciones (según los valores de ABC) 2-20 veces superiores a las observadas en humanos. Se desconoce la relación de las anomalías renales en particular la de fosfaturia con la toxicidad ósea.
Uso pediátrico:
Pacientes pediátricos de 12 años o más: La seguridad de Tenofovir disoproxil fumarato en pacientes pediátricos de 12 a <18 años está respaldada por los datos de un estudio aleatorizado en el que se administró Tenofovir disoproxil fumarato a sujetos infectados por el VIH-1 que habían recibido tratamiento previo. En este estudio, el perfil farmacocinético de Tenofovir disoproxil fumarato fue similar al perfil cuya seguridad y eficacia se demostró en ensayos clínicos realizados en adultos.
En el estudio 321, 87 sujetos de 12 a < 18 años con tratamiento previo recibieron Tenofovir disoproxjl fumarato (n = 45) o placebo (n = 42) en combinación con un régimen de fondo optimizado durante 48 semanas. La media basal del conteo de células CD4 fue de 374 células/mm3 y la media basal de ARN- VIH-1 plasmático fue de 4.6 log10 copias/ml. Al inicio, 90% de los sujetos presentaron sustituciones asociadas con la resistencia a los INTR en sus aislados de VIH-1. En general, el ensayo no mostró ninguna diferencia en la respuesta virológica entre los grupos de tratamiento con Tenofovir disoproxil fumarato y con placebo. Los análisis de subgrupos sugieren que la ausencia de una diferencia en la respuesta virológica podría atribuirse a desequilibrios entre los grupos de tratamiento en cuanto a la susceptibilidad viral basal a Tenofovir disoproxil fumarato y al régimen de fondo optimizado.
Aunque los cambios en el ARN- VIH-1 en estos sujetos con alto grado de administración de tratamientos previos fueron menores a los esperados, la comparabilidad de los datos farmacocinéticos y de seguridad respecto de los observados en adultos apoya el uso de Tenofovir disoproxil fumarato en pacientes pediátricos = 12 años que pesan = 35 kg y cuyos aislados de VIH-1 se prevé que sean sensibles a Tenofovir disoproxil fumarato (véanse Precauciones generales, Reacciones secundarias y adversas, y Farmacología clínica).
No se han establecido la seguridad ni la eficacia en pacientes pediátricos menores de 12 años.
Uso geriátrico:
Los estudios clínicos con Tenofovir disoproxil fumarato no incluyeron un número suficiente de sujetos de 65 años o más como para poder determinar si tienen respuestas al medicamento diferentes a las de sujetos más jóvenes. En general, la selección de la dosis para los pacientes de edad avanzada debe efectuarse con precaución, tomando en cuenta la mayor frecuencia de disminución de las funciones hepática, renal o cardiaca, así como las enfermedades concomitantes u otros tratamientos farmacológicos.
Pacientes con disfunción renal:
Se recomienda que se modifique el intervalo de dosificación de Tenofovir disoproxil fumarato en pacientes con depuración de creatinina < 50 ml/min o en pacientes con ERFT que requieran diálisis (véanse Dosis y vía de administración, Farmacología clínica).
Información para los pacientes:
Se debe informar a los pacientes de lo siguiente:
• Tenofovir disoproxil fumarato no cura la infección por el VIH-1 y es posible que los pacientes continúen presentando enfermedades asociadas con esta infección, incluidas infecciones oportunistas. Los pacientes deben permanecer bajo el cuidado de un médico cuando utilizan Tenofovir disoproxil fumarato.
• No se ha comprobado que el uso de Tenofovir disoproxil fumarato reduzca el riesgo de transmisión de la infección por el VIH-1 o el VHB a otras personas por medio del contacto sexual o de la contaminación con sangre. Se debe aconsejar a los pacientes que tomen mayores recaudos de seguridad al tener relaciones sexuales, y que usen preservativos de látex o poliuretano para reducir la probabilidad del contacto sexual con cualquier líquido corporal, como el semen, las secreciones vaginales o la sangre. Se debe aconsejar a los pacientes que nunca reutilicen ni compartan las agujas.
• Se desconocen los efectos a largo plazo de Tenofovir disoproxil fumarato.
• Las tabletas de Tenofovir disoproxil fumarato son sólo para administración por vía oral.
• No debe suspenderse la administración de Tenofovir disoproxil fumarato sin informar primero al médico.
• Si tiene una infección por el VIH-1, con o sin infección concomitante por el VHB, es importante que tome Tenofovir disoproxil fumarato con un tratamiento combinado.
• Es importante tomar Tenofovir disoproxil fumarato siguiendo un esquema de dosificación regular y evitar omitir dosis.
• Se ha notificado la aparición de acidosis láctica y hepatomegalia severa con esteatosis, incluso casos mortales. El tratamiento con Tenofovir disoproxil fumarato debe suspenderse en cualquier paciente que presente síntomas clínicos que indiquen la presencia de acidosis láctica o de hepatotoxicidad pronunciada (incluso náuseas, vómitos, molestias gástricas poco habituales o inesperadas y debilidad) (véase Precauciones generales).
• Los pacientes con VIH-1 deben realizarse una prueba para detectar la presencia del virus de la hepatitis B (VHB) antes de iniciar el tratamiento antirretroviral (véase Precauciones generales).
• Se han notificado exacerbaciones agudas severas de la hepatitis en pacientes infectados con el VHB o coinfectados con el VHB y el VIH-1 que suspendieron la administración de Tenofovir disoproxil fumarato (véase Precauciones generales).
• En pacientes con hepatitis B crónica, es importante obtener un análisis de los anticuerpos contra el VIH antes de iniciar la administración de Tenofovir disoproxil fumarato (véase Precauciones generales).
• Se han notificado casos de disfunción renal, entre los que se incluyen insuficiencia renal aguda y síndrome de Fanconi. Se debe evitar administrar Tenofovir disoproxil fumarato con el uso reciente o concomitante de un medicamento nefrotóxico (véase Precauciones generales). Puede ser necesario ajustar el intervalo de dosificación de Tenofovir disoproxil fumarato en los pacientes con disfunción renal (véase Dosis y vía de administración).
• Tenofovir disoproxil fumarato no debe administrarse concomitantemente con los productos de dosis fijas combinadas emtricitabina + tenofovir disoproxil fumarato o efavirenz + emtricitabina + tenofovir disoproxil fumarato, ya que es un componente de estos productos (véase Precauciones generales).
• Tenofovir disoproxil fumarato no debe administrarse en combinación con avefovir dipidoxil (véase Precauciones generales).
• Se ha observado disminución de la densidad mineral ósea con el uso de Tenofovir disoproxil fumarato en los pacientes infectados por el VIH. Se debe plantear la vigilancia de la densidad mineral ósea en los pacientes que tengan antecedentes de fracturas óseas patológicas o con riesgo de osteopenia (véase Precauciones generales).
• Se desconoce la duración óptima del tratamiento para la hepatitis B crónica. Tampoco se conoce la relación entre la respuesta y la prevención a largo plazo de los desenlaces como el carcinoma hepatocelular.
DOSIS Y VÍA DE ADMINISTRACIÓN:
Vía de administración: Oral.
Dosis recomendada en adultos:
Para el tratamiento del VIH-1 o la hepatitis B crónica: La dosis es una tableta de 300 mg de Tenofovir disoproxil fumarato administrada por vía oral una vez al día con o sin alimentos.
Se desconoce la duración óptima del tratamiento para la hepatitis B crónica.
Dosis recomendada en pacientes pediátricos(= 12 años y = 35 kg):
Para el tratamiento del VIH-1 en pacientes pediátricos de 12 años o más con un peso corporal ≥ 35 kg (≥ 77 lb): La dosis es una tableta de 300 mg de Tenofovir disoproxil fumarato administrado por vía oral una vez al día con o sin alimentos.
Ajuste de la dosis en caso de disfunción renal en adultos: Se produjo un aumento significativo de las exposiciones al fármaco cuando se administró Tenofovir disoproxil fumarato a sujetos con disfunción renal de moderada a severa (véase Interacciones medicamentosas y de otro género). Por lo tanto, se debe ajustar el intervalo de dosificación de Tenofovir disoproxil fumarato a los pacientes con depuración de creatinina basal < 50 ml/min, utilizando las recomendaciones que figuran en la tabla 17. Estas recomendaciones de intervalos de dosificación se basan en modelos de datos farmacocinéticos de dosis únicas en sujetos no infectados por el VIH ni por el VHB con diferentes grados de disfunción renal, incluso enfermedad renal terminal que requiere hemodiálisis.
No se han evaluado clínicamente la seguridad ni la eficacia de estas recomendaciones de ajuste de intervalos de dosificación en pacientes con disfunción renal moderada o severa; por lo tanto, se deben monitorear rigurosamente la respuesta clínica al tratamiento y la función renal en estos pacientes (véase Precauciones generales).
No es necesario ajustar la dosis para los pacientes con disfunción renal leve (depuración de creatinina de 50 a 80 ml/min). Debe realizarse una vigilancia sistemática de la depuración de creatinina calculada y del fósforo sérico en todos los pacientes con disfunción renal leve (véase Precauciones generales).
Tabla 17. Ajuste de la dosificación para pacientes con alteración en la depuración de creatinina
|
Depuración de creatinina (ml/min)a |
Pacientes con hemodiálisis |
|||
|
≥ 50 |
30-49 |
10-29 |
||
|
Intervalo de dosificación recomendado de 300 mg |
Cada 24 horas |
Cada 48 horas |
Cada 72 a 96 horas |
Cada 7 días o después de un total de aproximadamente 12 horas de diálisisb |
a Se calculó con el peso corporal ideal (magro).
b Por lo general, una vez a la semana, suponiendo tres sesiones de hemodiálisis a la semana, de aproximadamente 4 horas de duración. Tenofovir disoproxil fumarato debe administrarse después de terminar la diálisis.
No se ha evaluado la farmacocinética del tenofovir en pacientes con depuración de creatinina < 10 ml/min sin hemodiálisis; por consiguiente, no hay disponible ninguna recomendación de dosis para estos pacientes.
No hay datos disponibles que permitan hacer recomendaciones sobre la dosis a utilizar en pacientes pediátricos de 12 años o más con disfunción renal.
MANIFESTACIONES Y MANEJO DE LA SOBREDOSIFICACIÓN O INGESTA ACCIDENTAL:
Sobredosis: Se dispone de experiencia clínica limitada con dosis superiores a la dosis terapéutica de 300 mg de Tenofovir disoproxil fumarato. En el estudio 901, se administró tenofovir disoproxil fumarato en dosis de 600 mg por vía oral a 8 sujetos durante 28 días. No se reportaron reacciones adversas severas. Se desconocen los efectos de dosis más altas.
En caso de sobredosis, se debe monitorear al paciente para detectar si hay indicios de toxicidad, y se debe aplicar el tratamiento de apoyo estándar, según sea necesario.
El tenofovir se elimina eficientemente mediante hemodiálisis, con un coeficiente de extracción de aproximadamente 54%. Después de una dosis única de 300 mg de Tenofovir disoproxil fumarato, una sesión de hemodiálisis de cuatro horas eliminó aproximadamente 10% de la dosis de tenofovir administrada.
PRESENTACIÓN:
Caja con frasco con 30 tabletas de 300 mg e instructivo anexo.
RECOMENDACIONES SOBRE ALMACENAMIENTO:
Consérvese el frasco bien cerrado, a no más de 25ºC.
LEYENDAS DE PROTECCIÓN:
Literatura exclusiva para médicos. Léase instructivo anexo. Su venta requiere receta médica. No se deje al alcance de los niños. No se use en el embarazo, la lactancia ni en menores de 12 años. Prohibida la venta fraccionada del producto. Contiene un desecante NO INGERIBLE, consérvese dentro del envase. Este medicamento contiene Azul No. 1, que puede producir reacciones de hipersensibilidad.
Reporte las sospechas de reacción adversa a los correos: farmacovigilancia@cofepris.gob.mx y
farmacovigilancia@heterodrugs.com
Hecho y Acondicionado en India por:
Hetero Labs Limited
Unit-III, Formulation Plot nº 22-110, IDA,
Jeedimetla, Hyderabad, 500 055,
Telangana, India.
Representante Legal e Importador en México:
AMAROX PHARMA, S.A. de C.V.
Av. de los 50 Metros No. 402,
Col. CIVAC, C.P. 62578,
Jiutepec, Morelos, México.
Reg. Núm. 030M2019, SSA IV


