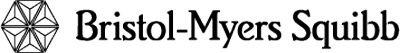
SPRYCEL
DASATINIB
Tabletas
Frasco(s),30 Tabletas,100 mg
Frasco(s),60 Tabletas,20 mg
Frasco(s),60 Tabletas,50 mg
Frasco(s),60 Tabletas,70 mg
FORMA FARMACÉUTICA Y FORMULACIÓN:
Cada TABLETA contiene:
Dasatinib 20, 50, 70 y 100 mg
Excipiente cbp 1 tableta
INDICACIONES TERAPÉUTICAS: SPRYCEL® está indicado para el tratamiento de adultos con leucemia mieloide crónica (LMC) en fase crónica recientemente diagnosticada.
SPRYCEL® está indicado para el tratamiento de adultos con leucemia mieloide crónica (LMC) en fase crónica, acelerada o blástica mieloide o linfoide con resistencia o intolerancia a la terapia previa (incluyendo imatinib).
SPRYCEL® también está indicado para el tratamiento de adultos con leucemia linfoblástica aguda positiva al cromosoma Filadelfia (LLA Ph+) con resistencia o intolerancia a la terapia previa.
FARMACOCINÉTICA Y FARMACODINAMIA:
Farmacodinamia: Dasatinib inhibe la actividad de la cinasa BCR-ABL y las cinasas de la familia SRC, además de otras cinasas oncogénicas incluyendo c-KIT, cinasas Efrina, y receptor PDGFβ.
Dasatinib es un potente, inhibidor subnanomolar de la cinasa BCR-ABL con potencia a una concentración de 0.6-0.8 nM. Se une tanto a la conformación inactiva y activa de la enzima BCR-ABL.
Mecanismo de acción: In vitro, dasatinib es activo en líneas celulares leucémicas que representan variantes de enfermedad sensible y resistente a imatinib. Estos estudios no clínicos demuestran que dasatinib puede superar la resistencia a imatinib resultante de la sobreexpresión BCR-ABL, mutaciones del dominio de la cinasa BCR-ABL, activación de vías alternas de señalamiento que involucran a las cinasas de la familia SRC (LYN, HCK), y sobreexpresión génica de resistencia a múltiples drogas. Adicionalmente, dasatinib inhibe las cinasas de la familia SRC a una concentración subnanomolar.
In vivo, en experimentos separados utilizando modelos murinos de LMC, dasatinib previno la progresión de LMC en fase crónica a fase blástica y prolongó la supervivencia de ratones portadores de líneas celulares derivadas de LMC en varios sitios, incluyendo el SNC.
Farmacocinética: La farmacocinética de dasatinib se ha evaluado en 235 sujetos adultos sanos y en 84 pacientes con leucemia.
Absorción: Dasatinib se absorbe rápidamente en pacientes después de su administración oral. Las concentraciones pico se observaron entre 0.5 y 6 horas. Después de su administración oral, el aumento en la exposición media (ABCτ) es aproximadamente proporcional al incremento en la dosis variando entre 15 mg a 240 mg una vez al día. La vida media terminal general de dasatinib es aproximadamente 3 a 5 horas en pacientes.
Datos de sujetos sanos tratados con una dosis única de 100 mg de dasatinib 30 minutos después del consumo de un alimento rico en grasas, indicaron un aumento promedio de 14% en el ABC de dasatinib. El consumo de un alimento bajo en grasas 30 minutos antes de dasatinib resultó en un aumento promedio de 21% en el ABC de dasatinib. Los efectos observados de los alimentos no fueron clínicamente relevantes.
Distribución: El volumen de distribución aparente de dasatinib en los pacientes es de 2505 L, lo cual sugiere que el fármaco presenta una extensa distribución en el espacio extravascular. A concentraciones clínicamente relevantes de dasatinib la unión a proteínas plasmáticas in vitro fue de aproximadamente 96%.
Metabolismo: Dasatinib sufre un extenso metabolismo en los seres humanos mediante múltiples enzimas involucradas en la generación de metabolitos. La enzima CYP3A4 fue la principal enzima responsable del metabolismo de dasatinib. En sujetos sanos que recibieron 100 mg de dasatinib marcado con [14C], dasatinib inalterado representó 29% de la radiactividad circulante en plasma. La concentración plasmática y actividad in vitro medida indican que es improbable que los metabolitos de dasatinib desempeñen un papel importante en la farmacología observada del producto.
Eliminación: La eliminación tiene lugar preponderantemente a través de las heces principalmente como metabolitos. Luego de la administración de una dosis oral única de dasatinib marcado con [14C], aproximadamente el 89% de la dosis se eliminó en los siguientes 10 días, recuperándose el 4% y 85% de la radioactividad en la orina y en las heces, respectivamente. Dasatinib intacto recuperado en la orina y en las heces representó un 0.1% y un 19% de la dosis administrada respectivamente, recuperándose el resto de la dosis en forma de metabolitos.
Poblaciones especiales:
Género: Análisis farmacocinéticos de los datos demográficos indican que no existen efectos clínicamente relevantes del género sobre la farmacocinética de SPRYCEL®.
Pacientes geriátricos: Análisis farmacocinéticos de los datos demográficos indican que no existe ningún efecto clínicamente relevante de la edad sobre la farmacocinética de SPRYCEL®.
Insuficiencia hepática: El efecto de insuficiencia hepática en la farmacocinética de dosis únicas de dasatinib se evaluó en 8 sujetos con una insuficiencia hepática moderada que recibieron una dosis de 50 mg y 5 sujetos con insuficiencia hepática severa que recibieron una dosis de 20 mg, en comparación con sujetos sanos con características similares que recibieron una dosis de 70 mg de dasatinib. La media de Cmáx y ABC de dasatinib ajustada para la dosis de 70 mg redujo en 47% y 8%, respectivamente, en sujetos con insuficiencia hepática moderada en comparación con sujetos con función hepática normal. En sujetos con insuficiencia hepática severa, la media de Cmáx y ABC ajustada para la dosis de 70 mg redujo en 43% y 28%, respectivamente, en comparación con los sujetos con función hepática normal. (Ver Dosis y vía de administración, Deterioro hepático).
Pacientes pediátricos: La farmacocinética de SPRYCEL® no se ha evaluado en pacientes pediátricos.
Insuficiencia renal: No existen estudios clínicos de SPRYCEL® en pacientes con deterioro de la función renal. Menos del 4% de dasatinib y sus metabolitos son eliminados vía renal. (Ver Dosis y vía de administración, Deterioro renal).
Estudios clínicos:
LMC en Fase Crónica Recientemente Diagnosticada: Se realizó un estudio abierto, multicéntrico, internacional, aleatorizado, Fase III, en pacientes adultos con LMC en fase crónica recientemente diagnosticada. Los pacientes fueron aleatorizados para recibir 100 mg de SPRYCEL® una vez al día o 400 mg de imatinib una vez al día. El criterio primario de valoración fue la tasa de respuesta citogenética completa confirmada (cCCyR) a los 12 meses. Los criterios secundarios de valoración incluyeron tiempo en cCCyR (medida de durabilidad de la respuesta), tiempo hasta cCCyR, tasa de respuesta molecular mayor (MMR), tiempo hasta MMR, supervivencia libre de progresión (PFS), y supervivencia global (OS). Otros resultados relevantes de eficacia incluyeron tasas de CCyR y de respuesta molecular completa (CMR).
Se aleatorizaron un total de 519 pacientes a un grupo de tratamiento: 259 a SPRYCEL® y 260 a imatinib. Las características basales estaban bien balanceadas entre los dos grupos de tratamiento con respecto a edad (la mediana de edad fue de 46 años para el grupo de SPRYCEL® y de 49 años para el grupo de imatinib, con 10% y 11% de los pacientes de 65 años de edad o mayores respectivamente), género (mujeres 44% y 37%, respectivamente) y raza (Caucásicos 51% y 55%; Asiáticos 42% y 37%, respectivamente). En la basal, la distribución de las Puntuaciones de Hasford fue similar en los grupos de tratamiento de SPRYCEL® y de imatinib (riesgo bajo: 33% y 34%; riesgo intermedio 48% y 47%; alto riesgo: 19% y 19%, respectivamente). La Puntuación de Desempeño ECOG también fue similar en los grupos de tratamiento de SPRYCEL® e imatinib (ECOG 0 = 82% y 79%; ECOG 1 = 18% y 20%; y ECOG 2 = 0 y 1%, respectivamente).
Con un mínimo de 12 meses de seguimiento, 85% de los pacientes aleatorizados al grupo de SPRYCEL® y 81% de los pacientes aleatorizados al grupo de imatinib aún estaban recibiendo tratamiento de primera línea. La suspensión por progresión de la enfermedad ocurrió en 3% de los pacientes tratados con SPRYCEL® y en 5% de los pacientes tratados con imatinib. Con un seguimiento mínimo de 36 meses, 71% de los pacientes del grupo asignado a SPRYCEL® y 69% de los pacientes del grupo asignado a imatinib estuvieron recibiendo tratamiento de primera línea. La descontinuación debida a progresión de la enfermedad ocurrió en 7% de los pacientes tratados con SPRYCEL® y 7% de los pacientes tratados con imatinib.
En la Tabla 1 se presentan los resultados de eficacia. Una proporción estadísticamente significativa mayor de los pacientes tratados con SPRYCEL® lograron una cCCyR en comparación con los pacientes del grupo de imatinib en los primeros 12 meses de tratamiento. Se demostró de manera consistente la eficacia de SPRYCEL® entre los diferentes subgrupos, incluyendo edad, género, y puntuación basal de Hasford.
Tabla 1. Resultados de eficacia en un estudio Fase III de pacientes con LMC
en fase crónica recientemente diagnosticada
|
SPRYCEL® n = 259 |
Imatinib n = 260 |
Valor-P |
|
|
Tasa de respuesta (95% CI) |
|||
|
Respuesta citogenética |
|||
|
a los 12 meses |
|||
|
cCCyRa |
76.8% (71.2-81.8) |
66.2% (60.1-71.9) |
p < 0.007* |
|
CCyRb |
85.3% (80.4-89.4) |
73.5% (67.7-78.7) |
– |
|
a los 24 meses |
|||
|
cCCyRa |
80.3% |
74.2% |
– |
|
CCyRb |
87.3% |
82.3% |
– |
|
a los 36 meses |
|||
|
cCCyRa |
82.6% |
77.3% |
– |
|
CCyRb |
88.0% |
83.5% |
– |
|
a los 48 meses |
|||
|
cCCyRa |
82.6% |
78.5% |
– |
|
CCyRb |
87.6% |
83.8% |
– |
|
a los 60 meses |
|||
|
cCCyRa |
83.0% |
78.5% |
– |
|
CCyRb |
88.0% |
83.8% |
– |
|
Respuesta molecular mayorc |
|||
|
a los 12 meses |
52.1% (45.9-58.3) |
33.8% (28.1-39.9) |
p < 0.00003* |
|
a los 24 meses |
64.5% (58.3-70.3) |
50% (43.8-56.2) |
– |
|
a los 36 meses |
69.1% (63.1-74.7) |
56.2% (49.9-62.3) |
– |
|
a los 48 meses |
75.7% (70.0-80.8) |
62.7% (56.5-68.6) |
– |
|
a los 60 meses |
76.4% (70.8-81.5) |
64.2% (58.1-70.1) |
p = 0.0021 |
|
Cociente de riesgo |
|||
|
a los 12 meses (99.99% CI) |
|||
|
Tiempo hasta cCCyR |
1.55 (1.0-2.3) |
p < 0.0001* |
|
|
Tiempo hasta MMR |
2.01 (1.2-3.4) |
p < 0.0001* |
|
|
Durabilidad de cCCyR |
0.7 (0.4-1.4) |
p < 0.035 |
|
|
a los 24 meses (95% CI) |
|||
|
Tiempo hasta cCCyR |
1.49 (1.22-1.82) |
– |
|
|
Tiempo hasta MMR |
1.69 (1.34-2.12) |
– |
|
|
Durabilidad de cCCyR |
0.77 (0.55-1.10) |
– |
|
|
a los 36 meses (95% CI) |
|||
|
Tiempo hasta cCCyR |
1.48 (1.22-1.80) |
– |
|
|
Tiempo hasta MMR |
1.59 (1.28-1.99) |
– |
|
|
Durabilidad de cCCyR |
0.77 (0.53-1.11) |
– |
|
|
a los 48 meses (95% CI) |
|||
|
Tiempo hasta cCCyR |
1.45 (1.20-1.77) |
– |
|
|
Tiempo hasta MMR |
1.55 (1.26-1.91) |
– |
|
|
Durabilidad de cCCyR |
0.81 (0.56-1.17) |
– |
|
|
a los 60 meses (95% CI) |
|||
|
Tiempo hasta cCCyR |
1.46 (1.20-1.77) |
p = 0.0001 |
|
|
Tiempo hasta MMR |
1.54 (1.25-1.89) |
p < 0.0001 |
|
|
Durabilidad de cCCyR |
0.79 (0.55-1.13) |
p = 0.1983 |
|
a La respuesta citogenética completa confirmada (cCCyR) se define como la respuesta observada en dos ocasiones consecutivas (al menos con 28 días de diferencia).
b La CCyR no confirmada se basa en una sola evaluación citogenética de médula ósea.
c La respuesta molecular mayor (en cualquier momento) se definió como cocientes de BCR-ABL ≤ 0.1% mediante RQ-PCR en muestras de sangre periférica estandarizadas en base a la escala internacional. Estas tasas acumuladas están representando un seguimiento mínimo del periodo especificado.
* Ajustado para puntuación de Hasford e indicó significancia estadística al nivel nominal pre-definido de significancia.
IC = intervalo de confianza.
Después de 60 meses de seguimiento la mediana de tiempo hasta cCCyR fue de 3.1 meses en 214 pacientes con respuesta a SPRYCEL® y de 5.8 meses en 204 pacientes con respuesta a imatinib. La mediana de tiempo hasta MMR después de 60 meses de seguimiento fue de 9.3 meses en 196 pacientes con respuesta a SPRYCEL® y de 15.0 meses en 163 pacientes con respuesta a imatinib. Las tasas de cCCyR en los grupos de tratamiento de SPRYCEL® y de imatinib, respectivamente, a los 3 meses (54% y 30%), 6 meses (70% y 56%), 9 meses (75% y 63%), 24 meses (80% y 74%), 36 meses (83% y 77%), 48 meses (83% y 79%) y 60 meses (83% y 79%) fueron consistentes con el criterio primario de valoración. Las tasas de MMR en los grupos de tratamiento de SPRYCEL® e imatinib, respectivamente, a los 3 meses (8% y 0.4%), 6 meses (27% y 8%), 9 meses (39% y 18%), 12 meses (46% y 28%), 24 meses (64% y 46%) y 36 meses (67% y 55%), 48 meses (73% y 60%) y 60 meses (76% y 64%) también fueron consistentes con el criterio primario de valoración. Con un mínimo de 60 meses de seguimiento, la tasa de CMR (esto es, una reducción de al menos 4.5 log desde el valor basal estandarizado del cociente BCR-ABL ≤ 0.0032%) en cualquier momento fue de 44% y de 34% en los grupos de tratamiento de SPRYCEL® e imatinib, respectivamente.
Las tasas de MMR en cualquier momento, en cada grupo de riesgo determinado por la puntuación Hasford, fue mayor en el grupo de SPRYCEL® en comparación con el grupo de imatinib (riesgo bajo: 90% y 69%; riesgo intermedio: 71% y 65%; riesgo alto: 67% y 54%, respectivamente).
Adicionalmente, más sujetos tratados con SPRYCEL® (84%) alcanzaron respuesta molecular temprana (definida como niveles de BCR-ABL ≤ 10% a 3 meses) comparados con sujetos tratados con imatinib (64%). Los sujetos en ambos brazos de estudio que alcanzaron respuesta molecular temprana tuvieron un menor riesgo de transformación, mayor tasa de supervivencia libre de progresión (PFS) y mayor tasa de supervivencia global (OS), como se muesta en las Tablas 2 y 3.
Tabla 2. Sujetos tratados con SPRYCEL®
con BCR-ABL ≤ 10% y > 10% a 3 meses
|
N = 235 |
Sujetos con BCR-ABL ≤ 10% a 3 meses |
Sujetos con BCR-ABL > 10% a 3 meses |
|
Número de sujetos (%) |
198 (84.3) |
37 (15.7) |
|
Transformación a 60 meses, n/N (%) |
6/198 (3.0) |
5/37 (13.5) |
|
Tasa de PFS a 60 meses (95% IC) |
92.0% (89.6, 95.2) |
73.8% (52.0, 86.8) |
|
Tasa de OS a 60 meses (95% IC) |
93.8% (89.3, 96.4) |
80.6% (63.5, 90.2) |
Tabla 3. Sujetos tratados con imatinib
con BCR-ABL ≤ 10% y > 10% a 3 meses
|
N = 235 |
Sujetos con BCR-ABL ≤ 10% a 3 meses |
Sujetos con BCR-ABL > 10% a 3 meses |
|
Número de sujetos (%) |
154 (64.4) |
85 (35.6) |
|
Transformación a 60 meses, n/N (%) |
5/154 (3.2) |
13/85 (15.3) |
|
Tasa de PFS a 60 meses (95% IC) |
93.5% (87.8, 96.6) |
79.3% (67.3, 87.3) |
|
Tasa de OS a 60 meses (95% IC) |
95.4% (90.5, 97.8) |
80.5% (70.1, 87.6) |
La tasa de supervivencia libre de progresión (PFS) por tiempo específico se muestra gráficamente en la Figura 1. La tasa de PFS fue consistentemente mayor en los pacientes tratados con SPRYCEL® que alcanzaron un nivel de BCR-ABL ≤ 10% a los 3 meses, que en aquellos que no lo hicieron.
Figura 1. Gráfica para la supervivencia libre de progresión para SPRYCEL® por nivel de BCR-ABL (≤ 10% o > 10% a los 3 meses en un estudio fase III de pacientes con LMC en fase crónica recientemente diagnosticada.
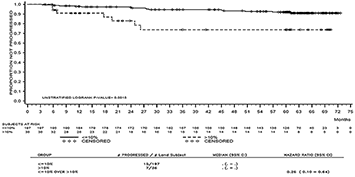
La tasa de supervivencia global (OS) por tiempo específico se muestra gráficamente en la Figura 2. La tasa de OS fue consistentemente mayor en los pacientes tratados con SPRYCEL® que alcanzaron un nivel de BCR-ABL ≤ 10% a los 3 meses, que en aquellos que no lo hicieron.
Figura 2. Gráfica para supervivencia global de SPRYCEL® por nivel de BCR-ABL (≤ 10% o > 10%) a los 3 meses en un estudio fase III de pacientes con LMC en fase crónica recientemente diagnosticada.
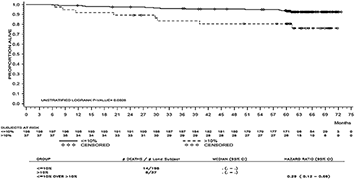
El tiempo hasta MMR se muestra gráficamente en la Figura 3. El tiempo hasta MMR fue consistentemente menor en los sujetos tratados con SPRYCEL® en comparación con los sujetos tratados con imatinib.
Figura 3. Tiempo hasta la respuesta molecular mayor (MMR) estimado por Kaplan-Meier, en un estudio fase III de pacientes con LMC en fase crónica recientemente diagnosticada.
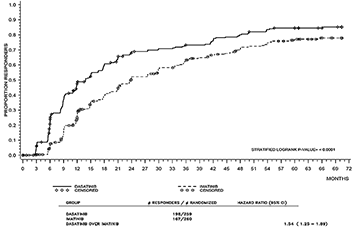
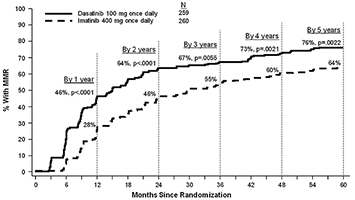
Las tasas de MMR por tiempo específico se muestran gráficamente en la Figura 4. Las tasas de MMR fueron consistentemente mayores en los sujetos tratados con SPRYCEL® en comparación con los sujetos tratados con imatinib.
Figura 4. Tasas de MMR a través del tiempo – total de sujetos aleatorizados en un estudio fase III de pacientes con LMC en fase crónica recientemente diagnosticada.
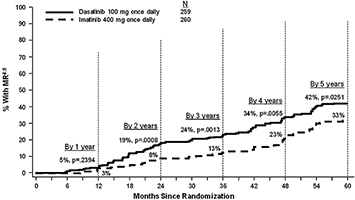
Las tasas de MR4.5 a través del tiempo se muestran gráficamente en la Figura 5. Las tasas de MR4.5 a través del tiempo fueron consistentemente mayores en los sujetos tratados con SPRYCEL® en comparación con los sujetos tratados con imatinib.
Figura 5. Tasas de MR4.5 a través del tiempo – total de sujetos aleatorizados en un estudio fase III de pacientes con LMC en fase crónica recientemente diagnosticada.
La progresión de la enfermedad se definió como aumento de células sanguíneas a pesar del manejo terapéutico apropiado, pérdida de CHR, CyR o CCyR parcial, progresión a fase acelerada o blástica, o muerte. La tasa estimada de PFS a los 60 meses fue de 88.9% (IC: 84.0% - 92.4%) y de 89.2% (IC: 84.3%-92.7%) para los grupos de tratamiento de dasatinib e imatinib, respectivamente.
La transformación a fase blástica o acelerada ocurrió con menos frecuencia con SPRYCEL® (n = 8; 3.1%) que con imatinib (n = 15; 5.8%). Las tasas estimadas de sobrevida a los 60 meses para los pacientes tratados con dasatinib e imatinib fueron de 90.9% (IC: 86.6% - 93.8%) y 89.6% (IC: 85.2% - 92.8%), respectivamente. Con un mínimo de 60 meses de seguimiento, no existen diferencias entre SPRYCEL® e imatinib en la OS (HR 1.01, 95% IC: 0.58 – 1.73, p = 0.9800) o en la PFS (HR 1.00, 95% IC: 0.58 – 1.72, p = 0.9998).
En el estudio fase III de LMC en fase crónica recientemente diagnósticada, se llevó a cabo la secuencia BCR-ABL en muestras de sangre en pacientes que suspendieron la terapia con dasatinib o imatinib. Entre los pacientes tratados con dasatinib las mutaciones detectadas fueron T315I, F317I/L y V299L.
Con base en los datos in vitro, dasatinib, imatinib y nilotinib no parecen ser activos contra la mutación T315I.
Estudios de LMC y LLA Ph+ fase III de optimización de dosis: Se realizó un estudio de fase III, aleatorizado, abierto en pacientes con LMC en fase crónica, cuya enfermedad fue resistente o intolerante a imatinib, para evaluar la eficacia de SPRYCEL® administrado una vez al día en comparación con SPRYCEL® administrado dos veces al día. La variable primaria fue MCyR en pacientes con LMC en fase crónica resistente o intolerante a imatinib.
La principal variable secundaria fue MCyR por dosis diaria total en la misma población. Un total de 670 pacientes, de los cuales 497 tuvieron enfermedad resistente a imatinib fueron aleatorizados al grupo de SPRYCEL® 100 mg una vez al día, 140 mg una vez al día, 50 mg dos veces al día o 70 mg dos veces al día. La mediana de la duración del tratamiento fue aproximadamente 22 meses (rango < 1-31 meses).
Los resultados de eficacia se presentan en las Tablas 4 y 5. Se alcanzó eficacia en todos los grupos de tratamiento con SPRYCEL® con el esquema una vez al día, lo que demuestra una eficacia comparable (no inferioridad) con el esquema dos veces al día en la variable primaria de eficacia (diferencia en MCyR 1.9%; intervalo de confianza 95% [6.8% - 10.6%]); sin embargo, el esquema de 100 mg una vez al día demostró una mejora en la eficacia y tolerabilidad.
Tabla 4. Eficacia de SPRYCEL® en un estudio fase III de optimización de dosis: LMC en fase crónica resistente o intolerante a imatinib (resultados a 2 años)
|
Todos los pacientes |
n = 167 |
|
Pacientes resistentes a imatinib |
n = 124 |
|
Tasa de respuesta hematológicab (%) (95% CI) |
|
|
RHC |
92% (86-95) |
|
Respuesta citogenéticac (%) (95% CI) |
|
|
RCyM |
|
|
Todos los pacientes |
63% (56-71) |
|
Pacientes resistentes a imatinib |
59% (50-68) |
|
RCyC |
|
|
Todos los pacientes |
50% (42-58) |
|
Pacientes resistentes a imatinib |
44% (35-53) |
|
Respuesta molecular mayor en pacientes que alcanzaron RCyCd (%) (95% CI) |
|
|
Todos los pacientes |
69% (58-79) |
|
Pacientes resistentes a imatinib |
72% (58-83) |
a Resultados reportados en una dosis de inicio recomendada de 100 mg una vez al día.
b Criterios de respuesta hematológica (todas las respuestas confirmadas después de 4 semanas):
Respuesta hematológica completa (RHC) (LMC crónica): LCC (leucocitos) ≤ LNS institucional, plaquetas < 450,000/mm3, sin blastos o promielocitos en sangre periférica, < 5% mielocitos más metamielocitos en sangre periférica, basófilos en sangre periférica < 20%, y sin afección extramedular.
c Criterios de respuesta citogenética: completa (0% Ph+ metafases) o parcial (> 0% - 35%). RCyM (0% - 35%) combina tanto respuestas parcial como completa.
d Criterios de respuesta molecular mayor: Definida como BCR-ABL/transcriptos control ≤ 0.1% mediante RQ-PCR en muestras de sangre periférica.
También fue evaluada la eficacia en pacientes que fueron intolerantes a imatinib. En esta población de pacientes que recibieron 100 mg una vez al día, 77% alcanzaron una MCyR y 67% una CCyR, con un mínimo de 2 años de seguimiento.
Tabla 5. Eficacia a largo plazo de SPRYCEL® en un estudio fase III
de optimización de dosis: LMCa en fase crónica resistente o intolerante a imatinib
|
Periodo mínimo de seguimiento |
||||
|
1 año |
2 años |
5 años |
7 años |
|
|
Respuesta molecular mayor |
||||
|
Todos los sujetos |
NA |
37% (57/154) |
44% (71/160) |
46% (73/160) |
|
Pacientes resistentes a imatinib |
NA |
35% (41/117) |
42% (50/120) |
43% (51/120) |
|
Pacientes intolerantes a imatinib |
NA |
43% (16/37) |
53% (21/40) |
55% (22/40) |
|
Supervivencia libre de progresiónb |
||||
|
Todos los sujetos |
90% (86, 95) |
80% (73, 87) |
51% (41, 60) |
42% (33, 51) |
|
Pacientes resistentes a imatinib |
88% (82, 94) |
77% (68, 85) |
49% (39, 59) |
39% (29, 49) |
|
Pacientes intolerantes a imatinib |
97% (92, 100) |
87% (76, 99) |
56% (37, 76) |
51% (32, 67) |
|
Supervivencia global |
||||
|
Todos los sujetos |
96% (93, 99) |
91% (86, 96) |
78% (72, 85) |
65% (56, 72) |
|
Pacientes resistentes a imatinib |
94% (90, 98) |
89% (84, 95) |
77% (69, 85) |
63% (53, 71) |
|
Pacientes intolerantes a imatinib |
100% (100, 100) |
95% (88, 100) |
82% (70, 94) |
70% (52, 82) |
a Resultados reportados en una dosis de inicio recomendada de 100 mg una vez al día.
b La progresión fue definida como incremento en la cuenta de LCC, pérdida de RHC o RCyM, aumento ≤ 30% en Ph+ metafases, enfermedad FA/FB confirmada o muerte. La PFS fue analizada en un principio de intención a tratar y a los pacientes se les dio seguimiento para detectar eventos incluyendo la terapia subsecuente.
Se realizó un estudio de Fase III, aleatorizado, abierto en pacientes con LMC en fase avanzada o LLA Ph+, cuya enfermedad fue resistente a o quienes fueron intolerantes a imatinib, para evaluar la eficacia de SPRYCEL® administrado una vez al día en comparación con SPRYCEL® administrado dos veces al día.
En este estudio, la variable primaria fue la respuesta hematológica mayor (RHMa). Un total de 611 pacientes fue aleatorizado ya sea a SPRYCEL® 140 mg una vez al día o 70 mg dos veces al día. La mediana de la duración del tratamiento fue aproximadamente 6 meses (rango < 1-31 meses).
El esquema una vez al día demostró una eficacia comparable (no inferioridad) con el esquema dos veces al día en la variable primaria de eficacia (diferencia en RHMa 0.8%; IC 95% [-7.1%-8.7%]); sin embargo, el esquema de 140 mg una vez al día demostró mejoría en la seguridad y tolerabilidad. Las tasas de respuesta se presentan en la Tabla 6.
Tabla 6. Eficacia de SPRYCEL® en el estudio fase III de optimización de dosis: LMC
en fase avanzada y LLA Ph+ (resultados a 2 años)a
|
Acelerada n = 158 |
Blástica mieloide n = 75 |
Blástica linfoide n = 33 |
LLA Ph+ n = 40 |
|
|
RHMab |
66% |
28% |
42% |
38% |
|
(95% CI) |
(59-74) |
(18-40) |
(26-61) |
(23-54) |
|
RHCb |
47% |
17% |
21% |
33% |
|
(95% CI) |
(40-56) |
(10-28) |
(9-39) |
(19-49) |
|
SELb |
19% |
11% |
21% |
5% |
|
(95% CI) |
(13-26) |
(5-20) |
(9-39) |
(1-17) |
|
RCyMc |
39% |
28% |
52% |
70% |
|
(95% CI) |
(31-47) |
(18-40) |
(34-69) |
(54-83) |
|
RCyC |
32% |
17% |
39% |
50% |
|
(95% CI) |
(25-40) |
(10-28) |
(23-58) |
(34-66) |
a Resultados reportados en una dosis de inicio recomendada de 140 mg una vez al día.
b Criterios de respuesta hematológica (todas las respuestas confirmadas después de 4 semanas): RHMa = RHC + sin evidencia de leucemia (SEL). RHC: LCC (leucocitos) ≤ LNS institucional, CAN neutrófilos ≥ 1,000/mm3, plaquetas ≥ 100,000/mm3; sin blastos o promielocitos en sangre periférica, blastos en médula ósea ≤ 5%, < 5% mielocitos más metamielocitos en sangre periférica, basófilos en sangre periférica < 20%, y sin afección extramedular. SEL: mismos criterios de RHC pero CAN ≥ 500/mm3 y < 1,000/mm3, o plaquetas ≥ 20,000/mm3 y ≤ 100,000/mm3.
c RCyM combina respuestas tanto completa (0% Ph+ metafases) como parcial (> 0%-35%).
IC = intervalo de confianza; LNS = límite superior del rango normal (límite normal superior).
En pacientes con LMC en fase acelerada tratados con el esquema de 140 mg una vez al día, no se alcanzó la duración mediana de la RHMa y la mediana de la supervivencia global; la mediana de la PFS fue 25 meses.
En pacientes con LMC en fase blástica mieloide tratados con el esquema de 140 mg una vez al día, la duración promedio de la RHMa fue 8 meses, la mediana de la PFS fue 4 meses, y la mediana de la supervivencia global fue de 8 meses. En pacientes con LMC en fase blástica linfoide, la duración promedio de la RHMa fue 5 meses, la mediana de la PFS fue 5 meses, y la mediana de la sobrevida general fue de 11 meses.
En pacientes con LLA Ph+ tratados con el esquema de 140 mg una vez al día, la mediana de la duración de RHMa fue 5 meses, la mediana de la PFS fue de 4 meses, y la mediana de la supervivencia global fue de 7 meses.
CONTRAINDICACIONES: El uso de SPRYCEL® está contraindicado en pacientes con hipersensibilidad a dasatinib o a cualquier otro componente de SPRYCEL®.
Embarazo y lactancia.
RESTRICCIONES DE USO DURANTE EL EMBARAZO Y LA LACTANCIA:
Toxicidad embriofetal: Dasatinib puede ocasionar daño fetal cuando se administra a mujeres embarazadas. Ha habido reportes post comercialización de abortos espontáneos y anormalidades fetales e infantiles de mujeres que han tomado SPRYCEL® durante el embarazo. En estudios no clínicos, a concentraciones plasmáticas menores que aquellas observadas en humanos que reciben dosis terapéuticas de SPRYCEL®, se observó toxicidad fetal en ratas y conejos. Se observó ocurrencia de muerte fetal en ratas. (Ver Precauciones en relación con efectos de carcinogénesis, mutagénesis, teratogénesis y sobre la fertilidad).
El uso de SPRYCEL® no se recomienda en mujeres embarazadas o que consideren quedar embarazadas. Si se administra SPRYCEL® durante el embarazo, o si la paciente queda embarazada durante el tratamiento con SPRYCEL®, la paciente debe ser advertida sobre el posible riesgo para el feto.
Los efectos potenciales de SPRYCEL® en el esperma se han evaluados en un estudio oral de fertilidad y desarrollo embrionaro temprano en ratas. Dasatinib no es tóxico para la reproducción en ratas macho en exposiciones clínicamente relevantes (ver Precauciones en relación con efectos de carcinogénesis, mutagénesis, teratogénesis y sobre la fertilidad). Sin embargo los datos de evaluación de toxicidad reproductiva en pacientes varones que toman SPRYCEL® son limitados.
Los pacientes hombres y mujeres sexualmente activos en edad fértil que tomen SPRYCEL® deben usar un método anticonceptivo adecuado.
Lactancia: No se sabe si SPRYCEL® se excreta en leche humana. Las mujeres que toman SPRYCEL® no deben amamantar.
REACCIONES SECUNDARIAS Y ADVERSAS: Las siguientes Reacciones adversas se discuten con mayor detalle en otras secciones de este documento.
• Mielosupresión (consulte las secciones Dosis y vía de aministración y Precauciones generales).
• Eventos relacionados con sangrado (consulte la sección Precauciones generales).
• Retención de líquidos (consulte la sección Precauciones generales).
• Prolongación del intervalo QT (consulte la sección Precauciones generales).
• Reacciones adversas cardiacas (consulte la sección Precauciones generales).
Experiencia derivada de estudios clínicos: Los datos descritos a continuación reflejan la exposición a SPRYCEL® a todas las dosis estudiadas en 324 pacientes con LMC en fase crónica recientemente diagnosticada y en 2,388 pacientes resistentes o intolerantes a imatinib con LMC en fase crónica o avanzada o LLA Ph+. La mediana de la duración del tratamiento en 2,712 pacientes tratados con SPRYCEL® fue 19.2 meses (rango 0-93.2 meses).
En el estudio fase III de LMC en fase crónica recientemente diagnosticada, la mediana de la duración del tratamiento fue aproximadamente de 60 meses tanto para SPRYCEL® (rango 0.03-72.7 meses) como para imatinib (rango 0.3-74.6 meses). La mediana de la duración del tratamiento en 1,618 pacientes con LMC en fase crónica fue 29 meses (rango 0-92.9 meses). En 1,094 pacientes con LMC en fase avanzada o en LLA Ph+, la mediana de la duración del tratamiento para pacientes fue 6.2 meses (rango 0-93.2 meses).
La mayoría de los pacientes tratados con SPRYCEL® experimentaron reacciones adversas en algún momento. En el total de la población de 2,712 pacientes tratados con SPRYCEL®, 520 (19%) experimentaron reacciones adversas que llevaron a la suspensión del tratamiento.
En el estudio fase III de LMC en fase crónica recientemente diagnosticada, el tratamiento fue suspendido por reacciones adversas en 14% de los pacientes que estaban recibiendo SPRYCEL®, y en 7% de los pacientes que estaban recibiendo imatinib con un seguimiento mínimo de 60 meses. Entre los 1618 pacientes tratados con SPRYCEL® con LMC en fase crónica, fueron reportadas reacciones adversas que llevaron a la suspensión del tratamiento en 329 (20.3%) pacientes; y entre los 1,094 pacientes tratados con SPRYCEL® con enfermedad en fase avanzada, fueron reportadas reacciones adversas que llevaron a la suspensión del tratamiento en 191 (17.5%) pacientes.
La mayoría de los pacientes intolerantes a imatinib con LMC en fase crónica fueron capaces de tolerar el tratamiento con SPRYCEL®. En estudios clínicos de LMC en fase crónica con seguimiento mínimo de 24 meses, 10 de los 215 pacientes intolerantes a imatinib tuvieron la misma toxicidad no hematológica de Grado 3 o 4 con SPRYCEL® que aquella que tuvieron previamente con imatinib; 8 de los 10 pacientes fueron manejados con disminución de la dosis y fueron capaces de continuar el tratamiento con SPRYCEL®.
Las reacciones adversas reportadas en ≥ 10% de los pacientes, y otras reacciones adversas de interés, en un estudio fase III de LMC en fase crónica recientemente diagnosticada, con una mediana de seguimiento de aproximadamente 60 meses, se presentan en la Tabla 7. En este estudio, se reportó derrame pleural en 73 pacientes (28%) que estaban recibiendo SPRYCEL®. La mediana de tiempo hasta la aparición de los eventos de derrame pleural Grado 1 o 2 fue 114 semanas (rango 4-299 semanas). Menos del 3% de los eventos de derrame pleural fueron Grado 3 o 4. Con atención médica apropiada, 58 pacientes (80% de aquellos con derrame pleural) pudieron continuar con SPRYCEL® (ver Precauciones generales, Precauciones y advertencias específicas del producto, Retención de líquidos).
Tabla 7. Reacciones adversas reportadas en ≥ 10% de los pacientes en un estudio fase III (LMC en fase crónica recientemente diagnosticada, 60 meses de seguimiento mínimo)
|
Término preferido |
Todos los grados |
Grado 3/4 |
||
|
SPRYCEL® n = 258 |
Imatinib n = 258 |
SPRYCEL® n = 258 |
Imatinib n = 258 |
|
|
Porcentaje (%) de pacientes |
||||
|
Retención de líquidos |
39 |
45 |
5 |
1 |
|
Derrame pleural |
28 |
1 |
3 |
0 |
|
Edema superficial localizado |
14 |
38 |
0 |
< 1 |
|
Hipertensión pulmonar |
5 |
1 |
< 1 |
0 |
|
Edema generalizado |
4 |
7 |
0 |
0 |
|
Derrame pericárdico |
4 |
1 |
1 |
0 |
|
Insuficiencia cardiaca congestiva/disfunción cardiacaa |
2 |
1 |
< 1 |
< 1 |
|
Edema pulmonar |
1 |
0 |
0 |
0 |
|
Diarrea |
22 |
23 |
1 |
1 |
|
Dolor músculo esquelético |
14 |
17 |
0 |
< 1 |
|
Erupciónb |
14 |
18 |
0 |
2 |
|
Cefalea |
13 |
11 |
0 |
0 |
|
Dolor abdominal |
11 |
8 |
0 |
< 1 |
|
Fatiga |
11 |
12 |
< 1 |
0 |
|
Náusea |
10 |
25 |
0 |
0 |
|
Mialgia |
7 |
12 |
0 |
0 |
|
Artralgia |
7 |
10 |
0 |
< 1 |
|
Hemorragiac |
7 |
7 |
1 |
1 |
|
Sangrado gastrointestinal |
2 |
1 |
1 |
0 |
|
Otro sangradod |
6 |
6 |
0 |
1 |
|
Vómito |
5 |
12 |
0 |
0 |
|
Espasmo muscular |
5 |
21 |
0 |
< 1 |
a Incluye insuficiencia cardiaca aguda, insuficiencia cardiaca congestiva, cardiomiopatía, disfunción diastólica, disminución de la fracción de eyección y disfunción ventricular izquierda.
b Incluye eritema, eritema multiforme, erupción, erupción generalizada, erupción macular, erupción papular, erupción pustular, exfoliación de piel, y erupción vesicular.
c Reacción adversa de interés especial con < 10% de frecuencia.
d Incluye hemorragia conjuntival, hemorragia de oído, equimosis, epistaxis, hemorragia ocular, sangrado gingival, hematoma, hematuria, hemoptisis, hematoma intra-abdominal, petequias, hemorragia escleral, hemorragia uterina, y hemorragia vaginal.
Una comparación de las tasas acumuladas de las reacciones adversas seleccionadas en el estudio fase III de pacientes con LMC en fase crónica recientemente diagnosticada con un seguimiento mínimo de 1 a 5 años, se muestran en la Tabla 8.
Tabla 8. Reacciones adversas seleccionadas reportadas en un estudio fase III (LMC en fase crónica recientemente diagnosticada) (n = 258)
|
Término preferido |
Mínimo 1 año |
Mínimo 5 años |
||
|
Todos |
Grado 3/4 |
Todos |
Grado 3/4 |
|
|
Porcentaje (%) de pacientes |
||||
|
Retención de líquidos |
19 |
1 |
39 |
5 |
|
Derrame pleural |
8 |
0 |
28 |
3 |
|
Edema superficial localizado |
9 |
0 |
14 |
0 |
|
Edema facial |
6 |
0 |
10 |
0 |
|
Hipertensión pulmonar |
1 |
0 |
5 |
<1 |
|
Edema generalizado |
2 |
0 |
4 |
0 |
|
Edema pericárdico |
1 |
<1 |
4 |
1 |
|
Insuficiencia cardiaca congestiva/disfunción cardiacaa |
2 |
<1 |
2 |
1 |
|
Edema pulmonar |
<1 |
0 |
1 |
0 |
|
Diarrea |
17 |
<1 |
22 |
1 |
|
Dolor musculoesquelético |
11 |
0 |
14 |
0 |
|
Erupciónb |
11 |
0 |
14 |
0 |
|
Cefalea |
12 |
0 |
13 |
0 |
|
Fatiga |
8 |
<1 |
11 |
<1 |
|
Náusea |
8 |
0 |
10 |
0 |
|
Mialgia |
6 |
0 |
7 |
0 |
|
Artralgia |
5 |
0 |
7 |
0 |
|
Hemorragiac |
5 |
<1 |
7 |
1 |
|
Sangrado gastrointestinal |
1 |
<1 |
2 |
1 |
|
Otro sangradod |
4 |
0 |
6 |
0 |
|
Vómito |
5 |
0 |
5 |
0 |
|
Espasmo musculare |
4 |
0 |
5 |
0 |
a Incluye insuficiencia cardiaca aguda, insuficiencia cardiaca congestiva, cardiomiopatía, disfunción diastólica, disminución de la fracción de eyección, y disfunción ventricular izquierda.
b Incluye eritema, eritema multiforme, erupción, erupción generalizada, erupción macular, erupción papular, erupción pustular, exfoliación de piel, y erupción vesicular.
c Reacción adversa de interés especial con <10% de frecuencia.
d Incluye hemorragia conjuntival, hemorragia de oído, equimosis, epistaxis, hemorragia ocular, sangrado gingival, hematoma, hematuria, hemoptisis, hematoma intra-abdominal, petequias, hemorragia escleral, hemorragia uterina, y hemorragia vaginal.
e Al término de los 60 meses de análisis “la inflamación muscular” fue reasignada a “espasmo muscular”.
Las reacciones adversas reportadas en múltiples estudios clínicos de SPRYCEL® que fueron consideradas por lo menos posiblemente relacionadas con SPRYCEL® se listan por clase de sistemas y órganos y por frecuencia. La frecuencia de las reacciones adversas se definió utilizando la siguiente convención: muy común (≥ 1/10); común (≥ 1/100 a < 1/10); poco común (≥ 1/1,000 a < 1/100); rara (≥ 1/10,000 a < 1/1,000). Estos eventos se incluyen con base en su relevancia clínica.
Infecciones e infestaciones:
Muy comunes: infección (incluyendo bacteriana, viral, micótica, no especificada).
Común: neumonía (incluyendo bacteriana, viral y micótica), infección/inflamación de vías respiratorias superiores, infección por virus del herpes, infección enterocolítica, sepsis (incluyendo reportes de resultados fatales poco comunes).
Trastornos de la sangre y sistema linfático:
Muy comunes: mielosupresión (incluyendo anemia, neutropenia, trombocitopenia).
Comunes: neutropenia febril.
Poco comunes: linfadenopatía, linfopenia.
Raros: aplasia pura de células rojas.
Trastornos del sistema inmune:
Poco comunes: hipersensibilidad (incluyendo eritema nodoso).
Trastornos endocrinos:
Poco comunes: hipotiroidismo.
Raros: hipertiroidismo, tiroiditis.
Trastornos del metabolismo y la nutrición:
Comunes: alteraciones del apetitoa, hiperuricemia.
Poco comunes: síndrome de lisis tumoral, deshidratación, hipoalbuminemia, hipercolesterolemia.
Raros: diabetes mellitus.
Trastornos psiquiátricos:
Comunes: insomnio, depresión.
Poco comunes: ansiedad, labilidad del afecto, estado de confusión, disminución de la líbido.
Trastornos del sistema nervioso:
Muy comunes: cefalea.
Comunes: neuropatía (incluyendo neuropatía periférica), mareos, disgeusia, somnolencia.
Poco comunes: hemorragia del SNCb, amnesia, temblor, síncope, trastorno del equilibrio.
Raros: convulsión, enfermedad vascular cerebral, ataque isquémico transitorio, neuritis óptica, parálisis del nervio VII, demencia, ataxia.
Trastornos oculares:
Comunes: alteraciones visuales (incluyendo alteración visual, visión borrosa y disminución de la agudeza visual), ojo seco.
Poco comunes: alteraciones visuales, conjuntivitis, fotofobia, aumento del lagrimeo.
Trastornos del oído y del laberinto:
Comunes: acúfenos.
Poco comunes: pérdida de la audición, vértigo.
Trastornos cardiacos:
Comunes: derrame pericárdico, arritmia (incluyendo taquicardia), insuficiencia cardiaca congestiva/disfunción cardiacac, palpitaciones.
Poco comunes: QT prolongado, angina de pecho, cardiomegalia, pericarditis, arritmia ventricular (incluyendo taquicardia ventricular), infarto al miocardio (incluyendo resultados fatales), onda T anormal en electrocardiograma, incremento de troponina.
Raros: Cor pulmonale, miocarditis, síndrome coronario agudo, paro cardiaco, prolongación de PR en electrocardiograma, enfermedad de la arteria coronaria, pleuropericarditis.
Trastornos vasculares:
Muy comunes: hemorragiad.
Comunes: enrojecimiento, hipertensión.
Poco comunes: hipotensión, tromboflebitis.
Raros: trombosis venosa profunda, embolismo, lividez reticular.
Trastornos respiratorios, torácicos y mediastinales:
Muy comunes: derrame pleural, disnea.
Comunes: edema pulmonar, infiltración pulmonar, neumonitis, hipertensión pulmonar, tos.
Poco comunes: hipertensión arterial pulmonar, asma, broncoespasmo, disfonía.
Raros: embolismo pulmonar, síndrome de dificultad respiratoria aguda.
Trastornos gastrointestinales:
Muy comunes: diarrea, náusea, vómito, dolor abdominal.
Comunes: hemorragia gastrointestinal, inflamación de la mucosa (incluyendo mucositis/estomatitis), dispepsia, distensión abdominal, estreñimiento, gastritis, colitis (incluyendo colitis neutropénica), trastorno de tejido blando oral.
Poco comunes: ascitis, disfagia, fisura anal, úlcera gastrointestinal superior, esofagitis, pancreatitis, enfermedad de reflujo gastro-esofágico.
Raros: gastroenteropatía perdedora de proteínas, íleo, pancreatitis aguda, fístula anal.
Trastornos hepatobiliares:
Poco comunes: colestasis, colecistitis, hepatitis.
Trastornos de la piel y tejido subcutáneo:
Muy comunes: erupción cutáneae.
Comunes: prurito, alopecia, acné, piel seca, hiperhidrosis, urticaria, dermatitis (incluyendo eccema).
Poco comunes: trastorno de la pigmentación, úlcera cutánea, condiciones bulosas, fotosensibilidad, trastorno ungular, dermatosis neutrofílica, paniculitis, síndrome de eritrodisestesia palmoplantar, alteraciones del cabello.
Raros: vasculitis leucocitoclástica, fibrosis de la piel.
Trastornos músculo-esqueléticos y de tejido conectivo:
Muy comunes: dolor músculo-esquelético.
Comunes: artralgia, mialgia, debilidad muscular, rigidez músculo-esquelética, espasmo muscular.
Poco comunes: rabdomiólisis, osteonecrosis, tendinitis, inflamación muscular, artritis.
Trastornos renales y urinarios:
Poco comunes: frecuencia urinaria, falla renal, proteinuria.
Raros: insuficiencia renal.
Embarazo, puerperio y enfermedades perinatales:
Raros: aborto.
Trastornos del sistema reproductivo y mama:
Poco comunes: ginecomastia, trastorno menstrual.
Trastornos generales y condiciones del sitio de administración:
Muy comunes: edema periféricof, fatiga, pirexia, edema facialg.
Comunes: astenia, dolor, dolor torácico, edema generalizadoh, escalofrío.
Poco comunes: malestar, otros edemas superficialesi.
Raros: trastornos al caminar.
Investigaciones:
Comunes: aumento de peso, disminución de peso.
Poco comunes: aumento de creatinina fosfoquinasa en sangre, aumento de gamma-glutamiltransferasa.
Lesiones, envenenamiento y complicaciones del procedimiento:
Comunes: contusión.
a Incluye disminución del apetito, saciedad temprana, aumento del apetito.
b Incluye hemorragia del sistema nervioso central, hematoma cerebral, hemorragia cerebral, hematoma extradural, hemorragia intracraneal, evento vascular cerebral hemorrágico, hemorragia subaracnoidea, hematoma subdural y hemorragia subdural.
c Incluye aumento de péptido natriurético cerebral, disfunción ventricular, disfunción ventricular izquierda, disfunción ventricular derecha, insuficiencia cardiaca, insuficiencia cardiaca aguda, insuficiencia cardiaca crónica, insuficiencia cardiaca congestiva, cardiomiopatía, cardiomiopatía congestiva, disfunción diastólica, fracción de eyección disminuida, insuficiencia ventricular, insuficiencia ventricular izquierda, insuficiencia ventricular derecha e hipocinesia ventricular.
d Excluye sangrados gastrointestinales y de SNC; estas RAMs se reportan bajo las clases de sistemas y órganos gastrointestinal y sistema nervioso, respectivamente.
e Incluye erupción cutánea por medicamento, eritema, eritema multiforme, eritrosis, dermatosis exfoliativa, eritema generalizado, dermatosis genital, dermatosis por calor, milia, miliaria, psoriasis pustulosa, erupción cutánea, dermatosis eritematosa, dermatosis folicular, dermatosis generalizada, dermatosis maculosa, dermatosis maculopapulosa, dermatosis papular, dermatosis pruriginosa, dermatosis pustulosa, dermatosis vesicular, exfoliación de la piel, irritación de la piel, erupción cutánea tóxica, urticaria vesiculosa y dermatosis vasculítica.
f Incluye edema gravitacional, edema localizado, edema periférico.
g Incluye edema conjuntival, edema ocular, inflamación ocular, edema palpebral, edema facial, edema labial, edema macular, edema bucal, edema orbital, edema periorbital, inflamación facial.
h Incluye sobrecarga de líquidos, retención de líquidos, edema gastrointestinal, edema generalizado, edema, edema debido a enfermedad cardiaca, derrame perirrenal, edema posterior al procedimiento, edema visceral.
i Incluye inflamación genital, edema en el sitio de incisión, edema genital, edema peniano, inflamación peniana, edema escrotal, inflamación cutánea, inflamación testicular, inflamación vulvovaginal.
En el estudio fase III de optimización de dosis en pacientes con LMC en fase crónica resistentes o intolerantes a imatinib, la mediana de la duración global del tratamiento fue aproximadamente 30 meses (rango < 1-93 meses), con una mediana de duración de 37 meses (rango < 1-91 meses) en el grupo de 100 mg una vez al día. Las tasas acumulativas de las reacciones adversas seleccionadas que fueron reportadas en la dosis inicial recomendada de 100 mg una vez al día, se muestran en la Tabla 9.
Tabla 9. Reacciones adversas seleccionadas reportadas en el estudio fase III de optimización de dosis
(LMC en fase crónica intolerantes o resistentes a imatinib)a
|
Término preferido |
Mínimo de 2 años de seguimiento |
Mínimo de 5 años de seguimiento |
Mínimo de 7 años de seguimiento |
|||
|
Todos los grados |
Grado ¾ |
Todos los grados |
Grado ¾ |
Todos los grados |
Grado ¾ |
|
|
Porcentaje (%) de pacientes |
||||||
|
Diarrea |
27 |
2 |
28 |
2 |
28 |
2 |
|
Retención de líquidos |
34 |
4 |
42 |
6 |
48 |
7 |
|
Edema superficial |
18 |
0 |
21 |
0 |
22 |
0 |
|
Derrame pleural |
18 |
2 |
24 |
4 |
28 |
5 |
|
Edema generalizado |
3 |
0 |
4 |
0 |
4 |
0 |
|
Derrame pericárdico |
2 |
1 |
2 |
1 |
3 |
1 |
|
Hipertensión pulmonar |
0 |
0 |
0 |
0 |
2 |
1 |
|
Hemorragia |
11 |
1 |
11 |
1 |
12 |
1 |
|
Sangrado gastrointestinal |
2 |
1 |
2 |
1 |
2 |
1 |
a Resultados reportados en el estudio fase III de optimización de dosis en la población con dosis inicial recomendada de 100 mg una vez al día (n = 165).
En un estudio fase III de optimización de la dosis en pacientes con LMC en fase avanzada y LLA Ph+, la mediana de la duración del tratamiento fue 14 meses (rango < 1-36 meses) para la LMC en fase acelerada, 3 meses (rango < 1-32 meses) para LMC en fase blástica mieloide, 4 meses (< 1-22 meses) para LMC en fase blástica linfoide y 3 meses (< 1-29 meses) para LLA Ph+. Las reacciones adversas seleccionadas que fueron reportadas a la dosis inicial recomendada de 140 mg una vez al día, se muestran en la Tabla 10. También se estudió un régimen de dosis de 70 mg dos veces al día. El régimen de 70 mg dos veces al día mostró un perfil de eficacia comparable con el régimen de 140 mg una vez al día, pero un perfil de seguridad menos favorable.
Tabla 10. Reacciones adversas al medicamento seleccionadas reportadas en un estudio fase III
de optimización de dosis (LMC en fase avanzada y LLA Ph+)
|
Término preferido |
140 mg una vez al díaa n = 304 |
|
|
Todos los grados |
Grado ¾ |
|
|
Porcentaje (%) de pacientes |
||
|
Diarrea |
28 |
3 |
|
Retención de líquidos |
33 |
7 |
|
Edema superficial |
15 |
< 1 |
|
Derrame pleural |
20 |
6 |
|
Edema generalizado |
2 |
0 |
|
Insuficiencia cardiaca congestiva/disfunción cardiacab |
1 |
0 |
|
Derrame pericárdico |
2 |
1 |
|
Edema pulmonar |
1 |
1 |
|
Hemorragia |
23 |
8 |
|
Sangrado gastrointestinal |
8 |
6 |
a Resultados reportados del estudio fase III de optimización de dosis en la población con dosis inicial recomendada de 140 mg una vez al día (n = 304) al final del estudio con 2 años de seguimiento.
b Incluye disfunción ventricular, insuficiencia cardiaca, insuficiencia cardiaca congestiva, cardiomiopatía, cardiomiopatía congestiva, disfunción diastólica, descenso de la fracción de eyección e insuficiencia ventricular.
Experiencia posterior a la comercialización: Las siguientes reacciones adversas adicionales han sido identificadas durante el uso posterior a la aprobación de SPRYCEL®. Ya que estas reacciones son reportadas voluntariamente en una población de tamaño incierto, no siempre es posible estimar confiablemente su frecuencia o establecer una relación causal con la exposición al medicamento.
|
Infecciones e infestaciones: |
reactivación de hepatitis B |
|
Trastornos cardiacos: |
fibrilación auricular/flutter auriculara |
|
Trastornos respiratorios, torácicos y mediastínicos: |
enfermedad pulmonar intersticial |
|
Trastornos de la piel y del tejido subcutáneo: |
síndrome de Stevens-Johnsonb |
|
Renales y urinarios: |
Síndrome nefrótico |
a Típicamente reportado en pacientes ancianos o en pacientes con factores de confusión incluyendo trastornos cardiacos o cardiovasculares subyacentes o concurrentes significativos u otras comorbilidades significativas (p. ej., infección/sepsis grave, anormalidades electrolíticas).
b En el ajuste posterior a la comercialización, se han reportado casos aislados de síndrome de Stevens-Johnson. No pudo ser determinado si estas reacciones adversas mucocutáneas estaban directamente relacionadas con SPRYCEL® o a las medicaciones concomitantes.
Anormalidades de laboratorio: La Tabla 11 muestra los hallazgos de laboratorio de un estudio clínico en pacientes con LMC en fase crónica recientemente diagnosticada. No se presentaron suspensiones del tratamiento con SPRYCEL® por parámetros bioquímicos de laboratorio.
Tabla 11. Anomalías de laboratorio grados CTC 3/4 en un estudio fase III de pacientes con LMC en fase crónica recientemente diagnosticada
|
SPRYCEL® (n = 258) |
Imatinib (n = 258) |
|
|
Porcentaje (%) de los pacientes |
||
|
Parámetros hematológicos |
||
|
Neutropenia |
29 |
24 |
|
Trombocitopenia |
22 |
14 |
|
Anemia |
13 |
9 |
|
Parámetros bioquímicos |
||
|
Hipofosfatemia |
7 |
31 |
|
Hipocalemia |
0 |
3 |
|
Hipocalcemia |
4 |
3 |
|
SGPT elevada (ALT) |
< 1 |
2 |
|
SGOT elevada (AST) |
< 1 |
1 |
|
Bilirrubina elevada |
1 |
0 |
|
Creatinina elevada |
1 |
1 |
Grados CTC: neutropenia (grado 3 ≥ 0.5-< 1.0 x 109/L, grado 4 < 0.5 x 109/L); trombocitopenia (grado 3 ≥ 25-< 50 x 109/L, grado 4 < 25 x 109/L); anemia (hemoglobina grado 3 ≥ 65-< 80 g/L; grado 4 < 65 g/L); creatinina elevada (grado 3 > 3-6 x límite superior del rango normal (ULN), grado 4 > 6 x ULN); bilirrubina elevada (grado 3 > 3-10 x ULN, grado 4 > 10 x ULN); SGOT o SGPT elevada (grado 3 > 5-20 x ULN, grado 4 > 20 x ULN); hipocalcemia (grado 3 < 7.0-6.0 mg/dL, grado 4 < 6.0 mg/dL); hipofosfatemia (grado 3 < 2.0-1.0 mg/dL, grado 4 < 1.0 mg/dL); hipocalemia (grado 3 < 3.0-2.5 mmol/L, grado 4 < 2.5 mmol/L).
La Tabla 12 muestra los hallazgos de laboratorio de los estudios clínicos de pacientes con LMC con resistencia o intolerancia a imatinib que recibieron 24 meses de seguimiento.
Tabla 12. Anomalías de laboratorio grados CTC 3/4 en estudios de pacientes con LMC
resistentes o intolerantes al tratamiento previo con imatiniba
|
Fase crónicab (n = 165) |
Fase aceleradac (n = 157) |
Fase blástica mieloidec (n = 74) |
Fase blástica linfoidec (n = 33) |
LLA Ph+c (n = 135) |
|
|
Porcentaje (%) de los pacientes |
|||||
|
Parámetros hematológicos |
|||||
|
Neutropenia |
35 |
58 |
77 |
79 |
75 |
|
Trombocitopenia |
23 |
63 |
78 |
85 |
71 |
|
Anemia |
13 |
47 |
74 |
52 |
42 |
|
Parámetros de química sanguínea |
|||||
|
Hipofosfatemia |
10 |
13 |
12 |
18 |
21 |
|
Hipocalemia |
2 |
7 |
11 |
15 |
16 |
|
Hipocalcemia |
< 1 |
4 |
9 |
12 |
9 |
|
SGPT (ALT) elevada |
0 |
2 |
5 |
3 |
7 |
|
SGOT (AST) elevada |
< 1 |
0 |
4 |
3 |
4 |
|
Bilirrubina elevada |
< 1 |
1 |
3 |
6 |
2 |
|
Creatinina elevada |
0 |
2 |
8 |
0 |
0 |
a Resultados de un estudio fase III de optimización de dosis reportados a los 2 años de seguimiento.
b Resultados del estudio CA180-034 a dosis inicial recomendada de 100 mg una vez al día.
c Resultados del estudio CA180-035 a dosis inicial recomendada de 140 mg una vez al día.
Grados CTC: neutropenia (grado 3 ≥ 0.5–< 1.0 × 109/L, grado 4 < 0.5 × 109/L); trombocitopenia (grado 3 ≥ 25–< 50 × 109/L, grado 4 < 25 × 109/L); anemia (hemoglobina grado 3 ≥ 65–< 80 g/L, grado 4 < 65 g/L); creatinina elevada (grado 3 > 3–6 × límite superior del rango normal [LNS], grado 4 > 6 × LNS); bilirrubina elevada (grado 3 > 3–10 × LNS, grado 4 > 10 × LNS); SGOT o SGPT elevada (grado 3 > 5–20 × LNS, grado 4 > 20 × LNS); hipocalcemia (grado 3 < 7.0–6.0 mg/dL, grado 4 < 6.0 mg/dL); hipofosfatemia (grado 3 < 2.0–1.0 mg/dL, grado 4 < 1.0 mg/dL); hipocalemia (grado 3 < 3.0-2.5 mmol/L, grado 4 < 2.5 mmol/L).
La mielosupresión fue un efecto comúnmente reportado en todas las poblaciones de pacientes. En pacientes con LMC en fase crónica recientemente diagnosticada, la mielosupresión se reportó con menos frecuencia que en los pacientes con LMC en fase crónica resistentes o intolerantes al tratamiento previo con imatinib. La frecuencia de neutropenia grado 3 o 4, trombocitopenia y anemia fue más elevada entre los pacientes con LMC en fase avanzada o LLA Ph+ que en LMC en fase crónica.
Por lo general, los pacientes que padecieron una mielosupresión severa exhibieron una recuperación después de la interrupción o la reducción de las dosis; la suspensión permanente del tratamiento ocurrió en 2% de los pacientes con LMC en fase crónica recientemente diagnosticada en el estudio fase III, y en un 5% de los pacientes con resistencia o intolerancia al tratamiento previo con imatinib en el estudio fase III.
Se reportaron elevaciones grado 3 o 4 en los niveles de transaminasas o de bilirrubina además de hipocalcemia, hipocalemia, e hipofosfatemia entre los pacientes con LMC en todas sus fases; sin embargo, la frecuencia de dichos reportes fue mayor entre los pacientes con LMC en fases blásticas mieloides o linfoides y con LLA Ph+. Las elevaciones en los niveles de transaminasas o de bilirrubina usualmente fueron manejadas a través de la interrupción o la reducción de las dosis. En general los niveles disminuidos de calcio no se asociaron con síntomas clínicos. Los pacientes que desarrollaron hipocalcemia grado 3 o 4 con frecuencia se recuperaron al recibir suplementos orales de calcio.
PRECAUCIONES EN RELACIÓN CON EFECTOS DE CARCINOGÉNESIS, MUTAGÉNESIS, TERATOGÉNESIS Y SOBRE LA FERTILIDAD: Dasatinib no fue mutagénico en ensayos celulares bacterianos in vitro (prueba de Ames) y no fue genotóxico en el estudio in vivo de micronúcleos en rata. Dasatinib fue clastogénico in vitro para células ováricas de hámster chino en división.
En un estudio de carcinogenicidad de dos años, se administraron a ratas dosis orales de dasatinib a 0.3, 1 y 3 mg/kg/día. La dosis más alta resultó en una exposición al fármaco en plasma (ABC) en un nivel generalmente equivalente a la exposición humana al rango recomendado de dosis inicial de 100 mg a 140 mg al día. Se observó un incremento estadísticamente significativo en la incidencia combinada de carcinomas de células escamosas y papilomas en el útero y cuello uterino de altas dosis en mujeres y de adenoma de próstata en los hombres a dosis bajas. La relevancia de los resultados del estudio de carcinogenicidad en ratas para el ser humano no se conoce.
Dasatinib no afectó la fertilidad de los machos o las hembras en un estudio convencional de fertilidad y desarrollo embriónico temprano en ratas, pero provocó letalidad embrionaria a niveles de dosis que se aproximaron a las exposiciones clínicas humanas. En estudios de desarrollo embriofetal, dasatinib produjo de la misma manera la letalidad embrionaria asociada a una reducción en el tamaño de las camadas en ratas, así como también alteraciones esqueléticas fetales tanto en ratas como en conejos. Estos efectos se presentaron a dosis que no produjeron toxicidad materna, lo que indica que dasatinib es un tóxico reproductivo selectivo desde la implantación hasta que finaliza la organogénesis. En un estudio exploratorio de desarrollo peri y post natal, la exposición indirecta de crías de ratas a dasatinib (in utero o mediante la lactancia) iniciando desde el fin de la organogénesis hasta la lactancia temprana, fue incompatible con la supervivencia de las crías, incluso a exposiciones maternas que son subterapéuticas.
INTERACCIONES MEDICAMENTOSAS Y DE OTRO GÉNERO:
Efectos de otros medicamentos en SPRYCEL®:
Medicamentos que pueden elevar las concentraciones plasmáticas de dasatinib:
Inhibidores CYP3A4: Dasatinib es un sustrato CYP3A4. El uso concomitante de SPRYCEL® y substancias que inhiben CYP3A4 (p. ej., ketoconazol, itraconazol, eritromicina, claritromicina, ritonavir, atazanavir, indinavir, nelfinavir, saquinavir, telitromicina y jugo de toronja) pueden incrementar la exposición a dasatinib y deben evitarse. Se recomienda la selección de un medicamento concomitante alterno sin o con un potencial mínimo de inhibición CYP3A4. Si no puede evitarse la administración sistémica de un inhibidor potente de CYP3A4, el paciente debe ser monitoreado estrechamente por toxicidad.
Medicamentos que pueden disminuir las concentraciones plasmáticas de dasatinib:
Inductores CYP3A4: Medicamentos que inducen la actividad CYP3A4 (p. ej., dexametasona, fenitoína, carbamazepina, rifampicina, fenobarbital o Hypericum perforatum también conocida como Hierba de San Juan) pueden disminuir la exposición a dasatinib. No se recomienda el uso concomitante de inductores CYP3A4 potentes con SPRYCEL®. En pacientes para quienes está indicado el uso de inductores CYP3A4, se deben seleccionar agentes alternativos sin o con mínimo potencial de inducción de CYP3A4.
Antiácidos (hidróxido de aluminio/hidróxido de magnesio): Los datos no clínicos demuestran que la solubilidad de dasatinib es dependiente del pH. Si se requiere de tratamiento antiácido, la dosis de antiácido debe administrarse por lo menos 2 horas antes o 2 horas después de la administración de SPRYCEL®. Se debe evitar la administración simultánea de SPRYCEL® con antiácidos.
Antagonistas H2/inhibidores de la bomba de protones: Es probable que la supresión a largo plazo de la secreción ácida gástrica por antagonistas H2 o inhibidores de la bomba de protones (p. ej., famotidina y omeprazol) disminuya la exposición a dasatinib. En un estudio de 14 sujetos sanos, la administración de una dosis única de 100 mg de SPRYCEL® 22 horas después de 4 días con una dosis de 40 mg de omeoprazol a concentración estable disminuyó el ABC y Cmáx de dasatinib 43% y 42%, respectivamente. No se recomienda el uso concomitante de antagonistas H2 o inhibidores de la bomba de protones con SPRYCEL®. Se debe considerar el uso de antiácidos (al menos 2 horas antes o 2 horas después de la dosis de SPRYCEL®) en vez de antagonistas H2 o inhibidores de la bomba de protones en pacientes que reciben tratamiento con SPRYCEL®.
Efectos de SPRYCEL® en otros medicamentos:
Substratos CYP3A4: Dasatinib es un inhibidor de CYP3A4. El uso concomitante de dasatinib y un substrato CYP3A4 puede aumentar la exposición al substrato CYP3A4. Por lo tanto, los substratos CYP3A4 que se sabe tienen un índice terapéutico estrecho tales como alfentanilo, astemizol, terfenadina, cisaprida, ciclosporina, fentanilo, pimozida, quinidina, sirolimus, tacrolimus, o alcaloides ergóticos (ergotamina, dihidroergotamina), deben administrarse cuidadosamente en pacientes tratados con SPRYCEL®.
ALTERACIONES EN LOS RESULTADOS DE PRUEBAS DE LABORATORIO: Ver Reacciones secundarias y adversas, Anormalidades de laboratorio.
PRECAUCIONES GENERALES:
Precauciones y advertencias específicas del producto:
Mielosupresión: El tratamiento con SPRYCEL® ha sido asociado con trombocitopenia, neutropenia y anemia, las cuales ocurren tempranamente y con más frecuencia en pacientes con LMC en fases avanzadas o en LLA Ph+ que en pacientes con LMC en fase crónica.
En pacientes con LMC en fase crónica, se deben llevar a cabo biometrías hemáticas completas (CBCs por sus siglas en inglés) cada dos semanas durante 12 semanas, posteriormente cada 3 meses o conforme esté clínicamente indicado. En pacientes con LMC en fases avanzadas o en LLA Ph+, se deben llevar a cabo CBCs semanalmente los primeros 2 meses y posteriormente mensualmente, o conforme esté clínicamente indicado.
La mielosupresión es generalmente reversible y usualmente manejada a través de la suspensión temporal de la administración de SPRYCEL® o mediante la reducción de la dosis. (Ver Dosis y vía de administración, Dosis recomendada y Reacciones secundarias y adversas, Anormalidades del laboratorio).
Eventos relacionados con sangrado: En pacientes con LMC en fase crónica, se presentó hemorragia grave en 5 pacientes (1%) recibiendo SPRYCEL® a la dosis recomendada (n = 548).
En pacientes con LMC en fases avanzadas o en LLA Ph+, se presentaron hemorragias graves en el sistema nervioso central (SNC) incluyendo algunas fatales, en el 1% de los pacientes recibiendo SPRYCEL® a la dosis recomendada (n = 304). Se presentó hemorragia gastrointestinal grave incluyendo algunas fatales, en 6% de los pacientes, y generalmente requirió de interrupciones del tratamiento y transfusiones. En 2% de los pacientes, ocurrió hemorragia grave por otras causas.
La mayoría de los eventos hemorrágicos en estudios clínicos se relacionaron con trombocitopenia grave. Adicionalmente, ensayos plaquetarios in vitro e in vivo sugieren que el tratamiento con SPRYCEL® afecta reversiblemente la activación plaquetaria.
Se debe tener precaución si los pacientes requieren tratamiento que inhiba la función plaquetaria o anticoagulantes.
Retención de líquidos: El uso de SPRYCEL® es asociado con la retención de líquidos. Después de 5 años de seguimiento en el estudio fase III de LMC en fase crónica recientemente diagnosticada (n = 258), se reportó retención de líquidos grave en 13 pacientes (5%) recibiendo SPRYCEL® comparado con 2 pacientes (1%) recibiendo imatinib (n = 258). (Ver Reacciones secundarias y adversas, Experiencia derivada de estudios clínicos).
En todos los pacientes con LMC en fase crónica recientemente diagnosticada o con resistencia o intolerancia a imatinib (n = 548), se presentó retención de líquidos grave en 32 pacientes (6%) recibiendo SPRYCEL® a la dosis recomendada.
En pacientes con LMC en fases avanzadas o en LLA Ph+ recibiendo SPRYCEL® a la dosis aprobada (n = 304), se reportó una retención de líquidos grave en 8% de los pacientes, incluyendo derrame pleural grave y pericárdico en 7% y 1% de los pacientes, respectivamente. En estos pacientes, se reportó edema pulmonar grave e hipertensión pulmonar grave, cada uno en 1% de los pacientes.
Aquellos pacientes que desarrollan síntomas que sugieran derrame pleural u otra retención de líquidos tales como disnea nueva o empeorada durante el ejercicio o en reposo, dolor torácico pleurítico, o tos seca, deben ser evaluados rápidamente con una radiografía de tórax o diagnóstico de imagen adicional según sea apropiado. Los eventos de retención de líquidos se manejaron típicamente mediante medidas de cuidado asistencial que pueden incluir diuréticos o cursos cortos de esteroides. El derrame pleural grave puede llegar a requerir una toracocentesis y terapia con oxígeno. Se debe considerar la modificación de la dosis. (Ver Dosis y vía de administración, Dosis recomendada y Reacciones secundarias y adversas, Experiencia derivada de estudios clínicos).
Reacciones adversas cardiacas: Se estudió SPRYCEL® en un estudio aleatorizado de 519 pacientes con LMC en fase crónica recientemente diagnosticada, que incluyó pacientes con enfermedad cardiaca previa. Las reacciones adversas de insuficiencia cardiaca congestiva/disfunción cardiaca, derrame pericárdico, arritmias, palpitaciones, prolongación del intervalo QT e infarto al miocardio (incluyendo fatal), se reportaron en pacientes que estaban tomando SPRYCEL®. La mayoría de los pacientes que experimentaron eventos adversos cardiacos tenían historia médica previa o factores de riesgo de enfermedad cardiaca. Los pacientes con factores de riesgo o con historia de enfermedad cardiaca deben ser monitoreados cuidadosamente en busca de signos y síntomas consistentes con disfunción cardiaca, y deben ser evaluados y tratados de manera apropiada (Ver Reacciones secundarias y adversas, Experiencia derivada de estudios clínicos).
Hipertensión arterial pulmonar: Se ha reportado hipertensión arterial pulmonar (HAP) confirmada mediante cateterización cardiaca derecha en asociación con el tratamiento con SPRYCEL®. En estos casos, la HAP fue reportada después del inicio de la terapia con SPRYCEL®, inclusive después de más de un año de tratamiento. Los pacientes en quienes se reportó HAP durante el tratamiento con SPRYCEL® utilizaban medicamentos concomitantes o presentaban comorbilidades en adición a la neoplasia maligna subyacente.
Se deberá evaluar a los pacientes sobre signos y síntomas de enfermedad cardiopulmonar subyacente antes de iniciar la terapia con SPRYCEL®. Los pacientes que desarrollen disnea y fatiga después del inicio de la terapia deberán ser evaluados para detectar o descartar etiologías comunes incluyendo derrame pleural, edema pulmonar, anemia o infiltración pulmonar. Durante esta evaluación, deben seguirse las recomendaciones para el manejo de las reacciones adversas no hematológicas (Ver Dosis y vía de administración, Dosis recomendada). Si la reacción adversa es grave, el tratamiento debe ser discontinuado hasta que el evento se resuelva o mejore. Si no se encuentra una explicación, se deberá considerar un diagnóstico de HAP. Si se confirma HAP, el tratamiento con SPRYCEL® deberá ser discontinuado permanentemente. Se deberá efectuar un seguimiento acorde a los lineamientos de práctica estándar. Se han observado mejorías de los parámetros hemodinámicos y clínicos en los pacientes tratados con SPRYCEL® que han presentado HAP una vez cesada la terapia con SPRYCEL®.
Prolongación del intervalo QT: Datos in vitro sugieren que dasatinib tiene el potencial de prolongar la repolarización ventricular cardiaca (intervalo QT).
Después de 5 años de seguimiento en el estudio clínico fase III de LMC en fase crónica recientemente diagnosticada, 1 paciente (< 1%) en cada grupo de tratamiento: SPRYCEL® (n = 258) e imatinib (n = 258), presentó prolongación del intervalo QTc reportada como una reacción adversa. La mediana de cambios en el QTcF desde la basal fue de 3.0 mseg en los pacientes tratados con SPRYCEL® en comparación con 8.2 mseg en pacientes tratados con imatinib. Un paciente (< 1%) en cada grupo experimentó un QTcF > 500 mseg.
En 865 pacientes con leucemia tratados con SPRYCEL® en estudios clínicos de fase II, los cambios en la media del intervalo QTc desde la línea basal, utilizando el método de Fridericia (QTcF), fueron 4-6 mseg; los intervalos de confianza 95% superiores para todos los cambios de la media desde la línea basal fueron < 7 mseg. De los 2,182 pacientes con resistencia o intolerancia a la terapia previa con imatinib y que recibieron SPRYCEL® en estudios clínicos, 15 (1%) tuvieron prolongación del intervalo QT reportado como una reacción adversa. Veintiuno de estos pacientes (1%) experimentaron un QTcF >500 mseg.
SPRYCEL® debe ser administrado con precaución en aquellos pacientes que presenten o puedan llegar a desarrollar una prolongación del intervalo QT. Entre ellos se encuentran los pacientes con hipopotasemia o hipomagnesemia, los pacientes con síndrome de QT largo congénito, los pacientes tratados con medicinas contra la arritmia u otros productos medicinales que pueden provocar una prolongación del intervalo QT y los pacientes tratados con altas dosis acumulativas de antraciclinas. La hipopotasemia o la hipomagnesemia deben ser corregidas en forma previa a la administración de SPRYCEL®.
En los estudios clínicos de SPRYCEL® no se incluyeron pacientes con enfermedad cardiovascular no controlada o significativa.
Reactivación del virus de la hepatitis B: Los inhibidores de la tirosina cinasa (TKI’s, por sus siglas en inglés) de BCR-ABL han sido asociados con la reactivación del virus de la hepatitis B (VHB), incluyendo casos de reportes individuales para SPRYCEL®. En algunos casos, la reactivación del VHB ocurre en conjunto con otros TKI’s de BCR-ABL y puede resultar en insuficiencia hepática aguda o hepatitis fulminante, conduciendo a un trasplante de hígado o la muerte.
Se debe considerar la detección del VHB de acuerdo a las guías publicadas antes de iniciar el tratamiento con SPRYCEL®. Se recomienda la consulta con un médico experto en el tratamiento del VHB para pacientes que den positivo en la prueba serológica del VHB.
Los pacientes que son portadores del VHB y requieren tratamiento con TKI’s de BCR-ABL, deben ser monitoreados cuidadosamente en busca de signos clínicos y de laboratorio de una infección activa por VHB durante todo el tratamiento y durante varios meses después del término del tratamiento. En pacientes que desarrollan una reactivación del VHB mientras reciben SPRYCEL®, se recomienda una consulta inmediata con un médico experto en el tratamiento del VHB.
Reacciones dermatológicas graves: Se han reportado casos aislados de reacciones dermatológicas mucocutáneas graves, incluyendo Síndrome de Stevens-Johnson y eritema multiforme, con el uso de SPRYCEL®. SPRYCEL® debe ser suspendido permanentemente en pacientes que experimentaron reacciones mucocutáneas graves durante el tratamiento, si otra etiología no pudo ser identificada.
Lactosa: SPRYCEL® contiene 135 mg de lactosa monohidratada en una dosis diaria de 100 mg y 189 mg de lactosa monohidratada en una dosis diaria de 140 mg.
Uso pediátrico: No se ha establecido la seguridad y eficacia de SPRYCEL® en pacientes menores de 18 años de edad.
Uso geriátrico: No fueron observadas diferencias en RCCc y en RMM entre los pacientes mayores y los más jóvenes. De los 2,712 pacientes en estudios clínicos de SPRYCEL®, 617 (23%) tuvieron 65 años de edad o más, y 123 (5%) tuvieron 75 años de edad o más. Aunque el perfil de seguridad de SPRYCEL® en la población geriátrica fue similar que en la población más joven, los pacientes de 65 años de edad y mayores tienen mayor probabilidad de experimentar los eventos adversos más comunes como fatiga, derrame pleural, disnea, tos, hemorragia gastrointestinal menor y disturbios en el apetito, y tienen más probabilidad de experimentar los eventos reportados menos frecuentemente como distensión abdominal, mareo, derrame pericárdico, insuficiencia cardiaca congestiva y pérdida de peso, y deben ser monitoreados estrechamente.
Efectos en la habilidad para conducir y manejar maquinaria: No se han llevado a cabo estudios para evaluar los efectos en la habilidad para conducir y manejar maquinaria.
DOSIS Y VÍA DE ADMINISTRACIÓN:
Vía de administración: Oral.
Dosis recomendada: La dosis inicial recomendada de SPRYCEL® para la LMC en fase crónica es de 100 mg administrados por la vía oral una vez al día. La dosis inicial recomendada de SPRYCEL® para la LMC en fase acelerada, la LMC en fase blástica mieloide o linfoide o la LLA Ph+ es de 140 mg administrados por la vía oral una vez al día. Las tabletas no deben ser trituradas ni cortadas, sino que deben ser tragadas enteras. SPRYCEL® puede ser tomado con o sin alimentos, ya sea por la mañana o por la noche. El aumento o disminución de la dosis se debe ajustar de acuerdo a la respuesta y tolerancia individual.
En los estudios clínicos, el tratamiento con SPRYCEL® ha sido continuado hasta que se ha presentado una progresión de la enfermedad o hasta que el fármaco ha dejado de ser tolerado por el paciente. No se ha investigado el efecto de la suspensión del tratamiento en el resultado de la enfermedad a largo plazo después de lograr una respuesta citogenética (incluyendo una respuesta citogenética completa [RCyC] o una respuesta molecular profunda [RMM y RM4.5]).
Escalamiento de la dosis: En estudios clínicos realizados en pacientes con LMC o LLA Ph+, se permitió un incremento progresivo de la dosis hasta alcanzar 140 mg una vez al día (LMC en fase crónica) o 180 mg una vez al día (LMC en fase acelerada y blástica o LLA Ph+) en aquellos pacientes que no lograron una respuesta hematológica o citogenética con la dosis inicial recomendada.
Ajuste de la dosis debido a reacciones adversas:
Mielosupresión: En los estudios clínicos, la mielosupresión fue manejada a través de la interrupción de las dosis, la reducción de las dosis o la suspensión de la terapia del estudio. Se utilizaron transfusiones plaquetarias y eritrocitarias conforme fue requerido. Se han utilizado factores de crecimiento hematopoyético en pacientes que han presentado una mielosupresión resistente. Los lineamientos para las modificaciones de la dosis aparecen resumidos en la Tabla 13.
Tabla 13. Ajustes de la dosis para casos de neutropenia y trombocitopenia
|
LMC en fase crónica (dosis inicial: 100 mg una vez al día) |
CAN* < 0.5 × 109/L o Plaquetas < 50 × 109/L |
1. Interrumpa el tratamiento con SPRYCEL® hasta alcanzar un CAN ≥ 1.0 × 109/L y un conteo de plaquetas ≥50 × 109/L. 2. Reanude el tratamiento con SPRYCEL® utilizando la dosis inicial original. 3. De presentarse un conteo de plaquetas < 25 × 109/L o de volver a presentarse un CAN < 0.5 × 109/L durante > 7 días, repita el paso 1 y reanude el tratamiento con SPRYCEL® utilizando una dosis reducida de 70 mg una vez al día para el segundo episodio. Para un tercer episodio reduzca la dosis a 50 mg una vez al día (para pacientes recientemente diagnosticados) o suspenda el tratamiento con SPRYCEL® (para pacientes resistentes o intolerantes al tratamiento previo incluyendo imatinib). |
|
LMC en fase acelerada, LMC en fase blástica y LLA Ph+ (dosis inicial: 140 mg una vez al día) |
CAN < 0.5 × 109/L o Plaquetas < 10 × 109/L |
1. Verifique si es que la citopenia está relacionada con la leucemia (mediante un aspirado o biopsia de médula ósea). 2. Si la citopenia no está relacionada con la leucemia, interrumpa el tratamiento con SPRYCEL® hasta alcanzar un CAN ≥ 1.0 × 109/L y un conteo de plaquetas ≥ 20 × 109/L y reanude el tratamiento utilizando la dosis inicial original. 3. De presentarse una recurrencia de la citopenia, repita el paso 1 y reanude el tratamiento con SPRYCEL® utilizando una dosis reducida de 100 mg una vez al día (segundo episodio) o 70 mg una vez al día (tercer episodio). 4. Si la citopenia está relacionada con leucemia, considere incrementar la dosis a 180 mg una vez al día. |
* CAN: conteo absoluto de neutrófilos.
Reacciones adversas no hematológicas: En caso de desarrollarse una reacción adversa no hematológica de carácter severo con el uso de SPRYCEL®, el tratamiento deberá ser suspendido hasta que el evento remita o exhiba una mejoría. A partir de ese momento, el tratamiento puede ser reanudado según sea apropiado utilizando una dosis reducida dependiendo de la severidad y recurrencia del evento (Ver Precauciones generales, Precauciones y advertencias específicas del producto).
Deterioro renal: Actualmente no existen estudios clínicos de SPRYCEL® en pacientes con reducción de la función renal (el estudio en pacientes con LMC en fase crónica recientemente diagnosticada excluyó pacientes con una concentración de creatinina sérica > 3 veces el límite superior del rango normal, y estudios en pacientes con LMC en fase crónica resistentes o intolerantes a tratamiento previo con imatinib excluyeron pacientes con concentración de creatinina sérica > 1.5 veces el límite superior del rango normal). Ya que la depuración renal de dasatinib y sus metabolitos es menor a 4%, no se espera una disminución en la depuración corporal total en pacientes con insuficiencia renal.
Deterioro hepático: Con base en los hallazgos de un estudio farmacocinético de dosis únicas, los pacientes con insuficiencia hepática leve, moderada o grave pueden recibir la dosis de inicio recomendada (Ver Dosis y vía de administración, Dosis recomendada y Farmacocinética y farmacodinamia, Farmacocinética, Poblaciones especiales). Debido a las limitaciones de este estudio clínico, se recomienda tener precaución al administrar SPRYCEL® a pacientes con insuficiencia hepática.
Uso pediátrico: No se ha establecido la seguridad y eficacia de SPRYCEL® en pacientes menores de 18 años de edad.
Uso geriátrico: No se han observado diferencias farmacocinéticas relacionadas con la edad clínicamente relevantes. No es necesaria una recomendación de dosis específica para ancianos.
MANIFESTACIONES Y MANEJO DE LA SOBREDOSIFICACIÓN O INGESTA ACCIDENTAL: La experiencia con sobredosis de SPRYCEL® en estudios clínicos se limita a casos aislados. La mayor sobredosificación ingerida que ha sido registrada fue de 280 mg al día durante 1 semana en dos pacientes; ambos desarrollaron una disminución significativa en las cuentas plaquetarias. Ya que SPRYCEL® ha sido asociado con mielosupresión severa (Ver Precauciones generales, Precauciones y advertencias específicas del producto, Mielosupresión y Reacciones adversas y secundarias), los pacientes que ingieran una dosis mayor a la recomendada deben ser monitoreados estrechamente para detectar señales de mielosupresión y deben recibir el tratamiento de soporte adecuado.
PRESENTACIONES:
Frascos con 60 tabletas de 20 mg, 50 mg o 70 mg e instructivo anexo.
Frasco con 30 tabletas de 100 mg e instructivo anexo.
RECOMENDACIONES SOBRE ALMACENAMIENTO: Consérvese el frasco bien cerrado a no más de 30°C.
Manejo: SPRYCEL® tabletas consiste de una tableta comprimida (que contiene el principio activo), rodeada de una capa para prevenir la exposición del personal clínico y de farmacia al principio activo. Sin embargo, si las tabletas se rompen o trituran inadvertidamente, el personal clínico y de farmacia debe utilizar guantes para quimioterapia desechables para minimizar el riesgo de exposición dérmica. Cualquier producto o material de desecho no utilizado debe ser desechado conforme a los requerimientos locales.
LEYENDAS DE PROTECCIÓN:
Literatura exclusiva para médicos. Su venta requiere receta médica. Este medicamento deberá ser administrado únicamente por médicos especialistas en hematología u oncología y con experiencia en quimioterapia antineoplásica. No se use en el embarazo ni en la lactancia. No se deje al alcance de los niños.
Reporte las sospechas de reacción adversa a los correos:
farmacovigilancia@cofepris.gob.mx
Hecho en Estados Unidos por:
AstraZeneca Pharmaceuticals LP
4601 Highway 62 East
Mount Vernon, IN, 47620, EUA
Para:
Bristol-Myers Squibb Manufacturing Company.
Road 3, Km 77.5, Post Box 609, Humacao, PR-00791, EUA.
Importado y distribuido por:
Bristol-Myers Squibb de México, S. de R.L. de C.V.
Rancho 4 Milpas km 1 Carretera Tepotzotlán
La Aurora, MDC Fase II, Sección “D”
Col. Ex Hacienda San Miguel, C.P. 54715
Cuautitlán Izcalli, México, México
Representante legal:
BRISTOL-MYERS SQUIBB DE MÉXICO, S. de R.L. de C.V.
Avenida Insurgentes Sur No.1602, Piso 5
Col. Crédito Constructor, C.P. 03940
Benito Juárez, Ciudad de México, México
Reg. Núm. 104M2007, SSA IV
Fecha de CCDS: CCDS Dasatinib, 2 Dec 2016.
Fecha de aprobación registro sanitario: 19 Apr 2017.


