
SISABIN - Tableta
Sustancia(s):
- Abiraterona
Presentaciones:
- 1 Caja, 120 Tabletas, 250 mg
- 1 Caja, 60 Tabletas, 500 mg
FORMA FARMACÉUTICA Y FORMULACIÓN:
Cada TABLETA contiene:
Acetato de abiraterona 250 y 500 mg
Excipiente cbp 1 tableta
INDICACIONES TERAPÉUTICAS:
En el tratamiento de pacientes con cáncer de próstata metastásico de alto riesgo que no han recibido tratamiento hormonal (CPmHN) o en pacientes con cáncer de próstata metastásico hormono sensible (CPmHS) de alto riesgo de reciente diagnóstico en combinación con prednisona o prednisolona y terapia de privación androgénica (TPA).
En el tratamiento de pacientes con cáncer de próstata metastásico resistente a la castración que son asintomáticos o con síntomas leves después del fracaso de la terapia de privación androgénica, indicado en combinación con prednisona o prednisolona.
En el tratamiento de cáncer de próstata avanzado metastásico (cáncer de próstata resistente a la castración) en pacientes que han recibido una quimioterapia previa con un taxano, indicado en combinación con prednisona o prednisolona.
FARMACOCINÉTICA Y FARMACODINAMIA:
Farmacocinética: Tras la administración de acetato de abiraterona, se ha estudiado la farmacocinética de abiraterona y acetato de abiraterona en sujetos sanos, pacientes con cáncer de próstata avanzado metastásico y sujetos sin cáncer con insuficiencia hepática o renal. El acetato de abiraterona se convierte rápidamente in vivo en abiraterona, un inhibidor de la biosíntesis de andrógenos (ver sección Farmacodinamia).
Absorción: Después de la administración oral de acetato de abiraterona en ayunas, el tiempo para alcanzar la concentración máxima de abiraterona en plasma es de aproximadamente 2 horas.
La administración de acetato de abiraterona con alimentos, en comparación con la administración en ayunas, da como resultado un aumento de hasta 10 veces (AUC) y hasta 17 veces (Cmáx) en la exposición sistémica media de abiraterona, dependiendo del contenido de grasa de la comida. Dada la variación normal en el contenido y la composición de las comidas, tomar abiraterona con las comidas tiene el potencial de resultar en exposiciones muy variables. Por lo tanto, la abiraterona no debe tomarse con alimentos. Se debe tomar al menos dos horas después de comer y no se debe comer ningún alimento durante al menos una hora después de tomar abiraterona. Los comprimidos se deben tragar enteros con agua (ver sección Dosis y vía de administración).
Distribución: La unión a proteínas plasmáticas de la abiraterona 14C en plasma humano es del 99.8%. El volumen aparente de distribución es de aproximadamente 5,630 L, lo que sugiere que la abiraterona se distribuye ampliamente a los tejidos periféricos.
Biotransformación: Después de la administración oral de acetato de 14C-abiraterona en cápsulas, el acetato de abiraterona se hidroliza a abiraterona, que luego sufre metabolismo que incluye sulfatación, hidroxilación y oxidación principalmente en el hígado. La mayoría de la radioactividad circulante (aproximadamente 92%) se encuentra en forma de metabolitos de abiraterona. De 15 metabolitos detectables, 2 metabolitos principales, sulfato de abiraterona y sulfato de abiraterona N-óxido, cada uno representa aproximadamente el 43% de la radiactividad total.
Eliminación: La vida media de la abiraterona en plasma es de aproximadamente 15 horas, según los datos de sujetos sanos. Después de la administración oral de 1,000 mg de acetato de 14C-abiraterona, aproximadamente el 88% de la dosis radiactiva se recupera en las heces y aproximadamente el 5% en la orina. Los principales compuestos presentes en las heces son el acetato de abiraterona inalterado y abiraterona (aproximadamente 55 y 22% de la dosis administrada, respectivamente).
Deterioro hepático: La farmacocinética del acetato de abiraterona se examinó en sujetos con insuficiencia hepática leve o moderada preexistente (Child-Pugh Clase A y B, respectivamente) y en sujetos de control sanos. La exposición sistémica a la abiraterona después de una sola dosis oral de 1,000 mg aumentó en aproximadamente 11 y 260% en sujetos con insuficiencia hepática preexistente leve y moderada, respectivamente. La vida media de la abiraterona se prolonga a aproximadamente 18 horas en sujetos con insuficiencia hepática leve y a aproximadamente 19 horas en sujetos con insuficiencia hepática moderada.
En otro ensayo, se examinó la farmacocinética de la abiraterona en sujetos con insuficiencia hepática grave preexistente (n = 8) (Child-Pugh Clase C) y en 8 sujetos control sanos con función hepática normal. El ABC de la abiraterona aumentó en aproximadamente un 600% y la fracción de fármaco libre aumentó en un 80% en sujetos con insuficiencia hepática grave en comparación con sujetos con función hepática normal.
No es necesario ajustar la dosis en pacientes con insuficiencia hepática leve preexistente.
El uso de acetato de abiraterona debe evaluarse con precaución en pacientes con insuficiencia hepática moderada en los que el beneficio claramente debe superar el posible riesgo (ver secciones Dosis y vía de administración y Precauciones generales). El acetato de abiraterona no debe utilizarse en pacientes con insuficiencia hepática grave (ver secciones Dosis y vía de administración, Contraindicaciones y Precauciones generales).
Para los pacientes que desarrollan hepatotoxicidad durante el tratamiento, puede ser necesario suspender el tratamiento y ajustar la dosis (ver secciones Dosis y vía de administración y Precauciones generales).
Insuficiencia renal: La farmacocinética del acetato de abiraterona se comparó en pacientes con enfermedad renal en etapa terminal en un programa de hemodiálisis estable versus sujetos de control pareados con función renal normal. La exposición sistémica a la abiraterona después de una dosis oral única de 1000 mg no aumentó en sujetos con enfermedad renal terminal en diálisis. La administración en pacientes con insuficiencia renal, incluida la insuficiencia renal grave, no requiere reducción de la dosis (ver sección Dosis y vía de administración). Sin embargo, no existe experiencia clínica en pacientes con cáncer de próstata e insuficiencia renal grave. Se recomienda precaución en estos pacientes.
Farmacodinamia:
Grupo farmacoterapéutico: Terapia endocrina, otros antagonistas hormonales y agentes relacionados.
Código ATC: L02BX03
Mecanismo de acción: El acetato de abiraterona se convierte in vivo en abiraterona, un inhibidor de la biosíntesis de andrógenos. Específicamente, la abiraterona inhibe selectivamente la enzima 17α-hidroxilasa / C17,20-liasa (CYP17). Esta enzima se expresa y se requiere para la biosíntesis de andrógenos en los tejidos tumorales testiculares, suprarrenales y prostáticos. CYP17 cataliza la conversión de pregnenolona y progesterona en precursores de testosterona, DHEA y androstenediona, respectivamente, por 17α-hidroxilación y escisión del enlace C17,20. La inhibición de CYP17 también produce un aumento de la producción de mineralocorticoides en las glándulas suprarrenales (ver sección Precauciones generales).
El carcinoma prostático sensible a andrógenos responde al tratamiento que disminuye los niveles de andrógenos. Las terapias de deprivación androgénica, tales como el tratamiento con agonistas LHRH o la orquiectomía, disminuyen la producción androgénica en los testículos, pero no afecta la producción androgénica por las suprarrenales o en el tumor. El tratamiento con abiraterona disminuye la testosterona sérica a niveles indetectables (usando pruebas comerciales) al administrarse con agonistas LHRH (u orquiectomía).
Efectos farmacodinámicos: La abiraterona disminuye la testosterona sérica y otros andrógenos a niveles inferiores a los alcanzados por el uso de análogos de LHRH solos o por orquiectomía. Esto resulta de la inhibición selectiva de la enzima CYP17 requerida para la biosíntesis de andrógenos. El PSA sirve como biomarcador en pacientes con cáncer de próstata. En un estudio clínico de Fase 3 de pacientes que fallaron la quimioterapia previa con taxanos, el 38% de los pacientes tratados con acetato de abiraterona, frente al 10% de los pacientes tratados con placebo, tuvieron al menos un 50% de disminución desde los valores basales de PSA.
Eficacia clínica y seguridad: La eficacia se estableció en tres ensayos clínicos aleatorizados controlados con placebo de fase 3 multicéntricos (estudios 302 y 301) de pacientes con mHSPC y mCRPC. El estudio 302 reclutó pacientes sin tratamiento con docetaxel; mientras que el estudio 301 inscribió pacientes que habían recibido docetaxel previo. Los pacientes estaban usando un análogo de LHRH o fueron tratados previamente con orquiectomía. En el grupo de tratamiento activo, se administró abiraterona a una dosis de 1000 mg diarios en combinación con dosis bajas de prednisona o prednisolona 5 mg dos veces al día. Los pacientes control recibieron placebo y dosis bajas de prednisona o prednisolona 5 mg dos veces al día.
Los cambios en la concentración sérica de PSA de forma independiente no siempre predicen el beneficio clínico. Por lo tanto, en todos los estudios se recomendó que se mantuviera a los pacientes en sus tratamientos de estudio hasta que se cumplieran los criterios de interrupción, como se especifica a continuación para cada estudio.
En todos los estudios, el uso de espironolactona no estaba permitido, ya que la espironolactona se une al receptor de andrógenos y puede aumentar los niveles de PSA.
Estudio 302 (pacientes sin tratamiento previo con quimioterapia): Este estudio incluyó pacientes sin tratamiento previo con quimioterapia que eran asintomáticos o levemente sintomáticos y para quienes la quimioterapia aún no estaba clínicamente indicada. Se consideró asintomático una puntuación de 0-1 en el peor dolor del Inventario breve de dolor en forma breve (BPI-SF) en las últimas 24 horas, y una puntuación de 2-3 se consideró levemente sintomática.
En el estudio 302, (n = 1,088) la edad media de los pacientes inscritos fue de 71 años para los pacientes tratados con abiraterona más prednisona o prednisolona y 70 años para los pacientes tratados con placebo más prednisona o prednisolona. El número de pacientes tratados con abiraterona por grupo racial fue Caucásico 520 (95.4%), Negro 15 (2.8%), Asiático 4 (0.7%) y otros 6 (1.1%). El estado de rendimiento del Eastern Cooperative Oncology Group (ECOG) fue 0 para el 76% de los pacientes y 1 para el 24% de los pacientes en ambos brazos. El cincuenta por ciento de los pacientes sólo tenía metástasis óseas, un 31% adicional de pacientes tenía metástasis óseas y de tejidos blandos o ganglios linfáticos, y el 19% de los pacientes sólo tenía metástasis de tejidos blandos o ganglios linfáticos. Se excluyeron pacientes con metástasis viscerales. Los puntos finales de eficacia coprimarios fueron la supervivencia global y la supervivencia libre de progresión radiográfica (rPFS). Además de las medidas de criterio de valoración coprimarias, el beneficio también se evaluó utilizando el tiempo para el uso de opiáceos para el dolor por cáncer, el tiempo hasta el inicio de la quimioterapia citotóxica, el tiempo hasta el deterioro en el puntaje de rendimiento del ECOG en ≥1 punto y el tiempo hasta la progresión del PSA según el cáncer de próstata Criterios del Grupo de trabajo-2 (PCWG2). Los tratamientos del estudio se interrumpieron en el momento de la progresión clínica inequívoca. Los tratamientos también podrían suspenderse en el momento de la progresión radiográfica confirmada a discreción del investigador.
La supervivencia libre de progresión radiográfica (rPFS) se evaluó mediante el uso de estudios de imágenes secuenciales según los criterios PCWG2 (para lesiones óseas) y los criterios de evaluación de respuesta modificada en tumores sólidos (RECIST) (para lesiones de tejidos blandos). El análisis de rPFS utilizó una evaluación radiográfica de progresión revisada centralmente.
En el análisis planificado de rPFS hubo 401 eventos, 150 (28%) de los pacientes tratados con abiraterona y 251 (46%) de los pacientes tratados con placebo tenían evidencia radiográfica de progresión o habían muerto. Se observó una diferencia significativa en rPFS entre los grupos de tratamiento (ver Tabla 1 y Figura 1).
Tabla 1. Estudio 302: supervivencia libre de progresión radiográfica de pacientes tratados con abiraterona o placebo en combinación con prednisona o prednisolona más análogos de LHRH u orquiectomía previa
|
Abiraterone (N = 546) |
Placebo (N = 542) |
|
|
Supervivencia libre de progresión radiológica (SLPr) |
||
|
Progresión o muerte |
150 (28%) |
251 (46%) |
|
Mediana de SLPr en meses (IC del 95%) |
No alcanzada (11.66; NE) |
8.3 (8.12; 8.54) |
|
Valor p* |
<0.0001 |
|
|
Hazard ratio** (IC del 95%) |
0.425 (0.347; 0.522) |
|
NE = no estimado.
* El valor p se deriva de una prueba de log-rank estratificada por la puntuación de ECOG basal (0 o 1).
** La razón de riesgo <1 favorece a la abiraterona.
Figura 1. Curvas de Kaplan Meier de la supervivencia libre de progresión radiológica en los pacientes tratados con abiraterona o placebo en combinación con prednisona o prednisolona más análogos de la LHRH u orquiectomía previa
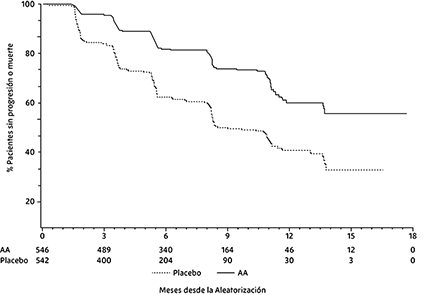
Sin embargo, los datos de los sujetos continuaron siendo recopilados hasta la fecha del segundo análisis intermedio de Supervivencia general (SG). La revisión radiográfica del investigador de rPFS realizada como un análisis de sensibilidad de seguimiento se presenta en la Tabla 2 y la Figura 2.
Seiscientos siete (607) sujetos tuvieron progresión radiográfica o murieron; 271 (50%) en el grupo de acetato de abiraterona y 336 (62%) en el grupo de placebo. El tratamiento con acetato de abiraterona disminuyó el riesgo de progresión radiográfica o muerte en un 47% en comparación con el placebo (HR = 0,530; IC del 95%: [0,451; 0,623], p <0,0001). La mediana de rPFS fue de 16,5 meses en el grupo de acetato de abiraterona y 8,3 meses en el grupo de placebo.
Tabla 2. Estudio 302: supervivencia libre de progresión radiográfica de pacientes tratados con abiraterona o placebo en combinación con prednisona o prednisolona más análogos de LHRH u orquiectomía previa (en el segundo análisis intermedio de OS-Investigator Review)
|
Abiraterona (N = 546) |
Placebo (N = 542) |
|
|
Supervivencia libre de progresión radiológica (SLPr) |
||
|
Progresión o muerte |
271 (50%) |
336 (62%) |
|
Mediana de rPFS en meses (IC 95%) |
16.5 (13.80; 16.79) |
8.3 (8.05; 9.43) |
|
Valor p* |
<0.0001 |
|
|
Cociente de riesgo** (IC 95%) |
0.530 (0.451; 0.623) |
|
* El valor p se deriva de una prueba de log-rank estratificada por la puntuación de ECOG de referencia (0 o 1).
** La relación de riesgo <1 favorece a la abiraterona.
Figura 2. Curvas de Kaplan Meier de la supervivencia libre e progresión radiológica en los pacientes tratados con abiraterona o placebo en combinación con prednisona o prednisolona más análogos de la LHRH u orquiectomía previa (En el segundo análisis intermedio de la SG - revisión del investigador)
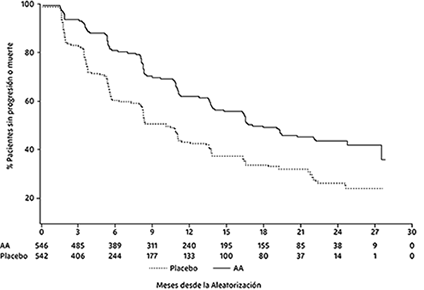
AA=Abiraterona
Se realizó un análisis provisional planificado (IA) para el sistema operativo después de que se observaron 333 muertes. El estudio no fue cegado según la magnitud del beneficio clínico observado y se ofreció a los pacientes en el grupo placebo el tratamiento con abiraterona. La supervivencia general fue más larga para la abiraterona que para el placebo, con una reducción del 25% en el riesgo de muerte (HR = 0.752; IC del 95%: [0.606; 0.934], p = 0.0097), pero la SG no fue madura y los resultados provisionales no alcanzaron el prelímite de detención especificado para la significación estadística (ver Tabla 1). Se siguió siguiendo la supervivencia después de esta IA.
El análisis final planificado para la SG se realizó después de observar 741 muertes (mediana de seguimiento de 49 meses). El sesenta y cinco por ciento (354 de 546) de los pacientes tratados con abiraterona, en comparación con el 71% (387 de 542) de los pacientes tratados con placebo, habían muerto. Se demostró un beneficio estadísticamente significativo de la SG a favor del grupo tratado con abiraterona con una reducción del 19.4% en el riesgo de muerte (HR = 0.806; IC del 95%: [0.697; 0.931], p = 0.0033) y una mejora en la mediana de la SG de 4,4 meses (abiraterona 34,7 meses, placebo 30,3 meses) (ver Tabla 3 y Figura 3). Esta mejora se demostró a pesar de que el 44% de los pacientes en el grupo de placebo recibieron abiraterona como terapia posterior.
Tabla 3. Estudio 302: Supervivencia global de pacientes tratados con abiraterona o placebo en combinación con prednisona o prednisolona más análogos de LHRH u orquiectomía previa
|
Abiraterona (N = 546) |
Placebo (N = 542) |
|
|
Análisis provisional de supervivencia |
||
|
Muertes (%) |
147 (27%) |
186 (34%) |
|
Análisis provisional de supervivencia (IC 95%) |
No alcanzado (NE; NE) |
27.2 (25.95; NE) |
|
Valor p* |
0.0097 |
|
|
Índice de riesgo** (IC 95%) |
0.752 (0.606; 0.934) |
|
|
Análisis final de supervivencia |
||
|
Muertes |
354 (65%) |
387 (71%) |
|
Mediana de supervivencia global en meses (IC 95%) |
34.7 (32.7; 36.8) |
30.3 (28.7; 33.3) |
|
Valor p* |
0.0033 |
|
|
Índice de riesgo** (IC 95%) |
0.806 (0.697; 0.931) |
|
NE = no estimado
* El valor p se deriva de una prueba de log-rank estratificada por la puntuación de ECOG de referencia (0 o 1).
** La relación de riesgo <1 favorece a la abiraterona.
Figura 3: Curvas de supervivencia de Kaplan Meier de los pacientes tratados con abiraterona o placebo en combinación con prednisona o prednisolona más análogos de la LHRH u orquiectomía previa, análisis final
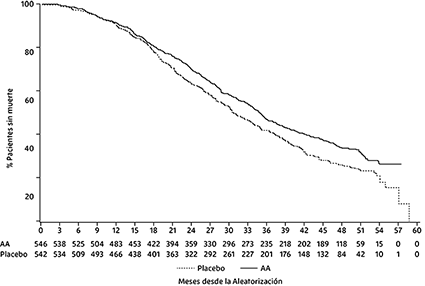
AA=Abiraterona
Además de las mejoras observadas en la supervivencia general y la rPFS, se demostró un beneficio para el tratamiento con abiraterona versus placebo en todas las medidas de criterio de valoración secundarias de la siguiente manera:
Tiempo hasta la progresión del PSA según los criterios de PCWG2: La mediana del tiempo hasta la progresión del PSA fue de 11.1 meses para los pacientes que recibieron abiraterona y 5.6 meses para los pacientes que recibieron placebo (HR = 0.488; IC 95%: [0.420; 0.568], p <0.0001). El tiempo hasta la progresión del PSA se duplicó aproximadamente con el tratamiento con abiraterona (HR = 0,488). La proporción de sujetos con una respuesta confirmada de PSA fue mayor en el grupo de abiraterona que en el grupo de placebo (62% frente a 24%; p <0,0001). En sujetos con enfermedad medible de tejidos blandos, se observó un aumento significativo en el número de respuestas tumorales completas y parciales con el tratamiento con abiraterona.
Tiempo para el uso de opiáceos para el dolor por cáncer: La mediana del tiempo para el uso de opiáceos para el dolor por cáncer de próstata en el momento del análisis final fue de 33,4 meses para los pacientes que recibieron abiraterona y de 23,4 meses para los pacientes que recibieron placebo (HR = 0.721; IC del 95%: [0.614; 0.846], p <0.0001).
Tiempo hasta el inicio de la quimioterapia citotóxica: La mediana del tiempo hasta el inicio de la quimioterapia citotóxica fue de 25.2 meses para los pacientes que recibieron abiraterona y 16.8 meses para los pacientes que recibieron placebo (HR = 0.580; IC 95%: [0.487; 0.691], p <0.0001).
Tiempo hasta el deterioro en la puntuación de rendimiento del ECOG en ≥1 punto: La mediana del tiempo hasta el deterioro en la puntuación de rendimiento del ECOG en ≥1 punto fue de 12.3 meses para los pacientes que recibieron abiraterona y 10.9 meses para los pacientes que recibieron placebo (HR = 0.821; IC del 95%: [0.714; 0.943], p = 0.0053).
Los siguientes criterios de valoración del estudio demostraron una ventaja estadísticamente significativa a favor del tratamiento con abiraterona:
Respuesta objetiva: La respuesta objetiva se definió como la proporción de sujetos con enfermedad medible que lograron una respuesta completa o parcial de acuerdo con los criterios RECIST (se requería que el tamaño basal de los ganglios linfáticos fuera ≥2 cm para ser considerado una lesión objetivo). La proporción de sujetos con enfermedad medible al inicio del estudio que tuvieron una respuesta objetiva fue del 36% en el grupo de abiraterona y del 16% en el grupo de placebo (p <0,0001).
Dolor: El tratamiento con abiraterona redujo significativamente el riesgo de progresión de la intensidad del dolor promedio en un 18% en comparación con el placebo (p = 0.0490). La mediana del tiempo hasta la progresión fue de 26.7 meses en el grupo de abiraterona y 18.4 meses en el grupo de placebo.
Tiempo de degradación en el FACT-P (puntaje total): El tratamiento con abiraterona disminuyó el riesgo de degradación del FACT-P (puntaje total) en un 22% en comparación con el placebo (p = 0,0028). La mediana del tiempo hasta la degradación en FACT-P (puntuación total) fue de 12.7 meses en el grupo de abiraterona y 8.3 meses en el grupo de placebo.
Estudio 301 (pacientes que habían recibido quimioterapia previa): Estudio 301 pacientes inscritos que habían recibido docetaxel previo. No se requirió que los pacientes mostraran progresión de la enfermedad con docetaxel, ya que la toxicidad de esta quimioterapia puede haber llevado a la interrupción. Los pacientes se mantuvieron en los tratamientos del estudio hasta que hubo una progresión del PSA (aumento confirmado del 25% sobre la línea de base/nadir del paciente) junto con la progresión radiográfica definida por el protocolo y la progresión sintomática o clínica. Los pacientes con tratamiento previo con ketoconazol para el cáncer de próstata fueron excluidos de este estudio. La variable principal de eficacia fue la supervivencia global.
La mediana de edad de los pacientes inscritos fue de 69 años (rango 39-95). El número de pacientes tratados con abiraterona por grupo racial fue de raza caucásica 737 (93.2%), negra 28 (3.5%), asiática 11 (1.4%) y otros 14 (1.8%). El once por ciento de los pacientes inscritos tenían una puntuación de rendimiento ECOG de 2; el 70% tenía evidencia radiográfica de progresión de la enfermedad con o sin progresión del PSA; el 70% había recibido una quimioterapia citotóxica previa y el 30% recibió dos. La metástasis hepática estuvo presente en el 11% de los pacientes tratados con abiraterona.
En un análisis planificado realizado después de que se observaron 552 muertes, el 42% (333 de 797) de los pacientes tratados con abiraterona en comparación con el 55% (219 de 398) de los pacientes tratados con placebo, habían muerto. Se observó una mejora estadísticamente significativa en la mediana de supervivencia global en pacientes tratados con abiraterona (ver Tabla 4).
Tabla 4. Supervivencia global de pacientes tratados con abiraterona o placebo en combinación con prednisona o prednisolona más análogos de LHRH u orquiectomía previa
|
Abiraterona (N = 797) |
Placebo (N = 398) |
|
|
Análisis primario de supervivencia |
||
|
Muertes (%) |
333 (42%) |
219 (55%) |
|
Mediana de supervivencia (meses) (IC 95%) |
14.8 (14.1;15.4) |
10.9 (10.2;12.0) |
|
Valor pa |
<0.0001 |
|
|
Índice de riesgo (IC 95%)b |
0.646 (0.543; 0.768) |
|
|
Análisis de supervivencia actualizado |
||
|
Muertes |
501 (63%) |
274 (69%) |
|
Supervivencia media (meses) (IC 95%) |
15.80 (14.8; 17.0) |
11.2 (10.4; 13.1) |
|
Índice de riesgo (IC 95%)b |
0.740 (0.638; 0.859) |
|
a El valor p se deriva de una prueba de log-rank estratificada por la puntuación del estado de rendimiento del ECOG (0-1 frente a 2), la puntuación del dolor (ausente frente al presente), el número de regímenes de quimioterapia previos (1 frente a 2) y el tipo de progresión de la enfermedad (sólo PSA versus radiografía).
b La razón de riesgo se deriva de un modelo de riesgos proporcionales estratificados. La relación de riesgo <1 favorece a la abiraterona.
En todos los puntos de tiempo de evaluación después de los primeros meses de tratamiento, una mayor proporción de pacientes tratados con abiraterona se mantuvo viva, en comparación con la proporción de pacientes tratados con placebo (ver Figura 4).
Figura 4. Curvas de supervivencia de Kaplan Meier de los pacientes tratados con abiraterona o placebo en combinación con prednisona o prednisolona más análogos de la LHRH u orquiectomía previa
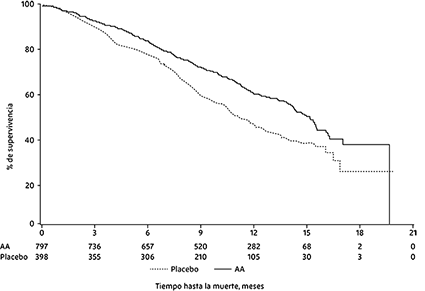
AA=Abiraterona
Los análisis de supervivencia de subgrupos mostraron un beneficio de supervivencia constante para el tratamiento con abiraterona (ver Figura 5).
Figura 5. Supervivencia global por subgrupos: Cociente de riesgo (HR) e intervalo de confianza del 95%
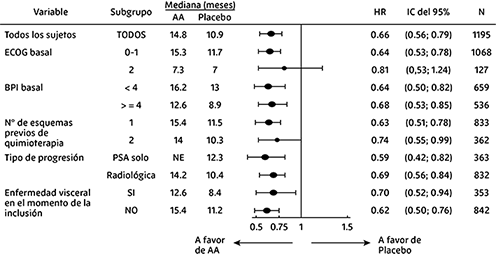
Además de la mejora observada en la supervivencia general, todos los criterios de valoración secundarios del estudio favorecieron a la abiraterona y fueron estadísticamente significativos después de ajustar para múltiples pruebas de la siguiente manera: Los pacientes que recibieron abiraterona demostraron una tasa de respuesta de PSA total significativamente más alta (definida como una reducción ≥50% del valor inicial), en comparación con los pacientes que recibieron placebo, 38% frente a 10%, p <0,0001.
La mediana del tiempo hasta la progresión del PSA fue de 10.2 meses para los pacientes tratados con abiraterona y 6.6 meses para los pacientes tratados con placebo (HR = 0.580; IC 95%: [0.462; 0.728], p <0.0001).
La mediana de supervivencia libre de progresión radiográfica fue de 5.6 meses para los pacientes tratados con abiraterona y 3.6 meses para los pacientes que recibieron placebo (HR = 0.673; IC 95%: [0.585; 0.776], p <0.0001).
Dolor: La proporción de pacientes con paliación del dolor fue estadísticamente significativamente mayor en el grupo de abiraterona que en el grupo de placebo (44 frente a 27%, p = 0.0002). Un respondedor para la paliación del dolor se definió como un paciente que experimentó al menos una reducción del 30% desde el inicio en el puntaje de intensidad de dolor peor BPI-SF en las últimas 24 horas sin ningún aumento en el puntaje de uso de analgésicos observado en dos evaluaciones consecutivas con cuatro semanas de diferencia. Sólo los pacientes con una puntuación de dolor basal de ≥4 y al menos una puntuación de dolor postbasal se analizaron (N = 512) para la paliación del dolor.
Una proporción menor de pacientes tratados con abiraterona tuvo progresión del dolor en comparación con los pacientes que tomaron placebo a los 6 (22 frente a 28%), 12 (30 frente a 38%) y 18 meses (35 frente a 46%). La progresión del dolor se definió como un aumento desde el inicio de ≥30% en el puntaje de intensidad de dolor peor BPI-SF durante las 24 horas anteriores sin una disminución en el puntaje de uso de analgésicos observado en dos visitas consecutivas, o un aumento de ≥30% en el uso de analgésicos puntuación observada en dos visitas consecutivas. El tiempo de progresión del dolor en el percentil 25 fue de 7.4 meses en el grupo de abiraterona, frente a 4.7 meses en el grupo de placebo.
Eventos relacionados con el esqueleto: Una proporción menor de pacientes en el grupo de abiraterona tuvo eventos relacionados con el esqueleto en comparación con el grupo de placebo a los 6 meses (18 frente a 28%), 12 meses (30 frente a 40%) y 18 meses (35 frente a 40%). El tiempo hasta el primer evento relacionado con el esqueleto en el percentil 25 en el grupo de abiraterona fue el doble que el del grupo control a los 9.9 meses versus los 4.9 meses. Un evento relacionado con el esqueleto se definió como una fractura patológica, compresión de la médula espinal, radiación paliativa al hueso o cirugía al hueso.
Población pediátrica: La Agencia Europea de Medicamentos ha renunciado a la obligación de presentar los resultados de los estudios con abiraterona en todos los subgrupos de la población pediátrica en cáncer de próstata avanzado. Consulte la sección Dosis y vía de administración para obtener información sobre el uso pediátrico.
CONTRAINDICACIONES:
• Hipersensibilidad al principio activo o a alguno de los excipientes.
• Menores de 18 años.
• Insuficiencia hepática grave (Child-Pugh Clase C).
• Abiraterona con prednisona o prednisolona está contraindicada en combinación con Ra-223.
• Mujeres; especialmente si están o pueden estar embarazadas o están amamantando.
RESTRICCIONES DE USO DURANTE EL EMBARAZO Y LA LACTANCIA:
Mujeres en edad fértil: Este medicamento no debe usarse en mujeres en edad fértil.
Anticoncepción en hombres y mujeres: No se sabe si la abiraterona o sus metabolitos están presentes en el semen. Se requiere un condón si el paciente participa en una actividad sexual con una mujer embarazada. Si el paciente tiene relaciones sexuales con una mujer en edad fértil, se requiere un condón junto con otro método anticonceptivo eficaz. Los estudios en animales han demostrado toxicidad reproductiva (ver sección Precauciones en relación con efectos de carcinogénesis, mutagénesis, teratogénesis y sobre la fertilidad).
El embarazo: Abiraterona no se usa en mujeres.
Amamantamiento: Abiraterona no se usa en mujeres.
REACCIONES SECUNDARIAS Y ADVERSAS:
A lo largo de esta sección, se presentan las reacciones adversas. Las reacciones adversas son eventos adversos que se consideran razonablemente asociados con el uso de acetato de abiraterona basado en la evaluación integral de la información de eventos adversos disponible. No se puede establecer una relación causal confiable con acetato de abiraterona en casos individuales. Además, como los estudios clínicos son llevados a cabo bajo condiciones muy variables, las tasas de reacciones adversas observadas en los estudios clínicos de un medicamento no pueden ser directamente comparados con las tasas en los estudios clínicos de otro medicamento y pueden no reflejar las tasas observadas en la práctica clínica.
En un análisis de reacciones adversas de estudios compuestos de Fase 3 con abiraterona, las reacciones adversas observadas en ≥10% de los pacientes fueron hipertensión, edema periférico, hipopotasemia, infección de vías urinarias y aumento de alanino aminotransferasa y/o aumento de aspartato aminotransferasa.
Abiraterona puede causar hipertensión, hipopotasemia y retención de líquidos como una consecuencia farmacodinámica de su mecanismo de acción. En estudios Fase 3 se observaron efectos mineralocorticoides esperados con mayor frecuencia en los pacientes tratados con abiraterona frente a los pacientes tratados con placebo: hipopotasemia 18 contra 8%, hipertensión 22 contra 16% y retención de líquidos (edema periférico) 23 contra 17%, respectivamente. En los pacientes tratados con Abiraterona vs. pacientes tratados con placebos se observó hipopotasemia grado 3 y 4 en 6 y 2% de los pacientes, se observó hipertensión grado 3 y 4 en 8 y 5% de los pacientes y se observó edema de retención de líquidos grado 3 y 4 en 1 y 1% de los pacientes, respectivamente. Los efectos mineralocorticoides generalmente se manejaron médicamente de forma exitosa. El uso concomitante de un corticosteroide disminuye la incidencia y severidad de estas reacciones adversas (ver Precauciones generales - Hipertensión, hipopotasemia y retención de líquidos por exceso de mineralocorticoides).
En un estudio Fase 3 de pacientes con cáncer de próstata avanzado metastásico (estudio 301) que se encontraban usando un agonista LHRH o fueron tratados previamente con orquiectomía, se administró abiraterona a una dosis de 1000 mg en combinación con una dosis baja de prednisona o prednisolona (10 mg diariamente) en el brazo del tratamiento activo; se administró placebo más una dosis baja de prednisona o prednisolona (10 mg diariamente) a los pacientes control. Los pacientes incluidos eran intolerantes o habían fallado a hasta dos esquemas previos de quimioterapia, uno de los cuales contenía un taxano. La duración promedio del tratamiento con abiraterona fue de 8 meses.
Las reacciones adversas que ocurrieron en una tasa de ≥1% (todos los grados) se muestran en la Tabla 5.
Tabla 5. Reacciones adversas en ≥1% de los pacientes en el Estudio 301a
|
Clase de Sistemas/órganos Reacción adversa |
Abiraterona 1000 mg diarios con prednisona o prednisolona (10 mg) n = 791b |
Placebo con prednisona o prednisolona (10 mg) n = 394b |
||||
|---|---|---|---|---|---|---|
|
Todos los grados % |
Grado 3 % |
Grado 4 % |
Todos los Grados % |
Grado 3 % |
Grado 4 % |
|
|
Trastornos generales y condiciones del sitio de administración |
||||||
|
Edema periférico |
25 |
1 |
<1 |
17 |
1 |
0 |
|
Trastornos metabólicos y de la nutrición |
||||||
|
Hipopotasemia |
17 |
3 |
<1 |
8 |
1 |
0 |
|
Hipertrigliceridemia |
1 |
<1 |
0 |
0 |
0 |
0 |
|
Infecciones e infestaciones |
||||||
|
Infección de vías urinarias |
12 |
2 |
0 |
7 |
1 |
0 |
|
Trastornos hepatobiliares |
||||||
|
Incremento de la alanina aminotransferasa |
3 |
1 |
0 |
1 |
<1 |
<1 |
|
Trastornos vasculares |
||||||
|
Hipertensión |
9 |
1 |
0 |
7 |
<1 |
0 |
|
Lesiones traumáticas, intoxicaciones y complicaciones de procedimientos |
||||||
|
Fracturasc |
6 |
1 |
<1 |
2 |
0 |
0 |
|
Trastornos cardiacos |
||||||
|
Insuficiencia cardiacac |
2 |
2 |
<1 |
1 |
0 |
<1 |
|
Angina de pecho |
1 |
<1 |
0 |
1 |
0 |
0 |
|
Arritmia |
1 |
0 |
0 |
0 |
0 |
0 |
|
Fibrilación auricular |
2 |
1 |
0 |
1 |
1 |
0 |
|
Taquicardia |
3 |
0 |
0 |
2 |
0 |
0 |
a Todos los pacientes estaban recibiendo un agonista LHRH o se habían sometido a una orquidectomía.
b n = pacientes evaluados para seguridad.
c Fracturas incluye todas las fracturas con excepción de fracturas patológicas.
d Insuficiencia cardiaca incluye también insuficiencia cardiaca congestiva, falla del ventrículo izquierdo y disminución de la fracción de eyección.
En un segundo estudio clínico controlado con placebo, multicéntrico, Fase 3 (estudio 302), en pacientes asintomáticos, o con síntomas leves, que no han recibido quimioterapia, con cáncer de próstata metastásico avanzado que estuvieron utilizando un agonista LHRH o fueron previamente tratados con orquiectomía, Abiraterona fue también administrado a dosis de 1000 mg diariamente en combinación con una dosis baja de prednisona o prednisolona 10 mg diariamente en el brazo de tratamiento activo. Se administró placebo y bajas dosis de prednisona o prednisolona (10 mg diariamente) a los pacientes control. La duración promedio del tratamiento con abiraterona en el estudio 302 fue de 13.8 meses.
Las reacciones adversas que ocurrieron en una tasa de ≥1% (todos los grados) se muestran en la Tabla 6.
Tabla 6. Reacciones adversas en ≥1% de los pacientes en un estudio 302a
|
Clase de sistema/órgano Reacción adversa |
Abiraterona 1000 mg diarios con prednisona o prednisolona (10 mg) n = 542b |
Placebo con prednisona o prednisolona (10 mg) n = 540b |
||||
|
Todos los grados % |
Grado 3 % |
Grado 4 % |
Todos los grados % |
Grado 3 % |
Grado 4 % |
|
|
Trastornos gastrointestinales |
||||||
|
Dispepsia |
11 |
0 |
0 |
5 |
<1 |
0 |
|
Trastornos hepatobiliares |
||||||
|
Aumento de alanina aminotransferasa |
12 |
5 |
1 |
5 |
1 |
<1 |
|
Aumento de aspartato aminotransferasa |
11 |
3 |
0 |
5 |
1 |
0 |
|
Trastornos renales y urinarios |
||||||
|
Hematuria |
10 |
1 |
0 |
6 |
1 |
0 |
a Todos los pacientes estaban recibiendo un agonista LHRH o se habían sometido a una orquiectomía.
b n = pacientes evaluados para seguridad.
Las reacciones adversas más frecuentes que dieron como resultado la descontinuación del medicamento en los datos combinados de estudios de Fase 3 fueron incremento de la alanina aminotransferasa, incremento de aspartato aminotransferasa e hipopotasemia (cada una en <1% de los pacientes que tomaron abiraterona).
La reacción adversa, insuficiencia suprarrenal, ocurrió en los estudios clínicos Fase 3 en una tasa de 0.3% en pacientes que tomaron abiraterona y en una tasa de 0.1% en los pacientes que tomaban placebo.
En los estudios Fase 3, 70% de los pacientes eran mayores de 65 años y 27% eran mayores de 75 años, para los pacientes que tomaron abiraterona. No se observaron diferencias generales en la seguridad entre los pacientes mayores y los más jóvenes.
Efectos cardiovasculares: Los tres estudios fase 3 excluyeron pacientes con hipertensión descontrolada, enfermedades cardiacas clínicamente significativas como evidencia de infarto al miocardio, eventos trombóticos arteriales en los 6 meses previos, angina severa o inestable, insuficiencia cardiaca Clase III o IV de la NYHA (estudio 301) o insuficiencia cardiaca de Clase II a IV (estudio 302) o fracción de eyección cardiaca <50%. Todos los pacientes incluidos (tanto tratados con activo como con placebo) recibieron tratamiento concomitante con terapia de deprivación androgénica predominantemente con el uso de agonistas de LHRH, la cual se ha asociado con diabetes, infarto al miocardio, accidente cerebrovascular y muerte cardiaca súbita. La incidencia de reacciones adversas cardiovasculares en los estudios Fase 3 en pacientes tomando abiraterona contra pacientes tomando placebo fueron los siguientes: fibrilación auricular, 2.6 contra 2.0%, taquicardia 1.9 contra 1.0%, angina de pecho 1.7 contra 0.8%, insuficiencia cardiaca 0.7 contra 0.2% y arritmia 0.7 contra 0.5%.
Hepatotoxicidad: Se ha reportado hepatotoxicidad asociada al fármaco con elevación de ALT, AST y bilirrubina total en pacientes tratados con abiraterona. En los estudios clínicos Fase 3, se reportó hepatotoxicidad Grado 3 y 4 (ej., incremento en ALT o AST >5 × LSN o incremento en la bilirrubina >1.5 × LSN) en aproximadamente 6% de los pacientes que recibieron abiraterona, típicamente durante los primeros 3 meses después de iniciar el tratamiento. En el estudio clínico 301, los pacientes cuyas cifras basales de ALT o AST eran elevadas, era más probable que experimentaran elevación en las pruebas de función hepática que aquellos que iniciaron con valores normales al inicio del estudio. Cuando se observaron elevaciones, ya sea de ALT o AST >5 × LSN, o elevaciones de la bilirrubina >3 × LSN, Abiraterona fue suspendido o descontinuado. En dos instancias ocurrieron incrementos marcados en las pruebas de función hepática (ver Precauciones generales – Hepatotoxicidad e insuficiencia hepática). Estos dos pacientes con función hepática basal normal, presentaron elevaciones de ALT o AST de 15 a 40 × LSN y elevaciones de bilirrubina de 2 a 6 × LSN. Posterior a la descontinuación de abiraterona ambos pacientes presentaron normalización en las pruebas de función hepática y un paciente fue retratado con abiraterona sin recurrencia de las elevaciones. En el estudio 302, se observaron elevaciones de Grado 3 o 4 en ALT o AST 35 (6.5%) pacientes tratados con abiraterona. Las elevaciones de aminotransferasa se resolvieron en todos menos en 3 pacientes (2 con nuevas metástasis hepáticas múltiples y 1 con elevación de AST aproximadamente 3 semanas después de la última dosis de Abiraterona). En los estudios clínicos Fase 3, las suspensiones del tratamiento debido al aumento de ALT y AST o función hepática anormal fueron reportadas en 1.1% de los pacientes tratados con Abiraterona y 0.6% de los pacientes tratados con placebo; no se reportaron muertes debido a eventos de hepatotoxicidad.
En los estudios clínicos, el riesgo de hepatotoxicidad fue mitigado mediante la exclusión de pacientes con hepatitis o alteraciones significativas de las pruebas de función hepática basales. En el estudio 301, los pacientes con ALT y AST basales ≥2.5 × LSN en ausencia de metástasis hepática y >5 × LSN en la presencia de metástasis hepática fueron excluidos. En el estudio 302, los pacientes con metástasis hepáticas no fueron elegibles y se excluyó a los pacientes con ALT y AST basales ≥2.5 × LSN. Las pruebas de función hepática anormales que se desarrollaron en pacientes que participaron en los estudios clínicos, se manejaron de forma estricta requiriendo interrupción del medicamento y permitiendo el retratamiento sólo después de que las pruebas de función hepática regresaran a la basal del paciente (ver Dosis y vía de administración – Insuficiencia hepática). Los pacientes con elevaciones de ALT o AST >20 × LSN, no fueron retratados. Se desconoce la seguridad del retratamiento en esos pacientes. No se entiende el mecanismo de hepatotoxicidad asociado con abiraterona.
Experiencia postcomercialización: A continuación, se describen las reacciones adversas identificadas durante la experiencia postcomercialización con base en reportes espontáneos con Abiraterona.
Las frecuencias se proporcionan de acuerdo a la siguiente convención: Poco común ≥1/1000 y <1/100, Raro ≥1/10000 y <1/1000, Muy raro <1/10000.
Clase de sistema/órgano:
Trastornos respiratorios, torácicos y mediastinales:
Raro: Alveolitis alérgica.
Trastornos musculoesqueléticos y del tejido conectivo:
Poco común: Rabdomiólisis, miopatía.
Trastornos hepatobiliares:
Raro: Hepatitis fulminante, insuficiencia hepática aguda.
Trastornos cardiacos:
Muy raro: Prolongación del intervalo QT y Torsade de pointes (observados en pacientes que desarrollaron hipercalcemia o tenían afecciones cardiovasculares subyacentes).
Trastornos del sistema inmunológico, hipersensibilidad:
Muy raras: Reacción anafiláctica (reacciones alérgicas graves que incluyen, entre otras, dificultad para tragar o respirar, hinchazón de la cara, labios, lengua o garganta, o erupción cutánea con comezón (urticaria)).
PRECAUCIONES EN RELACIÓN CON EFECTOS DE CARCINOGÉNESIS, MUTAGÉNESIS, TERATOGÉNESIS Y SOBRE LA FERTILIDAD:
Carcinogénesis y mutagénesis: Acetato de abiraterona no fue carcinogénico en un estudio de 6 meses en ratones transgénicos (Tg.rasH2). En un estudio de carcinogenicidad de 24 meses en ratas, el acetato de abiraterona incrementó la incidencia de neoplasias celulares intersticiales en los testículos. Este hallazgo se considera relacionado con la acción farmacológica de abiraterona y la rata específica. Acetato de abiraterona no fue carcinogénico en ratas hembra.
Acetato de abiraterona y abiraterona carecían de potencial genotóxico en el panel estándar de pruebas de genotoxicidad, incluyendo un ensayo de mutación inversa en bacterias in vitro (prueba de Ames), un ensayo de aberración cromosómica in vitro en mamíferos (utilizando linfocitos humanos) y un ensayo de micronúcleos in vivo en ratas.
Toxicidad reproductiva: En estudios de fertilidad en ratas macho y hembra, acetato de abiraterona disminuyó la fertilidad, lo que es completamente reversible de 4 a 16 semanas después de que acetato de abiraterona se detuvo.
En un estudio de toxicidad para el desarrollo en ratas, acetato de abiraterona afectó el embarazo, incluyendo la reducción de peso fetal y la supervivencia. Se observaron efectos en los genitales externos, aunque acetato de abiraterona no fue teratogénico.
En estos estudios de fertilidad y de toxicidad del desarrollo realizados en ratas, todos los efectos estaban relacionados con la actividad farmacológica de abiraterona.
Toxicología animal: En todos los estudios de toxicidad animal, los niveles de testosterona circulante se redujeron significativamente. Como resultado, se observó una reducción en los pesos de los órganos y cambios morfológicos y/o histopatológicos en los órganos reproductivos y en las glándulas suprarrenales, pituitaria y mamarias. Todos los cambios mostraron reversibilidad completa o parcial. Los cambios en los órganos reproductivos y órganos sensibles a andrógenos son consistentes con la farmacología de abiraterona. Todos los cambios hormonales relacionados con el tratamiento se invirtieron o mostraron resolverse después de un periodo de recuperación de 4 semanas.
Después del tratamiento crónico a partir de 13 semanas en adelante, se observó hiperplasia del conducto biliar/células ovales, asociada con niveles séricos elevados de fosfatasa alcalina y/o bilirrubina total, en los hígados de ratas y monos. Después de un periodo de recuperación de 4 semanas, los parámetros séricos se revirtieron, mientras que la hiperplasia del conducto biliar/células ovales persistió.
Se observaron cataratas en ratas después de 26 semanas de tratamiento. Estos cambios todavía estaban presentes después de un periodo de recuperación de 4 semanas. No se observaron cataratas en monos después de 39 semanas de tratamiento.
INTERACCIONES MEDICAMENTOSAS Y DE OTRO GÉNERO:
La administración con alimentos aumenta significativamente la absorción de acetato de abiraterona. No se ha establecido la eficacia y seguridad cuando se administra con alimentos, por lo tanto, este medicamento no debe tomarse con alimentos (ver secciones Dosis y vía de administración y Farmacocinética).
Interacciones con otros medicamentos:
Posibilidad de que otros medicamentos afecten las exposiciones a la abiraterona: En un estudio clínico de interacción farmacocinética de sujetos sanos pretratados con un fuerte inductor de CYP3A4 rifampicina, 600 mg diarios durante 6 días, seguido de una dosis única de acetato de abiraterona 1,000 mg, el AUC∞ plasmático medio de abiraterona se redujo en un 55%.
Deben evitarse los inductores potentes de CYP3A4 (p. ej., fenitoína, carbamazepina, rifampicina, rifabutina, rifapentina, fenobarbital, hierba de San Juan [Hypericum perforatum]) durante el tratamiento, a menos que no exista una alternativa terapéutica.
En un estudio clínico separado de interacción farmacocinética de sujetos sanos, la administración conjunta de ketoconazol, un inhibidor fuerte del CYP3A4, no tuvo un efecto clínicamente significativo sobre la farmacocinética de la abiraterona.
Potencial de afectar las exposiciones a otros medicamentos: La abiraterona es un inhibidor de las enzimas hepáticas metabolizadoras de fármacos CYP2D6 y CYP2C8. En un estudio para determinar los efectos del acetato de abiraterona (más prednisona) en una sola dosis del sustrato CYP2D6 dextrometorfano, la exposición sistémica (AUC) de dextrometorfano aumentó aproximadamente 2.9 veces. El AUC24 para el dextrorfano, el metabolito activo del dextrometorfano, aumentó aproximadamente un 33%.
Se recomienda precaución cuando se administra con medicamentos activados o metabolizados por CYP2D6, particularmente con medicamentos que tienen un índice terapéutico estrecho. Se debe considerar la reducción de la dosis de medicamentos con un índice terapéutico estrecho que son metabolizados por CYP2D6. Los ejemplos de medicamentos metabolizados por CYP2D6 incluyen metoprolol, propranolol, desipramina, venlafaxina, haloperidol, risperidona, propafenona, flecainida, codeína, oxicodona y tramadol (los últimos tres medicamentos que requieren CYP2D6 para formar sus metabolitos analgésicos activos).
En un ensayo de interacción farmacológica CYP2C8 en sujetos sanos, el AUC de pioglitazona aumentó en un 46% y los AUC para M-III y M-IV, los metabolitos activos de pioglitazona, cada uno disminuyó en un 10% cuando se administró pioglitazona junto con una dosis única de 1,000 mg de acetato de abiraterona. Los pacientes deben ser monitoreados para detectar signos de toxicidad relacionados con un sustrato CYP2C8 con un índice terapéutico estrecho si se usan concomitantemente. Los ejemplos de medicamentos metabolizados por CYP2C8 incluyen pioglitazona y repaglinida (ver sección Precauciones generales).
In vitro, se demostró que los metabolitos principales abiraterona sulfato y N-óxido abiraterona sulfato inhiben el transportador de absorción hepática OATP1B1 y, como consecuencia, pueden aumentar las concentraciones de medicamentos eliminados por OATP1B1. No hay datos clínicos disponibles para confirmar la interacción basada en el transportador.
Usar con productos que se sabe que prolongan el intervalo QT: Dado que el tratamiento de privación de andrógenos puede prolongar el intervalo QT, se recomienda precaución al administrar abiraterona con medicamentos que se sabe que prolongan el intervalo QT o medicamentos capaces de inducir torsades de pointes como la clase IA (por ejemplo, quinidina, disopiramida) o clase III (por ejemplo, amiodarona, sotalol, dofetilida, ibutilida) medicamentos antiarrítmicos, metadona, moxifloxacina, antipsicóticos, etc.
Usar con espironolactona: La espironolactona se une al receptor de andrógenos y puede aumentar los niveles de antígeno prostático específico (PSA). No se recomienda su uso con abiraterona (ver sección Farmacodinamia).
ALTERACIONES EN LOS RESULTADOS DE PRUEBAS DE LABORATORIO:
Abiraterona provoca hipocalcemia como consecuencia del incremento de los niveles de mineralocorticoides que resultan de la inhibición de CYP17. El uso concomitante de un corticosteroide disminuye la incidencia y severidad de esas reacciones adversas.
Ocurrieron incrementos marcados en enzimas hepáticas que llevaron a la descontinuación o modificación de la dosis en estudios clínicos controlados. Se ha reportado hepatotoxicidad asociada al fármaco con elevación de ALT, AST y bilirrubina total en pacientes tratados con abiraterona.
PRECAUCIONES GENERALES:
Hipertensión, hipocalemia, retención de líquidos e insuficiencia cardiaca debido al exceso de mineralocorticoides: La abiraterona puede causar hipertensión, hipocalemia y retención de líquidos (ver sección Reacciones secundarias y adversas) como consecuencia del aumento de los niveles de mineralocorticoides como resultado de la inhibición de CYP17 (ver sección Farmacodinamia). La administración conjunta de un corticosteroide suprime el impulso de la hormona adrenocorticotrópica (ACTH), lo que resulta en una reducción de la incidencia y la gravedad de estas reacciones adversas. Se requiere precaución en el tratamiento de pacientes cuyas afecciones médicas subyacentes pueden verse comprometidas por aumentos en la presión arterial, hipocalemia (por ejemplo, aquellos con glucósidos cardiacos) o retención de líquidos (por ejemplo, aquellos con insuficiencia cardiaca, angina de pecho severa o inestable, infarto de miocardio reciente o arritmia ventricular y aquellos con insuficiencia renal grave).
La abiraterona debe usarse con precaución en pacientes con antecedentes de enfermedad cardiovascular. Los estudios de Fase 3 realizados con abiraterona excluyeron pacientes con hipertensión no controlada, enfermedad cardiaca clínicamente significativa como se evidencia por infarto de miocardio o eventos trombóticos arteriales en los últimos 6 meses, angina severa o inestable, o Clase de Asociación del Corazón de Nueva York (NYHA) III o IV insuficiencia cardiaca (estudio 301) o insuficiencia cardiaca de clase II a IV (estudio 302) o medición de la fracción de eyección cardiaca <50%. En el estudio 302, se excluyeron los pacientes con fibrilación auricular u otra arritmia cardiaca que requiriera tratamiento médico. No se estableció la seguridad en pacientes con fracción de eyección del ventrículo izquierdo (FEVI) <50% o insuficiencia cardiaca NYHA Clase III o IV (en el estudio 301) o insuficiencia cardiaca NYHA Clase II a IV (en el estudio 302) (ver secciones Reacciones secundarias y adversas y Farmacodinamia).
Antes de tratar a pacientes con un riesgo significativo de insuficiencia cardiaca congestiva (por ejemplo, antecedentes de insuficiencia cardiaca, hipertensión no controlada o eventos cardiacos como cardiopatía isquémica), considere obtener una evaluación de la función cardiaca (por ejemplo, ecocardiograma). Antes del tratamiento con abiraterona, se debe tratar la insuficiencia cardiaca y optimizar la función cardiaca. La hipertensión, la hipocalemia y la retención de líquidos deben corregirse y controlarse. Durante el tratamiento, la presión arterial, el potasio sérico, la retención de líquidos (aumento de peso, edema periférico) y otros signos y síntomas de insuficiencia cardiaca congestiva deben controlarse cada 2 semanas durante 3 meses, luego mensualmente y corregir las anomalías. Se ha observado una prolongación del intervalo QT en pacientes que experimentan hipocalemia en asociación con el tratamiento con abiraterona.
Hepatotoxicidad e insuficiencia hepática: Se observaron aumentos marcados en las enzimas hepáticas que condujeron a la interrupción del tratamiento o la modificación de la dosis en estudios clínicos controlados (ver sección 4.8). Los niveles séricos de transaminasas y bilirrubina deben medirse antes de comenzar el tratamiento, cada dos semanas durante los primeros tres meses de tratamiento, y mensualmente a partir de entonces. Si se desarrollan síntomas o signos clínicos sugestivos de hepatotoxicidad, las transaminasas séricas deben medirse de inmediato. Si en algún momento el ALT o AST se eleva por encima de 5 veces el ULN, o la bilirrubina se eleva más allá de 3 veces el ULN el tratamiento debe interrumpirse de inmediato y la función hepática debe monitorearse de cerca. El nuevo tratamiento puede realizarse sólo después de que las pruebas de función hepática vuelvan a la línea base del paciente y a un nivel de dosis reducida (ver sección Reacciones secundarias y adversas).
Si los pacientes desarrollan hepatotoxicidad severa (ALT o AST 20 veces el ULN) en cualquier momento mientras están en tratamiento, el tratamiento debe suspenderse y los pacientes no deben ser tratados nuevamente.
Los pacientes con hepatitis viral activa o sintomática fueron excluidos de los ensayos clínicos; por lo tanto, no hay datos que respalden el uso de abiraterona en esta población.
No existen datos sobre la seguridad clínica y la eficacia de dosis múltiples de acetato de abiraterona cuando se administra a pacientes con insuficiencia hepática moderada o grave (Child-Pugh Clase B o C). El uso de abiraterona debe evaluarse con precaución en pacientes con insuficiencia hepática moderada, en quienes el beneficio claramente debe superar el posible riesgo (ver secciones: Dosis y vía de administración y Farmacocinética). La abiraterona no debe usarse en pacientes con insuficiencia hepática grave (ver secciones Dosis y vía de administración, Contraindicaciones y Farmacocinética).
Ha habido informes poco frecuentes posteriores a la comercialización de insuficiencia hepática aguda y hepatitis fulminante, algunos con resultados fatales (ver sección Reacciones secundarias y adversas).
Abstinencia de corticosteroides y cobertura de situaciones de estrés: Se recomienda precaución y se debe monitorear la insuficiencia adrenocortical si se retira a los pacientes de prednisona o prednisolona. Si se continúa con la abiraterona después de retirar los corticosteroides, se debe controlar a los pacientes para detectar síntomas de exceso de mineralocorticoides (ver información más arriba).
En pacientes con prednisona o prednisolona que están sometidos a un estrés inusual, se puede indicar un aumento de la dosis de corticosteroides antes, durante y después de la situación estresante.
Densidad ósea: La disminución de la densidad ósea puede ocurrir en hombres con cáncer de próstata avanzado metastásico. El uso de abiraterona en combinación con un glucocorticoide podría aumentar este efecto.
Uso previo de ketoconazol: Se pueden esperar tasas de respuesta más bajas en pacientes previamente tratados con ketoconazol para el cáncer de próstata.
Hiperglucemia: El uso de glucocorticoides podría aumentar la hiperglucemia, por lo tanto, el azúcar en la sangre debe medirse con frecuencia en pacientes con diabetes.
Hipoglucemia: Se han notificado casos de hipoglucemia cuando se administró abiraterona a pacientes con diabetes preexistente que recibieron pioglitazona o repaglinida (ver sección Interacciones medicamentosas y de otro género); por lo tanto, el azúcar en la sangre debe medirse con frecuencia en pacientes con diabetes.
Usar con quimioterapia: No se ha establecido la seguridad y eficacia del uso concomitante de abiraterona con quimioterapia citotóxica (ver sección Farmacodinamia).
Intolerancia a los excipientes: Este medicamento contiene lactosa. Los pacientes con intolerancia hereditaria a galactosa, insuficiencia de lactasa de Lapp o malabsorción de glucosa o galactosa no deben tomar este medicamento. Este medicamento también contiene más de 1 mmol (o 27.2 mg) de sodio por dosis de cuatro tabletas. A tener en cuenta por los pacientes con una dieta controlada de sodio.
Riesgos potenciales: La anemia y la disfunción sexual pueden ocurrir en hombres con cáncer de próstata metastásico, incluidos aquellos que reciben tratamiento con abiraterona.
Efectos del músculo esquelético: Se han notificado casos de miopatía y rabdomiólisis en pacientes tratados con abiraterona. La mayoría de los casos se desarrollaron dentro de los primeros 6 meses de tratamiento y se recuperaron después del retiro de abiraterona. Se recomienda precaución en pacientes tratados concomitantemente con medicamentos que se sabe que están asociados con miopatía/rabdomiólisis.
Interacciones con otros medicamentos: Deben evitarse los inductores potentes de CYP3A4 durante el tratamiento a menos que no exista una alternativa terapéutica, debido al riesgo de disminución de la exposición a abiraterona (ver sección Interacciones medicamentosas y de otro género).
Combinación de abiraterona y prednisona / prednisolona con Ra-223:
El tratamiento con abiraterona y prednisona/prednisolona en combinación con Ra-223 está contraindicado (ver sección Contraindicaciones) debido a un mayor riesgo de fracturas y una tendencia a una mayor mortalidad entre pacientes con cáncer de próstata asintomático o levemente sintomático, como se observó en ensayos clínicos.
Se recomienda que el tratamiento posterior con Ra-223 no se inicie durante al menos 5 días después de la última administración de abiraterona en combinación con prednisona/prednisolona.
DOSIS Y VÍA DE ADMINISTRACIÓN:
Vía de administración: Oral.
Dosis: La dosis recomendada de abiraterona es 1000 mg (dos tabletas de 500 mg o cuatro tabletas de 250 mg) como dosis única diaria que no debe tomarse con los alimentos. Abiraterona debe tomarse al menos dos horas después de los alimentos y no se deben tomar alimentos al menos una hora después de tomar abiraterona. Las tabletas deben de tragarse completas con agua (ver Propiedades farmacocinéticas - Absorción).
Dosis de prednisona y prednisolona: Para cáncer de próstata metastásico sin tratamiento hormonal previo (CPmHN) o cáncer de próstata sensible a hormonas (CPmHS) se utiliza abiraterona con 5 mg diarios de prednisona o prednisolona.
Para cáncer de próstata metastásico resistente a la castración (mCRPC), se utiliza abiraterona con 10 mg diarios de prednisona o prednisolona.
Monitoreo recomendado: Las transaminasas y bilirrubina séricas deben medirse previo al inicio del tratamiento con abiraterona, cada dos semanas durante los primeros tres meses de tratamiento y posteriormente de forma mensual. La presión sanguínea, niveles de potasio sérico y retención de líquidos deben monitorearse de forma mensual (ver Precauciones generales - Hipertensión, hipopotasemia y retención de líquidos por exceso de mineralocorticoides y Hepatotoxicidad e insuficiencia hepática).
Insuficiencia hepática: No se requiere ajuste de dosis en pacientes con insuficiencia hepática leve preexistente. No existen datos sobre eficacia clínica y seguridad de múltiples dosis de acetato de abiraterona cuando se administra a pacientes con insuficiencia hepática moderada o severa (Child Pugh Clase B o C). No se puede predecir un ajuste de dosis. Abiraterona debe usarse con precaución en pacientes con insuficiencia hepática moderada sólo si el beneficio sobrepasa claramente los posibles riesgos (ver Precauciones generales – Hepatotoxicidad e insuficiencia hepática y Propiedades farmacocinéticas – Poblaciones especiales). Abiraterona no debe usarse en pacientes con insuficiencia hepática severa (ver Precauciones generales - Hepatotoxicidad e insuficiencia hepática y Propiedades farmacocinéticas – Poblaciones especiales).
Para pacientes que desarrollan hepatotoxicidad durante el tratamiento con abiraterona (incrementos de alanina aminotransferasa (ALT) o aspartato aminotransferasa (AST) 5 veces por arriba del límite superior normal, o incrementos de bilirrubina 3 veces por arriba del límite superior normal) se debe suspender el tratamiento inmediatamente hasta que las pruebas de función hepática se regularicen (ver Precauciones generales - Hepatotoxicidad e insuficiencia hepática). El retratamiento posterior al regreso de las pruebas de función hepática a la basal del paciente puede darse a una dosis reducida de 500 mg (una tableta de 500 mg o dos tabletas de 250 mg) una vez al día. Para pacientes en retratamiento, las transaminasas y bilirrubina séricas deben monitorearse al menos cada dos semanas por tres meses y de forma mensual posteriormente. Si la hepatotoxicidad recurre con la dosis reducida de 500 mg diarios, descontinuar el tratamiento con abiraterona. Las dosis reducidas no deben tomarse con alimentos.
Si el paciente desarrolla hepatotoxicidad severa (ALT o AST 20 veces por arriba del límite superior normal) en cualquier momento durante el tratamiento, abiraterona debe descontinuarse y los pacientes no deben ser retratados con abiraterona.
Insuficiencia renal: No es necesario realizar ajustes de dosis en pacientes con insuficiencia renal (ver Propiedades farmacocinéticas – Poblaciones especiales).
MANIFESTACIONES Y MANEJO DE LA SOBREDOSIFICACIÓN O INGESTA ACCIDENTAL:
La experiencia humana de sobredosis con abiraterona es limitada.
No hay un antídoto específico. En caso de sobredosis, se debe suspender la administración y tomar medidas generales de apoyo, incluida la monitorización de arritmias, hipocalemia y de signos y síntomas de retención de líquidos. La función hepática también debe ser evaluada.
PRESENTACIONES:
Caja con 1 frasco con 120 tabletas de 250 mg con instructivo anexo.
Caja con 1 frasco con 60 tabletas de 500 mg con instructivo anexo.
RECOMENDACIONES SOBRE ALMACENAMIENTO:
Consérvese a no más de 25 °C.
Consérvese el frasco bien cerrado.
LEYENDAS DE PROTECCIÓN:
Léase instructivo anexo. Su venta requiere receta médica. Contiene un desecante. No ingerir, consérvese dentro del envase. No se deje al alcance de los niños. No se administre a menores de 18 años. El uso de SISABIN no está indicado para mujeres. Mujeres que están o pudieran estar embarazadas no deberán manipular SISABIN sin guantes. Este medicamento contiene lactosa, que puede producir reacciones de hipersensibilidad. Este medicamento deberá ser prescrito únicamente por médicos especialistas en el tratamiento de enfermedades oncológicas. Prohibida la venta fraccionada del producto.
Reporte las sospechas de reacción adversa al correo:
farmacovigilancia@cofepris.gob.mx
Propiedad de:
Sandoz GmbH
Biochemiestraꞵe 10, Kundl, 6250, Austria.
Representante legal:
SANDOZ, S.A. de C.V.
La Candelaria No. 186
Col. Atlántida, C.P. 04370
Coyoacán, Ciudad de México, México
Reg. Núm. 009M2024 SSA IV
®Marca Registrada
Clave de IPP:
213300404D0555/10Ene2024/IPPA_DRA-Sandoz