
SAYANA
MEDROXIPROGESTERONA
Suspensión inyectable
Caja, 1 Jeringa(s) prellenada(s), 104/0.65 mg/ml
FORMA FARMACÉUTICA Y FORMULACIÓN:
Suspensión
Cada dispositivo prellenado uniject contiene:
Acetato de medroxiprogesterona de depósito (AMPD)
104 mg
Vehículo cbp 0.65 mL
INDICACIONES TERAPÉUTICAS:
Anticoncepción:
La suspensión inyectable (SC) de AMPD está indicada para:
• Anticoncepción.
Ginecología:
La suspensión inyectable SC de AMPD está indicada para:
• Manejo del dolor asociado a la endometriosis.
FARMACOCINÉTICA Y FARMACODINAMIA:
Propiedades farmacodinámicas: El acetato de medroxiprogesterona (acetato de 17-alfa-hidroxi-6 metilprogesterona) es un derivado de la progesterona.
Mecanismo de acción: Acetato de medroxiprogesterona (AMP) es una progestina sintética (estructuralmente relacionada con la hormona progesterona endógena) que ha demostrado poseer varias acciones farmacológicas sobre el sistema endocrino:
• Inhibición de las gonadotrofinas hipofisarias (FSH yLH).
• Disminución de los niveles de ACTH e hidrocortisona en la sangre.
• Disminución de la testosterona circulante.
• Disminución de los niveles de estrógeno circulante (como resultado de la inhibición de FSH y la inducción enzimática de la reductasa hepática, dando lugar a un aumento de la eliminación de testosterona y la consecuente disminución de la conversión de andrógenos en estrógenos).
Todas estas acciones generan una cantidad de efectos farmacológicos, que se describen a continuación.
Anticoncepción: AMPD, cuando se administra en mujeres por vía parenteral en la dosis recomendada, inhibe la secreción de gonadotrofinas, la cual, a su vez, impide la maduración folicular y la ovulación provocando engrosamiento del moco cervical, el cual inhibe la entrada de los espermatozoides en el útero.
Ginecología: El acetato de medroxiprogesterona (AMP) administrado a mujeres por vía oral o parenteral en las dosis recomendadas con estrógeno endógeno adecuado, transforma el endometrio proliferativo en secretor. Se han advertido efectos androgénicos y anabólicos, pero el fármaco aparentemente está desprovisto de actividad estrogénica significativa. Mientras que la administración de AMPD por vía parenteral inhibe la producción de gonadotrofina, lo cual a su vez impide la maduración folicular y la ovulación, los datos disponibles indican que esto no ocurre cuando la dosis oral recomendada normalmente se administra como dosis única diaria.
Endometriosis: Es probable que la supresión de las concentraciones séricas de estradiol y una posible acción directa de AMPD SC en las lesiones de la endometriosis sean responsables del efecto terapéutico sobre el dolor asociado con la endometriosis.
Estudios clínicos:
Estudios de densidad mineral ósea (DMO):
Cambios de DMO en mujeres adultas: En un estudio clínico controlado no aleatorizado que compara mujeres adultas usuarias del anticonceptivo AMPD inyectable (150 mg IM) durante un máximo de 5 años con mujeres que eligieron no usar ningún anticonceptivo hormonal, 42 usuarias de AMPD completaron un tratamiento de 5 años realizando por lo menos un seguimiento de medición de la DMO después de suspender el AMPD. Entre las usuarias de AMPD, la DMO disminuyó durante los primeros 2 años de uso, con pequeños descensos en los años siguientes. Se observaron cambios medios en la DMO de la columna lumbar de -2.86%, -4.11%, -4.89%, -4.93% y -5.38% después de 1, 2, 3, 4 y 5 años, respectivamente. Las disminuciones medias en la DMO de la cadera completa y el cuello femoral fueron similares. No hubo cambios significativos en la densidad mineral ósea en las mujeres del grupo control durante el mismo periodo de tiempo.
Recuperación postratamiento de la DMO en mujeres adultas: En el mismo estudio poblacional hubo una recuperación parcial de la DMO hacia los valores basales durante un periodo de 2 años después de suspender el uso de la inyección de AMPD (150 mg IM).
Después de 5 años de tratamiento con AMPD inyectable (150 mg IM), el cambio porcentual de la media en la DMO basal fue de -5.4%, -5.2% y -6.1% en la columna vertebral, cadera total y cuello femoral respectivamente, mientras que las mujeres del grupo control no tratadas, durante el mismo intervalo de tiempo, mostraron cambios en la media basal de +/- 0.5% o menos en los mismos sitios del esqueleto. Dos años después de suspender la inyección de AMPD, aumentó la media de DMO en los 3 sitios del esqueleto, pero el déficit se mantuvo: -3.1%, -1.3% y -5.4% en la columna vertebral, cadera total y cuello femoral respectivamente. Al mismo tiempo, las mujeres del grupo control mostraron cambios en la media de la DMO basal de 0.5%, 0.9% y -0.1% en la columna vertebral, cadera total y cuello femoral, respectivamente.
Cambios en la DMO en mujeres adolescentes (de entre 12 y 18 años): El efecto del uso de AMPD inyectable (150 mg IM) sobre la DMO por más de 240 semanas (4.6 años) se evaluó en un estudio clínico abierto no comparativo de 159 mujeres adolescentes (12-18 años) que eligieron comenzar el tratamiento con AMPD; 114 de las 159 participantes utilizaron AMPD continuamente (4 inyecciones durante un periodo de 60 semanas) e hicieron mediciones de la DMO a la semana 60. La DMO disminuyó durante los primeros 2 años de uso con pocos cambios en los años subsecuentes. Después de 60 semanas de uso de AMPD, los cambios porcentuales de la media de la DMO basal fueron -2.5%, -2.8% y -3.0% en la columna vertebral, cadera total y cuello femoral, respectivamente. Un total de 73 sujetos continuaron usando el AMPD durante 120 semanas; los cambios porcentuales de la media de la DMO basales fueron -2.7%, -5.4% y -5.3% en la columna vertebral, cadera total y cuello femoral, respectivamente. Un total de 28 sujetos continuaron usando el AMPD durante 240 semanas; los cambios porcentuales de la media de la DMO basal fueron -2.1%, -6.4% y -5.4% en la columna vertebral, cadera total y cuello femoral, respectivamente.
Recuperación postratamiento de la DMO en adolescentes: En el mismo estudio, 98 participantes adolescentes recibieron al menos 1 inyección de AMPD y al menos una medición de la DMO después de suspender el uso de AMPD, con tratamiento de AMPD durante un máximo de 240 semanas (equivalente a 20 inyecciones de AMPD) y el seguimiento postratamiento que se extendió hasta por 240 semanas después de la última inyección de AMPD. La mediana del número de inyecciones recibidas durante la fase de tratamiento fueron 9. En el momento de la inyección final de AMPD, los cambios porcentuales de la DMO basales fueron -2.7%, -4-1% y -3.9% en la columna vertebral, cadera total y cuello femoral, respectivamente. Con el tiempo, estos déficits medios de DMO se recuperaron completamente después de la suspensión de AMPD. La recuperación completa requirió 1.2 años para la columna lumbar, 4.6 años para la cadera total y 4.6 años en el cuello femoral. La duración prolongada del tratamiento y el tabaquismo se asocian con una recuperación más lenta (ver sección Precauciones generales-Advertencias y Precauciones adicionales para el Uso o Formulación Específica, Anticoncepción/Endometriosis-Formulaciones Inyectables; Pérdida de Densidad Mineral Ósea DMO).
Relación de la incidencia de fracturas con el uso de AMPD inyectable (150 mg IM) o la no utilización por mujeres en edad reproductiva: Un estudio de cohorte retrospectivo para evaluar la asociación entre la AMPD inyectable y la incidencia de fracturas óseas se llevó a cabo en 312.395 usuarias de anticonceptivos en el Reino Unido. La tasa de incidencia de fracturas fue comparada antes y después del inicio de AMPD y también entre las usuarias de AMPD y mujeres que utilizaron otros anticonceptivos pero no tenían registrado el uso de AMPD. Entre las mujeres que usan el AMPD, el uso de AMPD no se asocia con un aumento del riesgo de fractura (proporción de la tasa de incidencia = 1.01, IC del 95%: 0.92 a 1.11, comparando el periodo de seguimiento del estudio con hasta 2 años de observación antes del uso de AMPD). Sin embargo, las usuarias de AMPD presentaban más fracturas en comparación con las que no lo usaban, no sólo después del primer uso de anticonceptivos (TIR = 1.23; IC del 95%: 1.16 a 1.30), sino también antes del primer uso de anticonceptivos (TIR = 1.28; IC del 95%: 1.07 a 1.53).
Además, las fracturas óseas en los sitios específicos característicos de las fracturas osteoporóticas por fragilidad (columna vertebral, cadera, pelvis) no fueron más frecuentes entre las usuarias de AMPD en comparación con las no-usuarias (TIR = 0.95, IC 95% 0.74 a 1.23), no hubo ninguna evidencia de que el uso prolongado de AMPD (2 años o más) confiere mayor riesgo de fractura en comparación con menos de 2 años de uso.
Estos datos demuestran que las usuarias de AMPD tienen un perfil de riesgo de fractura intrínsecamente diferente a las que no fueron usuarias por razones no relacionadas con el uso de AMPD.
El seguimiento más extenso en este estudio fue de 15 años; por lo tanto, los posibles efectos del AMPD que podrían extenderse más allá de 15 años de seguimiento no se pueden determinar.
Estudios sobre la endometriosis: Se demostró la eficacia de AMPD SC en la reducción del dolor asociado a la endometriosis en mujeres con los signos y síntomas de la enfermedad en dos estudios controlados con comparador activo. Cada estudio evaluó la reducción del dolor asociado a la endometriosis a lo largo de 6 meses de tratamiento y la recurrencia de los síntomas durante los 12 meses posteriores al tratamiento. Los sujetos tratados con AMPD SC durante 6 meses recibieron una dosis de 104 mg cada 3 meses (2 inyecciones), mientras que las mujeres tratadas con microesferas de leuprolida durante 6 meses recibieron una dosis de 11.25 mg cada 3 meses (2 inyecciones) o 3.75 mg mensuales (6 inyecciones). El estudio 268 se realizó en los EE. UU. y en Canadá e incluyó a 274 sujetos (136 bajo tratamiento con AMPD SC y 138 con leuprolida). El estudio 270 se llevó a cabo en América del Sur, Europa y Asia e incorporó a 299 sujetos (153 bajo tratamiento con AMPD SC y 146 con leuprolida).
Se evaluó la reducción del dolor utilizando una escala modificada de Biberoglu y Behrman que constaba de tres síntomas informados por las pacientes (dismenorrea, dispareunia y dolor pélvico no relacionado con la menstruación) y de dos signos evaluados durante el examen pélvico (sensibilidad pélvica e induración). Para cada categoría, una respuesta favorable se definió como la mejoría de al menos 1 unidad (la gravedad se evaluó en una escala de 0 a 3) con respecto a la puntuación inicial (Figura 1).
Figura 1. Porcentajes de sujetos que respondieron al final del tratamiento (mes 6 o última evaluación si es antes) en estudios 268 y 270
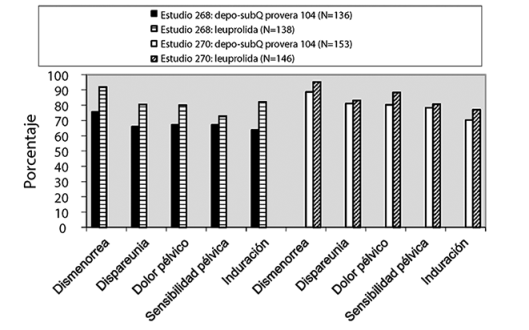
Respuesta favorable = reducción en la severidad de los síntomas o signos de ≥ 1 punto en una escala de 0 a 3, en comparación con el valor inicial.
Además, se combinaron las puntuaciones de cada una de las cinco categorías con el total (puntuación compuesta), lo cual se consideró una medición global de la mejoría general de la enfermedad. En el caso de los sujetos con puntuaciones iniciales para cada una de las 5 categorías, se consideró una disminución media de 4 puntos con respecto al valor inicial como una mejoría clínicamente significativa. En los dos estudios, en ambos grupos de tratamiento, los cambios medios en la puntuación compuesta cumplieron con el criterio definido por el protocolo de mejoría.
En los ensayos clínicos, el tratamiento con AMPD SC se limitó a seis meses. No están disponibles los datos sobre la persistencia del beneficio con un tratamiento más prolongado.
Los sujetos registraron diariamente la incidencia y severidad de los bochornos. De las usuarias de AMPD SC, el 28.6% informó haber experimentado bochornos moderados o graves al inicio del estudio, el 36.2% a los 3 meses y el 26.7% a los 6 meses. De las usuarias de leuprolida, el 32.8% informó haber experimentado bochornos moderados o graves al inicio del estudio, el 74.2% a los 3 meses y el 68.5% a los 6 meses.
Estudio Women’s Health Initiative (WHI): El ensayo de ECE (0.625 mg)/AMP (2.5 mg) de WHI incorporó 16,608 mujeres posmenopáusicas de entre 50 y 79 años con útero intacto al inicio del estudio para evaluar los riesgos y beneficios de la terapia combinada con placebo en la prevención de determinadas enfermedades crónicas. El criterio de valoración primario fue la incidencia de cardiopatía coronaria (CC) (infarto de miocardio no mortal y muerte por CC), con cáncer de mama invasivo como el resultado adverso primario estudiado. El estudio se suspendió prematuramente después de un seguimiento promedio de 5.2 años (duración prevista de 8.5 años) ya que, de acuerdo con la regla de interrupción predefinida, el aumento del riesgo de cáncer de mama y eventos cardiovasculares excedía los beneficios especificados incluidos en el "índice global" (ver Precauciones generales-Advertencias y precauciones adicionales para el uso o formulación específica, Anticoncepción/endometriosis-formulaciones inyectables, Cáncer de mama).
La terapia de combinación de ECE/AMP informó una disminución significativa de las fracturas osteoporóticas (23%) y totales (24%).
Estudio Million Women (MWS): El MWS fue un estudio prospectivo de cohorte que inscribió a 1,084,110 mujeres en el Reino Unido de entre 50 y 64 años de las cuales 828,923, con tiempo definido desde la menopausia, se incluyeron en los análisis principales de riesgo de cáncer de mama en relación con la TH. En forma general, el 50% de la población de estudio había utilizado TH en algún momento. La mayoría de las usuarias actuales de TH al inicio del estudio informaron ingerir preparados que contienen estrógeno solo (41%) o combinaciones de estrógenos y progestina (50%). La duración promedio del seguimiento fue de 2.6 años para el análisis de la incidencia de cáncer y 4.1 años para los análisis de mortalidad (ver Precauciones generales-Advertencias y precauciones adicionales para el uso o formulación específica, Cáncer de mama).
Estudios de reemplazo de estrógeno/progestina y del corazón (HERS): Los estudios HERS y HERS II fueron dos ensayos aleatorizados, prospectivos, de prevención secundaria sobre los efectos a largo plazo del régimen oral combinado continuo de ECE/AMP (0.625 mg de ECE más 2.5 mg de AMP) en mujeres posmenopáusicas con CC (ver Precauciones generales-Advertencias y precauciones adicionales para el uso o formulación específica, Trastornos cardiovasculares). 2763 mujeres posmenopáusicas con una edad media de 66.7 años y con útero intacto se inscribieron en este estudio. La duración promedio del seguimiento fue de 4.1 años para HERS y 2.7 años adicionales (para un total de 6.8 años) para HERS II (ver Precauciones generales-Advertencias y precauciones adicionales para el uso o formulación específica, Trastornos cardiovasculares).
Estudio Women’s Health Initiative Memory Study (WHIMS): El WHIMS, un estudio secundario del WHI, incorporó a 4532 mujeres postmenopáusicas predominantemente sanas de entre 65 a 79 años para evaluar los efectos de ECE/AMP (0.625 mg de ECE más 2.5 mg de AMP) o ECE solo (0.625 mg) sobre la incidencia de demencia probable en comparación con placebo. La duración promedio del seguimiento fue de 4.05 años para el ECE/AMP (ver Precauciones generales-Advertencias y precauciones adicionales para el uso o formulación específica, Demencia).
Propiedades farmacocinéticas:
Absorción: La absorción de AMP desde el sitio de inyección SC para conseguir niveles terapéuticos es relativamente rápida. El tmáx medio se alcanzó aproximadamente una semana después de la inyección. Las concentraciones máximas (Cmáx) de AMP generalmente oscilan entre 0.5 a 3.0 ng/mL con una Cmáx media de 1.5 ng/mL después de una única inyección SC.
Efecto del sitio de inyección: La inyección subcutánea de AMPD se administró en la cara anterior del muslo o en el abdomen para evaluar los efectos sobre el perfil de concentración/tiempo de AMP. Las concentraciones mínimas (Cmín; Día 91) de AMP fueron similares en los dos lugares de inyección, lo que sugiere que el sitio de inyección no afecta negativamente la eficacia anticonceptiva.
Distribución: El promedio de la unión a proteínas plasmáticas de AMP es del 86%. La unión de AMP se produce principalmente a la albúmina sérica; no se genera ninguna unión de AMP con la globulina unida a hormonas sexuales (GUHS).
Metabolismo: El AMP se metaboliza ampliamente en el hígado.
Eliminación: Las concentraciones residuales de AMP al final del intervalo de dosificación (3 meses) de AMPD subcutáneo son generalmente inferiores a 0.5 ng/mL, en consonancia con su aparente vida media terminal de aprox. 40 días después de la administración SC. La mayoría de los metabolitos de AMP se excretan en la orina como conjugados glucurónidos con sólo pequeñas cantidades excretadas como sulfatos.
Poblaciones especiales:
Raza: No hubo diferencias aparentes en la farmacocinética y/o dinámica de AMP después de la administración SC de AMPD entre las mujeres de todos los orígenes étnicos estudiados. Se ha evaluado la farmacocinética/dinámica de AMPD en mujeres asiáticas en un estudio separado.
Efecto del peso corporal: No es necesario ajustar la dosis de la inyección subcutánea de AMPD en función del peso corporal. El efecto del peso corporal sobre la farmacocinética del AMP se evaluó en un subgrupo de mujeres (n = 42, el índice de masa corporal [IMC] osciló entre 18.2 y 46.0 kg/m2). Los valores AUC0-91 para AMP fueron 68.5, 74.8 y 61.8 ng-día/mL en mujeres con categorías de IMC de ≤ 25 kg/m2, > 25 kg/m2 a ≤ 30 kg/m2 y > 30 kg/m2, respectivamente. La Cmáx media de AMP fue de 1.65 ng/mL en mujeres con IMC ≤ 25 kg/m2, 1.76 ng/mL en mujeres con IMC > 25 kg/m2 a ≤ 30 kg/m2 y 1.40 ng/mL en mujeres con IMC > 30 kg/m2, respectivamente. El intervalo de las concentraciones mínimas (Cmín) de AMP y las vidas medias fueron similares en los 3 grupos de IMC.
CONTRAINDICACIONES:
AMP está contraindicada en pacientes con las siguientes condiciones:
• Embarazo confirmado o sospechado.
• Sangrado vaginal no diagnosticado.
• Disfunción hepática grave.
• Hipersensibilidad conocida a AMP o a cualquiera de los componentes del fármaco.
Contraindicaciones adicionales para usos específicos:
Anticoncepción/ginecología: Conocimiento o sospecha de tumores malignos en la mama.
RESTRICCIONES DE USO DURANTE EL EMBARAZO Y LA LACTANCIA:
Embarazo: AMP está contraindicado en mujeres que están embarazadas.
Algunos informes sugieren, bajo determinadas circunstancias, una asociación entre la exposición intrauterina a fármacos progestacionales en el primer trimestre del embarazo y las anomalías genitales en fetos.
Los bebés de embarazos involuntarios que tienen lugar de 1 a 2 meses después de la inyección de AMPD suspensión inyectable pueden tener un mayor riesgo de bajo peso al nacer, que, a su vez, está asociado con un mayor riesgo de muerte neonatal. El riesgo atribuible es bajo, debido a que la ocurrencia de embarazos mientras se administra AMPD no es común. No hay información definitiva para las otras formulaciones de AMP (ver Farmacocinética y farmacodinamia).
Si la paciente se queda embarazada mientras se le administra este medicamento, se le debe informar sobre el daño potencial para el feto.
Lactancia: AMP y sus metabolitos se excretan en la leche materna. No hay evidencia que sugiera que esto representa algún peligro para el lactante (ver Farmacocinética y farmacodinamia, Distribución).
REACCIONES SECUNDARIAS Y ADVERSAS: Las reacciones adversas se reportan en base al Diccionario Médico para Actividades Regulatorias (MedDRA, por sus siglas en inglés) por clases de órgano sistema (SOC por sus siglas en inglés).
Anticoncepción:
|
Clasificación por órganos y sistemas |
Reacciones adversas |
|
Trastornos del sistema inmunológico |
Reacción anafiláctica*, reacción anafilactoide*, angioedema*, hipersensibilidad al fármaco*. |
|
Trastornos del metabolismo y de la nutrición |
Retención de líquidos, aumento del apetito, disminución del apetito. |
|
Trastornos psiquiátricos |
Depresión, insomnio, ansiedad, trastorno emocional, trastorno afectivo, irritabilidad, anorgasmia, disminución de la libido. |
|
Trastornos del sistema nervioso |
Migraña, mareos, cefalea. |
|
Trastornos del oído y del laberinto |
Vértigo. |
|
Trastornos vasculares |
Hipertensión, venas varicosas, bochornos. |
|
Trastornos gastrointestinales |
Dolor abdominal, náusea, distensión abdominal. |
|
Trastornos de la piel y del tejido subcutáneo |
Alopecia, acné, hirsutismo, lipodistrofia adquirida*, dermatitis, equimosis, cloasma, salpullido. |
|
Trastornos musculoesqueléticos y del tejido conectivo |
Dolor de espalda, espasmos musculares, dolor en las extremidades. |
|
Trastornos mamarios y del sistema reproductivo |
Menometrorragia, metrorragia, menorragia, quistes ováricos, dismenorrea, amenorrea, vaginitis, secreción vaginal, dispareunia, dolor pélvico, sequedad vulvovaginal, dolor de las mamas, síndrome premenstrual, sensibilidad en las mamas, aumento en el tamaño de las mamas. |
|
Trastornos generales y condiciones del sitio de administración |
Fatiga, reacción en el sitio de inyección*, atrofia persistente/ sangrado/ hoyuelos en el sitio de inyección*, nódulos/bultos en el sitio de inyección*, dolor/sensibilidad en el sitio de inyección*, decoloración en el sitio de inyección*. |
|
Investigaciones |
Enzimas hepáticas anormales, aumento de peso, examen citológico cervical anormal, disminución de peso. |
* RAM reportadas después de la comercialización.
Tenga en cuenta que entre las pacientes a las que se le administra la inyección de acetato de medroxiprogesterona (150 mg IM), ha habido informes de reacciones anafilácticas, eventos tromboembólicos y casos muy poco frecuentes de osteoporosis, entre ellos, fracturas osteoporóticas.
Ginecología–Dolor asociado a la endometriosis:
|
Clasificación por órganos y sistemas |
Reacciones adversas |
|
Trastornos del sistema inmunológico |
Reacción anafiláctica*, reacción anafilactoide*, angioedema*, hipersensibilidad al fármaco*. |
|
Trastornos psiquiátricos |
Depresión, insomnio, ansiedad, trastorno afectivo, irritabilidad, disminución de la libido. |
|
Trastornos del sistema nervioso |
Migraña, mareos, hormigueo, cefalea, hipersomnia, parestesia. |
|
Trastornos cardiacos |
Palpitaciones. |
|
Trastornos vasculares |
Bochornos. |
|
Trastornos gastrointestinales |
Náusea, distensión abdominal. |
|
Trastornos de la piel y del tejido subcutáneo |
Alopecia, acné, lipodistrofia adquirida*, dermatitis. |
|
Trastornos musculoesqueléticos y del tejido conectivo |
Artralgia, dolor en las extremidades. |
|
Trastornos mamarios y del sistema reproductivo |
Sangrado uterino disfuncional (irregular, aumento, disminución de manchado), metrorragia, menorragia, quiste ovárico, galactorrea, vaginitis, dolor pélvico, sequedad vulvovaginal, dolor en las mamas, sensibilidad en las mamas. |
|
Trastornos generales y condiciones del sitio de administración |
Fatiga, reacción en el sitio de inyección*, atrofia persistente/sangrado/ hoyuelos en el sitio de inyección*, nódulos/bultos en el sitio de inyección*, dolor/sensibilidad en el sitio de inyección*, decoloración en el sitio de inyección*. |
|
Investigaciones |
Aumento de peso, disminución de peso. |
* RAM reportadas después de la comercialización.
PRECAUCIONES EN RELACIÓN CON EFECTOS DE CARCINOGÉNESIS, MUTAGÉNESIS, TERATOGÉNESIS Y SOBRE LA FERTILIDAD: Se ha demostrado que la administración intramuscular a largo plazo de acetato de medroxiprogesterona (AMPD) genera tumores mamarios en perros beagle. No hubo evidencia de un efecto carcinogénico asociado con la administración oral de AMP a ratas y ratones. El acetato de medroxiprogesterona no fue mutagénico en una batería de ensayos de toxicidad genética in vitro o in vivo. El acetato de medroxiprogesterona en dosis altas es un fármaco antifertilidad y se prevé que dichas altas dosis impidan la fertilidad hasta el cese del tratamiento.
INTERACCIONES MEDICAMENTOSAS Y DE OTRO GÉNERO: La aminoglutetimida administrada concomitantemente con altas dosis orales de AMP puede deprimir significativamente las concentraciones séricas de acetato de medroxiprogesterona. Se debe advertir a las usuarias de altas dosis orales de AMP de la posibilidad de disminución de la eficacia con la administración de aminoglutetimida.
El acetato de medroxiprogesterona (AMP) se metaboliza principalmente in vitro por hidroxilación a través de CYP3A4. No se han realizado estudios específicos sobre interacciones medicamentosas que evalúen los efectos clínicos de los inductores o inhibidores de CYP3A4 sobre el AMP y, por lo tanto, se desconocen los efectos clínicos de estos últimos.
ALTERACIONES EN LOS RESULTADOS DE PRUEBAS DE LABORATORIO:
Se debe informar al patólogo (laboratorio) del uso de AMP de la paciente si el tejido endometrial o endocervical se somete a examen.
Se debe informar al médico/laboratorio que el uso de AMP puede disminuir los niveles de los siguientes biomarcadores endocrinos:
a. Esteroides urinarios/plasmáticos (por ejemplo, cortisol, estrógeno, pregnanediol, progesterona, testosterona).
b. Gonadotrofinas urinarias/plasmáticas (por ejemplo, LH y FSH).
c. Globulina vinculante de las hormonas sexuales.
Alteraciones de la secreción cervical, cambios en el apetito, alteraciones de la función hepática, elevación de glóbulos blancos y plaquetas, hipercalcemia, disminución de la tolerancia a la glucosa, aumento de la presión arterial.
PRECAUCIONES GENERALES:
General: Se debe investigar el sangrado vaginal imprevisto durante el tratamiento con AMP.
AMP puede causar cierto grado de retención de líquidos, por lo tanto, se debe tener precaución en el tratamiento de cualquier paciente con una afección médica preexistente que podría verse afectada negativamente por la retención de líquidos.
Durante el tratamiento con AMP, se debe controlar con detenimiento a aquellas pacientes con antecedentes de tratamiento para la depresión clínica.
Algunas pacientes que reciben AMP pueden evidenciar una disminución en la tolerancia a la glucosa. Se debe observar atentamente a las pacientes diabéticas durante la recepción de dicha terapia.
Se debe informar al patólogo (laboratorio) del uso de AMP por parte de la paciente si el tejido endometrial o endocervical se somete a examen.
Se debe informar al médico/laboratorio que el uso de AMP puede disminuir los niveles de los siguientes biomarcadores endocrinos:
a. Esteroides urinarios/plasmáticos (por ejemplo, cortisol, estrógeno, pregnanediol, progesterona, testosterona).
b. Gonadotrofinas urinarias/plasmáticas (por ejemplo, LH y FSH).
c. Globulina vinculante de las hormonas sexuales.
No se debe volver a administrar el fármaco en la espera del examen si se produce una pérdida repentina parcial o completa de la visión o si se desarrolla un inicio repentino de proptosis, diplopía o migraña. Si el examen revela papiledema o lesiones vasculares de la retina, no se debe volver a administrar el fármaco.
AMP no se ha asociado causalmente con la inducción de trastornos trombóticos o tromboembólicos; sin embargo, no se recomienda AMP en todas las pacientes con antecedentes de tromboembolismo venoso (TEV). Se recomienda la interrupción de AMP en pacientes que desarrollen TEV durante la terapia con AMP.
Advertencias y precauciones adicionales para el uso o formulación específica:
Anticoncepción/endometriosis-formulaciones inyectables:
Pérdida de la densidad mineral ósea (DMO): El uso de AMPD inyectable reduce los niveles de estrógeno sérico en mujeres premenopáusicas y se asocia con una pérdida estadísticamente significativa de la DMO a medida que el metabolismo óseo se acomoda a un nivel inferior de estrógeno. La pérdida ósea puede ser mayor al aumentar la duración de uso y puede no ser completamente reversible en algunas mujeres. Se desconoce si el uso de la inyección de AMPD durante la adolescencia y la adultez temprana, un periodo crítico de la creación ósea, reducirá la masa ósea máxima. Tanto en mujeres adultas como adolescentes, la disminución de la DMO durante el tratamiento parece ser sustancialmente reversible después de la interrupción de la inyección de AMPD y del aumento de la producción ovárica de estrógenos (ver Farmacocinética y farmacodinamia, Estudios clínicos; Estudios de densidad mineral ósea DMO).
Después de interrumpir la inyección en las adolescentes, la recuperación total de la DMO media en la columna lumbar requirió 1.2 años, 4.6 años en la cadera total y 4.6 años en el cuello femoral (ver sección Farmacocinética y Farmacodinamia-Estudios clínicos, Estudios de densidad mineral ósea [DMO] –Recuperación postratamiento de la DMO en adolescentes).
En adultos, se observó la DMO durante un periodo de 2 años después de que la inyección de AMPD fue descontinuada y la recuperación parcial de la media de la DMO basal se observó en la cadera total, el cuello femoral y columna lumbar (ver sección Farmacocinética y Farmacodinamia, Estudios clínicos, Estudios de densidad mineral ósea (DMO)– Cambios de DMO en Mujeres Adultas). Un gran estudio observacional de usuarias de anticonceptivos femeninos mostró que el uso de la inyección no tiene ningún efecto sobre el riesgo de fracturas osteoporóticas o no osteoporóticas en la mujer (ver sección Farmacocinética y Farmacodinamia-Estudios clínicos, estudios de densidad mineral ósea (DMO)– Relación de la incidencia de fracturas con el uso de AMPD inyectable (150 mg IM) o la no utilización por mujeres en edad reproductiva).
Cambios de DMO en mujeres adultas después de seis meses de tratamiento para la endometriosis: En dos estudios clínicos de 573 mujeres adultas con endometriosis, los efectos de la DMO después de 6 meses de tratamiento con AMPD SC se compararon con 6 meses de tratamiento con leuprolida. Luego, los sujetos se observaron fuera de la terapia, durante 12 meses adicionales.
La proporción de pacientes con una reducción de 5% o más en la DMO fue estadística y significativamente mayor en el grupo de leuprolida en comparación con AMPD SC en cada punto temporal (Tabla 1).
Tabla 1. Proporción de pacientes con una disminución del 5% o más con respecto al valor inicial después de 6 meses de terapia con AMPD SC o leuprolida y 6 meses después de la suspensión de la terapia (estudios 268 y 270 combinados)
|
Parámetro de DMO |
AMPD-SC n/N* (%) |
Leuprolida n/N* (%) |
Valor de p** |
|
Fin del tratamiento (6 meses de terapia) |
|||
|
Columna vertebral |
12/208 (5.8%) |
85/229 (37.1%) |
< 0.001 |
|
Cadera completa |
1/207 (0.5%) |
25/227 (11.0%) |
< 0.001 |
|
En la visita a los 12 meses (6 meses fuera de terapia) |
|||
|
Columna vertebral |
8/166 (4.8%) |
32/178 (18.0%) |
< 0.001 |
|
Cadera completa |
3/166 (1.8%) |
25/178 (14.0%) |
< 0.001 |
* n = número de pacientes con una disminución de la DMO ≥ 5%; N = total de observaciones.
** chi-cuadrado.
Otros métodos anticonceptivos o tratamientos endometriales se deben considerar en el análisis de riesgo/beneficio para el uso de la inyección de AMPD en mujeres con factores de riesgo de osteoporosis tales como:
• Consumo crónico de alcohol y/o tabaco.
• Ingesta crónica de fármacos que pueden reducir la masa ósea, por ejemplo, anticonvulsivos o corticosteroides.
• Índice de masa corporal (IMC) bajo o trastorno de la alimentación, por ejemplo, anorexia nerviosa o bulimia.
• Enfermedad ósea metabólica.
• Antecedentes familiares fuertes de osteoporosis.
Se recomienda que todas las pacientes tengan un consumo suficiente de calcio y vitamina D.
Anticoncepción: La mayoría de las mujeres a las que se administra AMPD solución inyectable experimentan la interrupción de los patrones de sangrado menstrual (por ejemplo, sangrado/manchado irregular o impredecible, sangrado escaso, abundante o continuo). A medida que continúa la administración de AMPD inyectable, una menor cantidad de mujeres experimenta sangrado irregular y una cantidad mayor desarrolla amenorrea.
La vigilancia a largo plazo de casos controlados de usuarias de la suspensión inyectable de AMPD encontró riesgo total leve o no incrementado de cáncer de mama y ningún aumento del riesgo total de cáncer de ovario, de hígado o cervical, pero sí detectó un efecto prolongado y protector en la reducción del riesgo de cáncer de endometrio. Las mujeres tuvieron una tendencia al aumento de peso durante el tratamiento con AMPD.
Hubo una tendencia a ganar peso para las mujeres, mientras se encontraban bajo terapia de AMPD.
Si se desarrolla ictericia, se debe considerar la posibilidad de no volver a administrar el fármaco.
Infecciones de transmisión sexual: Las mujeres deben recibir información en el sentido que la suspensión inyectable de AMPD no protege contra las infecciones de transmisión sexual (ITS) incluyendo infección por VIH (SIDA), igualmente, el AMPD es una inyección estéril, y usada de la manera indicada, no las expondrá a infecciones de transmisión sexual. Las prácticas sexuales más seguras incluyendo el uso correcto y consistente de condones reducen la transmisión de ITS a través del contacto sexual, incluyendo VIH.
Cáncer de mama: Ver a continuación.
Ginecología:
Tratamiento de los síntomas vasomotores menopáusicos/oposición de los efectos endometriales del estrógeno en mujeres menopáusicas en tratamiento con estrógenos (Terapia hormonal): No se estudiaron otras dosis de estrógenos conjugados por vía oral con acetato de medroxiprogesterona y otras combinaciones y formas de dosificación de la terapia hormonal (TH) en el ensayo Women’s Health Initiative (WHI) (ver Farmacocinética y farmacodinamia, Estudios clínicos, Estudio Women’s Health Initiative) y a falta de datos comparables, se debe suponer que estos riesgos sean similares.
Cáncer de mama: Se ha informado que la administración de la combinación de estrógeno/progestina por vía oral en mujeres posmenopáusicas aumenta el riesgo de cáncer de mama. Los resultados de un ensayo aleatorizado y controlado con placebo, el ensayo WHI y los estudios epidemiológicos (ver Farmacocinética y farmacodinamia, Estudios clínicos) han informado de un aumento del riesgo de cáncer de mama en mujeres que tomaban combinaciones de estrógeno/progestina como TH durante varios años. En el ensayo WHI de estrógenos conjugados de origen equino (ECE) más el ensayo AMP y estudios observacionales, el exceso de riesgo aumentó con la duración del tratamiento (ver Dosis y vía de administración). También se ha informado que la administración de estrógeno más progestina genera un aumento de mamografías anormales que requiere una evaluación más profunda.
En varios estudios epidemiológicos no se detectó un aumento de riesgo general de cáncer de mama entre las usuarias de progestágenos de depósito inyectables en comparación con las no usuarias. Sin embargo, se observó un aumento del riesgo relativo (por ejemplo, 2.0 en un estudio) en mujeres que utilizan actualmente progestágenos de depósito inyectables o los habían utilizado solo unos pocos años antes. No es posible deducir a partir de estos datos si este aumento de la tasa de diagnóstico de cáncer de mama entre las usuarias actuales se debe al aumento de la vigilancia entre las usuarias actuales, a los efectos biológicos de los progestágenos inyectables o a una combinación de motivos.
Trastornos cardiovasculares: No se deben administrar estrógenos con o sin progestinas para la prevención de la enfermedad cardiovascular. Varios ensayos aleatorizados y prospectivos sobre los efectos a largo plazo (ver Dosis y vía de administración) de un régimen combinado de estrógeno/progestina en mujeres posmenopáusicas han informado de un aumento del riesgo de eventos cardiovasculares tales como infarto de miocardio, enfermedad coronaria, accidente cerebrovascular (ACV) y tromboembolismo venoso.
Arteriopatía coronaria: No hay evidencias derivadas de ensayos controlados aleatorizados de beneficio cardiovascular con la combinación continua de estrógenos conjugados y acetato de medroxiprogesterona (AMP). Dos grandes ensayos clínicos [ECE/AMP de WHI y el Estudio del reemplazo de estrógeno/progestina y de corazón (HERS, por sus siglas en inglés)] (ver Farmacocinética y farmacodinamia, Estudios Clínicos) mostraron un posible aumento del riesgo de morbilidad cardiovascular en el primer año de tratamiento y ningún beneficio general.
En el ensayo ECE/AMP de WHI se observó un riesgo mayor de eventos de cardiopatía coronaria (CC) (definidos como infarto de miocardio no mortal y muerte por CC) en mujeres que recibieron ECE/AMP en comparación con mujeres que recibieron placebo (37 en comparación con 30 por 10,000 personas/años). El aumento en el riesgo de TEV se observó en el primer año y persistió durante el periodo de observación (ver Dosis y vía de administración).
Accidente cerebrovascular: En el ensayo ECE/AMP de WHI, se observó un riesgo mayor de accidente cerebrovascular en mujeres que recibieron ECE/AMP en comparación con mujeres que recibieron placebo (29 en comparación con 21 por 10,000 personas/años). El aumento en el riesgo se observó en el primer año y persistió durante el periodo de observación (ver Dosis y vía de administración).
Tromboembolismo venoso/embolia pulmonar: La TH se asocia con un mayor riesgo relativo de desarrollar tromboembolismo venoso (TEV), es decir, trombosis venosa profunda o embolia pulmonar. En el ensayo ECE/AMP de WHI, se observó una tasa dos veces mayor de TEV, lo que incluye trombosis venosa profunda y embolia pulmonar en mujeres que recibieron ECE/AMP en comparación con mujeres que recibieron placebo. El aumento en el riesgo se observó en el primer año y persistió durante el periodo de observación (ver Precauciones generales, Advertencias y precauciones adicionales para el uso o formulación específica).
Demencia: El estudio Women’s Health Initiative Memory (WHIMS, por sus siglas en inglés) (ver Farmacocinética y farmacodinamia, Estudios clínicos), un estudio auxiliar del WHI, ECE/AMP informó de un aumento del riesgo de probable demencia en mujeres posmenopáusicas de 65 años o mayores. Además, la terapia de ECE/AMP no previene el deterioro cognitivo leve (DCL) en estas mujeres. No es recomendable administrar terapia hormonal (TH) para evitar la demencia o el DCL en mujeres de 65 años o más.
Cáncer de ovario: En algunos estudios epidemiológicos, la administración actual de productos de estrógeno solo o estrógeno más progestina en mujeres posmenopáusicas durante cinco años o más, se ha asociado con un mayor riesgo de cáncer de ovario. Aquellas pacientes a las que se administró en el pasado productos de estrógeno solo o estrógeno más progestina no estuvieron en mayor riesgo de cáncer de ovario. Otros estudios no mostraron una asociación significativa. El ensayo ECE/AMP de WHI informó que el estrógeno más progestina aumentó el riesgo de cáncer de ovario, pero que este riesgo no fue estadísticamente significativo. En un estudio, las mujeres a las que se administra TRH tienen un mayor riesgo de cáncer de ovario mortal.
Recomendación de examen físico y de antecedentes: Se debe confeccionar una historia clínica y familiar completa antes de iniciar cualquier terapia hormonal. Los exámenes físicos previos al tratamiento y periódicos deben prestar una especial atención a la presión arterial, las mamas, el abdomen y los órganos pélvicos, lo que incluye la citología cervical.
Ginecología: Una anovulación prolongada con amenorrea y/o patrones menstruales erráticos puede desarrollarse luego de la administración de una o varias dosis inyectables de AMPD.
Efectos en la habilidad de conducir y usar máquinas: El efecto del acetato de medroxiprogesterona sobre la habilidad de conducir y usar maquinaria no ha sido evaluado sistemáticamente.
DOSIS Y VÍA DE ADMINISTRACIÓN: La suspensión inyectable se debe agitar bien antes de usar.
Anticoncepción: La suspensión inyectable subcutánea (SC) de AMPD debe agitarse vigorosamente justo antes de su administración, para asegurar que la dosis que se administra representa una suspensión uniforme.
Administración subcutánea (SC): La dosis recomendada es de 104 mg. La inyección SC de AMPD debe administrarse mediante inyección subcutánea en la cara anterior del muslo o en el abdomen, cada 3 meses (12 a 14 semanas). La dosis no necesita ajustarse al peso corporal, (ver Farmacocinética y farmacodinamia). La suspensión SC no está formulada para inyección intramuscular.
Autoinyección: El dispositivo prellenado uniject, AMPD SC 104 mg/0,65 mL, puede ser administrado por un profesional de la salud (HCP, por sus siglas en inglés) o, cuando se considere apropiado por el HCP, el paciente puede autoinyectarse.
La administración del dispositivo prellenado uniject, AMPD SC 104 mg/0,65 mL, debe iniciarse bajo la supervisión de un profesional de la salud (HCP). Después de una formación adecuada en la técnica de inyección y de administración, los pacientes pueden autoinyectarse con el dispositivo prellenado uniject, AMPD SC 104 mg/0,65 mL, si su HCP determina que es apropiado y con el seguimiento médico necesario.
Primera inyección: La primera inyección SC se debe administrar durante los primeros 5 días después del comienzo de un periodo menstrual normal; dentro de los 5 días después del parto si no se encuentra en periodo de lactancia; o bien, si solo ejercita la lactancia materna exclusiva, a las 6 semanas del parto o después.
Segunda inyección y subsecuentes: Si han transcurrido más de 14 semanas desde la última inyección SC, se deberá descartar la posibilidad de embarazo antes de administrar la siguiente inyección SC.
Cambio a partir de otros métodos anticonceptivos: Cuando se realiza el cambio a partir de otros métodos anticonceptivos, (AMPD SC) se debe administrar de modo que garantice la cobertura anticonceptiva continua basada en el mecanismo de acción de ambos métodos (por ejemplo, las pacientes que cambian a partir de anticonceptivos orales deben recibir su primera inyección de AMPD dentro los 7 días después de tomar la última píldora activa).
Ginecología: El uso de la terapia combinada de estrógeno/progestina en mujeres posmenopáusicas se debe limitar a la dosis efectiva más baja y de menor duración consistente con los objetivos del tratamiento y los riesgos para cada mujer en particular, lo que se debe evaluar periódicamente (ver Precauciones generales-Advertencias y precauciones adicionales para el uso o formulación específica).
Se recomiendan revisiones periódicas con una frecuencia y naturaleza adaptadas a cada mujer (ver Precauciones generales-Advertencias y precauciones adicionales para el uso o formulación específica).
A menos que exista un diagnóstico previo de endometriosis, no se recomienda añadir una progestina en mujeres sin útero intacto.
Endometriosis: AMPD inyectable por vía subcutánea 104 mg cada 3 meses durante al menos 6 meses.
Insuficiencia hepática: No hay estudios clínicos que hayan evaluado el efecto de la enfermedad hepática en la farmacocinética de AMP. Sin embargo, AMP se elimina casi exclusivamente mediante el metabolismo hepático y las hormonas esteroides pueden metabolizarse de manera escasa en pacientes con insuficiencia hepática grave (ver Contraindicaciones).
Insuficiencia renal: No hay estudios clínicos que hayan evaluado el efecto de la enfermedad renal en la farmacocinética de AMP. Sin embargo, ya que AMP se elimina casi exclusivamente mediante el metabolismo hepático, no será necesario ningún ajuste de dosis en mujeres con insuficiencia renal.
MANIFESTACIONES Y MANEJO DE LA SOBREDOSIFICACIÓN O INGESTA ACCIDENTAL: Las dosis orales de hasta 3 g al día se han tolerado bien. El tratamiento de la sobredosis es sintomático y de apoyo.
PRESENTACIÓN:
Caja de cartón con jeringa prellenada con 104 mg/0.65 mL e instructivo anexo.
RECOMENDACIONES SOBRE EL ALMACENAMIENTO: Consérvese a no más de 30 °C.
LEYENDAS DE PROTECCIÓN:
Su venta requiere receta médica. Literatura exclusiva para médicos. Léase instructivo anexo. No se deje al alcance de los niños. Agítese antes de usarse. No se administre si el cierre ha sido violado. Deséchese inmediatamente después de su uso. No use durante el embarazo. En caso de lactancia, consulte a su médico.
Reporte las sospechas de reacción adversa al correo:
farmacovigilancia@cofepris.gob.mx y
MEX.AEReporting@pfizer.com y a la línea de
Pfizer 800 401 2002.
Titular del Registro:
Pharmacia and Upjohn Company LLC
7000 Portage Road, Kalamazoo,
Michigan (MI) 49001, EUA
Representante Legal, Importador y Distribuidor:
Pfizer, S.A. de C.V.
Km. 63 Carretera México Toluca,
Zona Industrial, C.P. 50140,
Toluca, México, México
Reg. Núm. 473M2015 SSA IV
Clave de IPP: 23330022130112


