
SANDOSTATINA LAR - Suspensión inyectable
Sustancia(s):
- Octreótida
Presentaciones:
- 1 Caja , 1 Frasco(s) , 10 Miligramos
- 1 Caja , 1 Frasco(s) , 20 Miligramos
- 1 Caja , 1 Frasco(s) , 30 Miligramos
- 1 Caja , 1 Frasco(s) , 10 Miligramos
FORMA FARMACÉUTICA Y FORMULACIÓN:
El frasco ámpula contiene:
|
Acetato de octreótida equivalente a de Octreótida |
11.2 mg 10 mg |
22.4 mg 20 mg |
33.6 mg 30 mg |
|
Excipiente cbp |
355 mg |
625 mg |
900 mg |
|
La jeringa con diluyente contiene: |
2.5 ml |
2.5 ml |
2.5 ml |
INDICACIONES TERAPÉUTICAS:
Tratamiento de pacientes con acromegalia:
– Pacientes que estén adecuadamente controlados con el tratamiento subcutáneo con Sandostatina® S.C.
– Pacientes en quienes la cirugía o la radioterapia es inapropiada, inefectiva o no disponible, o en el periodo intermedio hasta que la cirugía pueda llevarse a cabo, o en el periodo intermedio hasta que la radioterapia se hace completamente efectiva.
– Pacientes que estén indispuestos a una cirugía.
Tratamiento de pacientes con síntomas asociados con tumores funcionales endocrinos gastro-entero-pancreáticos en quienes los síntomas se controlan adecuadamente con el tratamiento S.C. con Sandostatina®:
– Tumores carcinoides con características del síndrome carcinoide.
– VIPomas.
– Glucagonomas.
– Gastrinomas/síndrome de Zollinger-Ellison.
– Insulinomas, para control pre-operatorio de la hipoglucemia y para la terapia de mantenimiento.
– GRFomas.
Tratamiento de los pacientes con tumores neuroendocrinos avanzados funcionales o inactivos del intestino medio o sitio desconocido del tumor primario.
FARMACOCINÉTICA Y FARMACODINAMIA:
Propiedades farmacocinéticas: Después de la administración I.M. única de SANDOSTATINA® LAR®, la concentración de octreótida en suero alcanza un pico transitorio inicial dentro de la primera hora después de la administración, seguido por una reducción progresiva a un nivel bajo de octreótida no detectable dentro de 24 horas. Después de este pico inicial el día 1, la octreótida permanece en niveles subterapéuticos en la mayoría de los pacientes durante los siguientes 7 días. Posteriormente, las concentraciones de octreótida se incrementan nuevamente y alcanzan concentraciones estacionarias durante las siguientes 3 a 4 semanas. El nivel máximo durante el día 1 es más bajo que los niveles durante la fase estacionaria (no se presenta más de 0.5% de la liberación total del fármaco durante el día 1). Después de alrededor del día 42, la concentración de octreótida se reduce lentamente, concomitante con la fase de degradación terminal de la matriz del polímero de la forma farmacéutica.
En pacientes con acromegalia, las concentraciones estacionarias de octreótida después de dosis simples de 10 mg, 20 mg y 30 mg de SANDOSTATINA® LAR® son de 358 ng/L, 926 ng/L y 1,710 ng/L, respectivamente. Las concentraciones séricas de octreótida en el estado estable, alcanzadas después de 3 inyecciones en intervalos de 4 semanas, son mayores por un factor de aproximadamente 1.6 a 1.8 y llegan a 1,557 ng/L y 2,384 ng/L después de inyecciones múltiples de 20 mg y 30 mg de SANDOSTATINA® LAR®, respectivamente.
En pacientes con tumores carcinoides, la media (y la mediana) de las concentraciones séricas de octreótida después de inyecciones múltiples de 10 mg, 20 mg y 30 mg de SANDOSTATINA® LAR®, administradas en intervalos de 4 semanas se incrementaron también linealmente con la dosis y fueron de 1,231 (894) ng/L, 2,620 (2,270) ng/L y 3,928 (3,010) ng/L, respectivamente.
No ocurrió ninguna acumulación de octreótida más allá de lo esperado en los perfiles de liberación superpuestos a lo largo de hasta 28 inyecciones mensuales de SANDOSTATINA® LAR®.
El perfil farmacocinético de octreótida después de la inyección de SANDOSTATINA® LAR® refleja el perfil de liberación de la matriz del polímero y su biodegradación. Una vez liberado en la circulación sistémica, la octreótida se distribuye de acuerdo a sus propiedades farmacocinéticas conocidas, según se describen en la administración S.C. El volumen de distribución de la octreótida en el estado estable es de 0.27 L/kg y la depuración total del cuerpo es de 160 mL/min. La unión a proteínas plasmáticas llega a 65% y esencialmente el fármaco no se une a células sanguíneas.
Propiedades farmacodinámicas: La octreótida es un derivado octapéptido sintético de la somatostatina natural con efectos farmacológicos similares, aunque con una duración de la acción considerablemente prolongada. Inhibe la secreción, incrementada patológicamente, de la hormona de crecimiento (HC) y de los péptidos y serotonina producidos dentro del sistema endocrino gastro-entero-pancreático (GEP).
En animales, la octreótida es un inhibidor más potente de la HC, liberación del glucagón e insulina que la somatostatina, con una mayor selectividad por la HC y la supresión del glucagón.
En sujetos sanos, se ha demostrado que la octreótida y la somatostatina, inhiben:
• La liberación de la HC estimulada por la arginina, y la hipoglucemia inducida por el esfuerzo y la insulina.
• La liberación pospandrial de insulina, glucagón, gastrina, otros péptidos del sistema GEP y la liberación de insulina y glucagón estimulada por la arginina.
• Liberación de la hormona estimulante de la tiroides (TSH) estimulada por la hormona liberadora de tirotropina (TRH).
A diferencia de la somatostatina, la octreótida inhibe la HC preferentemente sobre la insulina y su administración no está seguida por una hipersecreción de rebote de las hormonas (es decir, HC en pacientes con acromegalia).
En pacientes con acromegalia, la SANDOSTATINA® LAR®, una formulación galénica de octreótida, adecuada para la administración repetida en intervalos de 4 semanas, da concentraciones séricas consistentes y terapéuticas de octreótida reduciendo así consistentemente la HC y normalizando las concentraciones séricas del IGF-1 en la mayoría de los pacientes. En la mayor parte de los pacientes, la SANDOSTATINA® LAR® reduce marcadamente los síntomas clínicos de la enfermedad, como dolor de cabeza, transpiración, parestesia, fatiga, osteoartralgia y síndrome del túnel carpiano. En pacientes sin tratamiento previo con acromegalia con adenoma de pituitaria que secreta HC, el tratamiento con SANDOSTATINA® LAR® resultó en una reducción del volumen del tumor de > 20% en una porción significativa de pacientes (50%).
Para pacientes con tumores funcionales del sistema endocrino gastro-entero-pancreático, el tratamiento con SANDOSTATINA® LAR® proporciona un control continuo de los síntomas relacionados con la enfermedad subyacente. El efecto de la octreótida en diferentes tipos de tumores gastro-entero-pancreáticos es el siguiente:
Tumores carcinoides: La administración de la octreótida puede resultar en un aumento de los síntomas, particularmente sofocos y diarrea. En muchos casos, esto está acompañado por reducción de las concentraciones plasmáticas de serotonina y de la excreción urinaria de ácido 5-hidroxiindol acético.
VIPomas: La característica bioquímica de estos tumores es la sobreproducción de péptido intestinal vasoactivo (VIP). En la mayoría de los casos, la administración de octreótida resulta en el alivio de la diarrea secretoria severa típica de la condición, con la consecuente mejora en la calidad de vida. Esto está acompañado por una mejora en los trastornos electrolíticos asociados, por ejemplo, hipocaliemia, lo que permite que se retire el suministro de líquidos enterales y parenterales y de electrólitos. En algunos pacientes, la lectura de la tomografía computarizada sugiere una reducción o arresto del avance del tumor o incluso una reducción del mismo, particularmente de metástasis hepática. La mejora clínica generalmente está acompañada por una reducción en los niveles plasmáticos del VIP, los cuales pueden caer dentro del rango de referencia normal.
Glucagonomas: La administración de octreótida resulta, en la mayoría de los casos, en una mejora sustancial del eritema migratorio necrolítico, el cual es característico de esta enfermedad. El efecto de octreótida sobre el estado de la diabetes mellitus leve, la cual frecuentemente ocurre, no es marcado y, en general, no resulta en una reducción de los requerimientos para la insulina o los agentes hipoglicémicos orales. La octreótida produce una mejora de la diarrea, y por lo tanto ganancia de peso en los pacientes afectados. Aunque la administración de octreótida con frecuencia lleva a una reducción inmediata en los niveles plasmáticos del glucagón, esta reducción generalmente no se mantiene durante un periodo prolongado de administración, a pesar de la mejora sintomática continua.
Gastrinomas/síndrome de Zollinger-Ellison: Aunque la terapia con los inhibidores de la bomba de protones o con los agentes bloqueadores de los receptores H2 controla la ulceración péptica recurrente que resulta de la hipersecreción crónica del ácido gástrico estimulada por la gastrina, dicho control puede ser incompleto. La diarrea también puede ser un síntoma prominente que no se alivia en todos los pacientes mediante esta terapia. La octreótida sola o en conjunto con los inhibidores de la bomba de protones o con los antagonistas de los receptores H2 puede reducir la hipersecreción del ácido gástrico y mejorar los síntomas, incluyendo la diarrea. Otros síntomas debidos posiblemente a la producción de péptidos por el tumor, por ejemplo, sofocos, también pueden aliviarse. Los niveles de gastrina plasmática caen en algunos pacientes.
Insulinomas: La administración de octreótida produce una caída en la insulina inmunorreactiva circulante. En pacientes con tumores operables, la octreótida puede ayudar a restaurar y mantener la normoglucemia antes de la cirugía. En pacientes con tumores benignos o malignos inoperables, el control glicémico puede mejorarse aún sin una reducción sostenida concomitante en los niveles de insulina circulantes.
GRFomas: Estos raros tumores están caracterizados por la producción del factor liberador de la hormona de crecimiento (GRF) solo o junto con otros péptidos activos. La octreótida mejora las características y síntomas de la acromegalia resultante. Esto se debe probablemente a la inhibición de la secreción de GRF y de HC, y puede seguir una reducción en el agrandamiento de la pituitaria.
Tumores neuroendocrinos avanzados del intestino medio o sitio desconocido del tumor primario: Un estudio Fase III, aleatorizado, doble ciego, controlado con placebo (PROMID) demostró que SANDOSTATINA® LAR® inhibe el crecimiento del tumor en pacientes con tumores neuroendócrinos avanzados del intestino medio.
Se aleatorizaron 85 pacientes para recibir SANDOSTATINA® LAR® 30 mg cada 4 semanas (n = 42) o placebo (n = 43) por 18 meses, o hasta que haya progreso del tumor o muerte.
Los criterios de inclusión principales fueron: Tratamiento inicial; tumores/carcinomas funcionalmente activos o inactivos, histológicamente confirmados, localmente inoperables o metastásicos bien diferenciados; con el tumor primario localizado en el intestino medio u origen desconocido, que se cree tener el origen en el intestino medio si se excluyó uno primario en el páncreas, pecho y otra parte.
El punto final primario fue el tiempo para el progreso del tumor o muerte relacionada con el tumor.
En la población de intento a tratar (ITT) (todos los pacientes aleatorizados), 26 y 41 progresos o muertes relacionadas con el tumor se observaron en los grupos de SANDOSTATINA® LAR® y placebo, respectivamente (HR = 0.32, 95% IC, 0.19 a 0.55; valor p = .000015).
En el análisis poblacional conservador de ITT (clTT) en donde se retiraron 3 pacientes en la aleatorización, 26 y 40 progresos o muertes relacionadas con el tumor se observaron en los grupos de SANDOSTATINA® LAR® y placebo, respectivamente (HR = 0.34; 95% IC, 0.20 a 0.59; valor p = 0.000072; Fig. 1). La mediana de tiempo para el progreso del tumor fue de 14.3 meses (95% IC, 11.0 a 28.8 meses) en el grupo de SANDOSTATINA® LAR® y 6.0 meses (95% IC, 3.7 a 9.4 meses) en el grupo de placebo.
En la población de análisis por protocolo (PP) en donde los pacientes se retiraron al final de la terapia de estudio, se observa progreso del tumor o muerte relacionada con el tumor en 19 y 38 receptores de SANDOSTATINA® LAR® y placebo, respectivamente (HR = 0.24; 95% IC, 0.13 a 0.45; valor p = .0000036).
Figura 1. Estimados Kaplan-Meier de TTP comparando SANDOSTATINA® LAR® con placebo (población ITT conservadora)
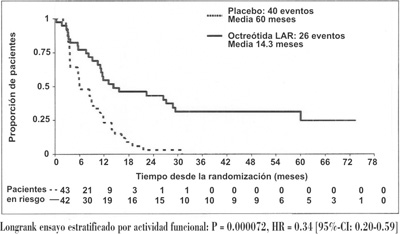
Longrank ensayo estratificado por actividad funcional: P= 0.000072, HR = 0.34 [95%-Cl: 0.20-0.59].
Tabla 1. Resultados TTP por poblaciones de análisis
|
Eventos TTP |
Mediana de meses TTP (95% IC) |
HR [95%IC] Valor p* |
|||
|
SANDOSTATINA® LAR® |
Placebo |
SANDOSTATINA® LAR® |
Placebo |
||
|
ITT |
26 |
41 |
NR |
NR |
0.32 [95% IC, 0.19 a 0.55] P=0.000015 |
|
clTT |
26 |
40 |
14.3 [95% IC, 11.0 a 28.8] |
6.0 [95% IC, 3.7 a 9.4) |
0.34 [95% IC, 0.20 a 0.59] P=0.000072 |
|
pp |
19 |
38 |
NR |
NR |
0.24 [95% IC, 0.13 a 0.45] P=0.0000036 |
NR=no reportado; HR=razón de peligro; TIP=tiempo para el progreso del tumor; ITT=intento a tratar; clTT=ITT conservador; PP=por protocolo.
* Prueba logrank estratificada por actividad funcional.
El efecto del tratamiento fue similar en pacientes con tumores funcionalmente activos (HR = 0.23, 95% IC, 0.09 a 0.57) e inactivos (HR = 0.25, 95% IC, 0.10 a 0.59).
Después de 6 meses de tratamiento, se observó una enfermedad estable en 66% de los pacientes en el grupo de SANDOSTATINA® LAR® y 37% de los pacientes en el grupo de placebo.
Se detuvo el reclutamiento con base al beneficio clínico significante de SANDOSTATINA® LAR® observado en este análisis interno pre-planeado.
La seguridad de SANDOSTATINA® LAR® en este ensayo fue consistente con su perfil de seguridad establecido.
CONTRAINDICACIONES: Hipersensibilidad conocida a la octreótida o a alguno de sus excipientes.
RESTRICCIONES DE USO DURANTE EL EMBARAZO Y LA LACTANCIA:
Embarazo: No hay estudios adecuados y bien controlados en mujeres embarazadas. En la experiencia posmercado, se han reportado los datos sobre un número limitado de embarazos expuestos en pacientes con acromegalia, sin embargo, en la mitad de los casos el resultado del embarazo se desconoce. La mayoría de las mujeres expuestas a la octreótida durante el embarazo fue a una dosis en un rango de 100 a 300 microgramos/día de Sandostatina® S.C. o 20 a 30 mg/mes de SANDOSTATINA® LAR®. En aproximadamente dos tercios de los casos con resultado conocido, las mujeres eligieron continuar con la terapia de octreótida durante sus embarazos. En la mayoría de los casos con resultado conocido, se reportaron recién nacidos normales pero también varios abortos espontáneos durante el primer trimestre, y unos cuantos abortos inducidos.
No hubo casos de anomalías congénitas o malformaciones debido al uso de la octreótida en los casos que se reportó el resultado del embarazo.
Los estudios en animales no indican efectos dañinos directos o indirectos con respecto al embarazo, desarrollo embrio/fetal, desarrollo del parto o posnatal, además de algún retraso transitorio del crecimiento fisiológico (ver Farmacocinética y farmacodinamia).
SANDOSTATINA® LAR® sólo debe prescribirse a mujeres embarazadas bajo circunstancias comprometedoras (ver Precauciones generales).
Lactancia: Se desconoce si la octreótida se excreta en la leche maternal humana. Los estudios en animales han mostrado la excreción de la octreótida en la leche materna. Las pacientes no deben amamantar durante el tratamiento con Sandostatina®.
REACCIONES SECUNDARIAS Y ADVERSAS: Las reacciones adversas más frecuentes que se reportan durante la terapia con octreótida incluyen desórdenes gastrointestinales, desórdenes del sistema nervioso, desórdenes hepatobiliares y desórdenes del metabolismo y de nutrición.
Las reacciones adversas más comúnmente reportadas en los ensayos clínicos con la administración de octreótida fueron diarrea, dolor abdominal, náusea, flatulencia, dolor de cabeza, colelitiasis, hiperglucemia y constipación. Otras reacciones adversas comúnmente reportadas fueron mareo, dolor localizado, lodo biliar, disfunción tiroidea (por ejemplo, disminución de la hormona estimulante de la tiroides [HET], disminución de T4-Total, y disminución de T4 Libre), heces líquidas, tolerancia a la glucosa dañada, vómito, astenia e hipoglucemia.
En casos raros, los efectos colaterales gastrointestinales simulan una obstrucción intestinal aguda, con distensión abdominal progresiva, dolor epigástrico severo, suavidad y guarda abdominal.
Aunque se puede incrementar la grasa fecal medida, no hay evidencia hasta la fecha de que el tratamiento a largo plazo con octreótida haya generado deficiencia nutricional debido a una mala absorción.
En casos muy raros, se ha reportado pancreatitis aguda en las primeras horas o días del tratamiento con Sandostatina® S.C. y se resolvió al retirar el medicamento. Además, se ha reportado pancreatitis inducida por colelitiasis en pacientes en tratamiento con Sandostatina® S.C.
Tanto en pacientes acromegálicos como con síndrome carcinoide, se observaron cambios en el ECG como prolongación de QT, cambios axiales, repolarización temprana, voltaje bajo, transición R/S, progreso temprano de la onda R, y cambios de la onda ST-T no específicos. La relación de estos eventos con el acetato de octreótida no estableció porque muchos de estos pacientes tenían enfermedades cardiacas subyacentes (ver Precauciones generales).
Las siguientes reacciones adversas al medicamento, enlistadas en la tabla 1, se han recolectado de los estudios clínicos con octreótida:
Las reacciones adversas al medicamento (tabla 2) se catalogan por frecuencia, siendo la más frecuente primero, utilizando la siguiente convención: muy común (³ 1/10); común (³ 1/100, < 1/10); no común (³ 1/1,000, < 1/100); raro (³ 1/10,000, < 1/1,000); muy raro (< 1/10,000), incluyendo los reportes aislados. Dentro de cada grupo de frecuencia, las reacciones adversas se catalogan en orden decreciente de seriedad.
Tabla 2. Reacciones adversas al medicamento reportadas en los estudios clínicos
|
Desórdenes gastrointestinales |
|
|
Muy común |
Diarrea, dolor abdominal, náusea, constipación, flatulencia. |
|
Común |
Dispepsia, vómito, distensión abdominal, esteatorrea, heces sueltas, decoloración de heces. |
|
Desórdenes del sistema nervioso |
|
|
Muy común |
Dolor de cabeza. |
|
Común |
Mareo. |
|
Desórdenes endocrinos |
|
|
Común |
Hipotiroidismo, disfunción de tiroides (por ejemplo, disminución de HET, disminución de T4 Total, y disminución de T4 Libre). |
|
Desórdenes hepatobiliares |
|
|
Muy común |
Colelitiasis. |
|
Común |
Colecistitis, lodo biliar, hiperbilirrubinemia. |
|
Desórdenes del metabolismo y de nutrición |
|
|
Muy común |
Hiperglucemia. |
|
Común |
Hipoglucemia, tolerancia a la glucosa dañada, anorexia. |
|
No común |
Deshidratación. |
|
Desórdenes generales y sitio de administración |
|
|
Muy común |
Dolor localizado en el sitio de inyección. |
|
Investigaciones |
|
|
Común |
Niveles elevados de transaminasa. |
|
Desórdenes de piel y tejido subcutáneo |
|
|
Común |
Prurito, salpullido, alopecia. |
|
Desórdenes respiratorios |
|
|
Común |
Disnea. |
|
Desórdenes cardiacos |
|
|
Común |
Bradicardia. |
|
No común |
Taquicardia. |
Posmercadeo: Las reacciones adversas espontáneas, mostradas en la tabla 3, se reportaron de manera voluntaria y no siempre es posible establecer una frecuencia confiable o relación causal con la exposición al medicamento.
Tabla 3. Reacciones adversas al medicamento derivadas de los reportes espontáneos
|
Desórdenes inmunes |
Anafilaxis, reacciones de alergia/hipersensibilidad. |
|
Desórdenes de piel y tejido subcutáneo |
Urticaria. |
|
Desórdenes hepatobiliares |
Pancreatitis aguda, hepatitis aguda sin colestasis, hepatitis colestática, colestasis, ictericia, ictericia colestática. |
|
Desórdenes cardiacos |
Arritmias. |
|
Investigaciones |
Incremento en los niveles de fosfatasa alcalina, incremento en los niveles de gama glutamil transferasa. |
PRECAUCIONES EN RELACIÓN CON EFECTOS DE CARCINOGÉNESIS, MUTAGÉNESIS, TERATOGÉNESIS Y SOBRE LA FERTILIDAD:
Toxicidad tras dosis únicas: En los estudios de toxicidad tras dosis únicas en el ratón, la DL50 de la octreótida fue de 72 mg/kg de peso por vía intravenosa y de 470 mg/kg de peso por vía subcutánea; en la rata, la DL50 por vía intravenosa fue de 18 mg/kg de peso. Los perros toleraron bien bolos intravenosos de acetato de octreótida de hasta 1 mg/kg de peso.
Toxicidad tras dosis repetidas: En un estudio de toxicidad en ratas, con autopsia en la semana 26, una inyección intramuscular de 2.5 mg de SANDOSTATINA® LAR® (en microesferas de 50 mg) cada 4 semanas durante 21 semanas no produjo signos patológicos relacionados con el fármaco. El único dato anatomopatológico de interés fue la miositis granulomatosa reversible que ocasionaron las microesferas en el lugar de la inyección, tanto en las ratas tratadas como en los animales de comparación. Tras una inyección intramuscular única de SANDOSTATINA® LAR® a ratas y conejos, las microesferas se biodegradaron por completo en 75 días en ambas especies.
Mutagenicidad: Ni la octreótida ni sus metabolitos presentaron un potencial mutagénico in vitro en sistemas analíticos validados de bacterias y células de mamífero. En células de hámster chino V79 se observó un aumento de la frecuencia de alteraciones cromosómicas in vitro, pero sólo con concentraciones elevadas y citotóxicas. En cambio, no se notó un aumento de las aberraciones cromosómicas en linfocitos humanos incubados con acetato de octreótida in vitro. In vivo, no se observó actividad clastógena en la médula ósea de ratones tratados con la octreótida por vía intravenosa (ensayo de micronúcleos) ni tampoco signos de genotoxicidad en ratones machos en un ensayo de reparación de ADN en cabezas de espermatozoides. Las microesferas carecieron de potencial mutágeno en un ensayo bacteriano in vitro validado.
Carcinogenicidad/toxicidad crónica: En los estudios en ratas tratadas con dosis diarias de hasta 1.25 mg/kg de peso de Sandostatina® S.C. se observaron fibrosarcomas en el lugar de la inyección al cabo de 52, 104 y 113/116 semanas, sobre todo en los machos. Las ratas de comparación también mostraron tumores locales; sin embargo, el desarrollo de estos tumores se atribuyó a una displasia fibrosa causada por el efecto irritante sostenido en los lugares de inyección, y potenciado por la acidez del vehículo de ácido láctico y manitol. Esta reacción hística inespecífica pareció limitarse a la rata. No se observaron lesiones neoplásicas ni en los ratones tratados con inyecciones subcutáneas diarias de hasta 2 mg/kg de peso de Sandostatina® durante 98 semanas ni en los perros tratados con inyecciones subcutáneas diarias durante 52 semanas.
El estudio de carcinogenia de 116 semanas en ratas tratadas con Sandostatina® S.C. también reveló la formación de adenocarcinomas en el endometrio uterino, cuya incidencia llegó a ser estadísticamente significativa con la dosis más alta de 1.25 mg/kg de peso diarios. Este hallazgo se acompañó de una elevada incidencia de endometritis, una disminución de la cantidad de cuerpos lúteos, una disminución de los adenomas mamarios y la presencia de dilatación luminal y glandular uterina, sugiriendo un cuadro de desequilibrio hormonal. La información disponible indica claramente que los hallazgos de tumores de carácter endocrino son específicos de la rata y no revisten importancia para la utilización del medicamento en el ser humano.
Toxicidad durante la reproducción: Los estudios de fecundidad y los estudios prenatales, perinatales y posnatales en ratas hembras no revelaron efectos adversos en la función reproductora ni en el desarrollo de las crías al administrar dosis subcutáneas diarias de hasta 1 mg/kg de peso. Se observó un cierto retraso del crecimiento fisiológico de las crías, de carácter pasajero y atribuible a la inhibición de la HC, por la excesiva actividad farmacodinámica.
INTERACCIONES MEDICAMENTOSAS Y DE OTRO GÉNERO: Se ha encontrado que la octreótida reduce la absorción intestinal de la ciclosporina y retarda la de cimetidina.
La administración concomitante de la octreótida y de bromocriptina incrementa la biodisponibilidad de esta última.
Los limitados datos publicados indican que los análogos de la somatostatina pueden reducir la depuración metabólica de compuestos que se sabe son metabolizados por las enzimas del citocromo P450, lo cual puede deberse a la supresión de la hormona de crecimiento. Como no se puede excluir este efecto con la octreótida, deben utilizarse con cautela los medicamentos metabolizados principalmente por la forma CYP3A4 que presentan un bajo índice terapéutico (por ejemplo, la quinidina y la terfenadina).
ALTERACIONES EN LOS RESULTADOS DE PRUEBAS DE LABORATORIO: Se han reportado casos muy raros de elevación de las concentraciones de fosfatasa alcalina, gamma-glutamiltransferasa y transaminasas.
PRECAUCIONES GENERALES:
Generales: Dado que los tumores hipofisarios secretores de la hormona del crecimiento pueden crecer y generar complicaciones graves (por ejemplo, defectos del campo visual), es esencial que todos los pacientes sean cuidadosamente monitoreados. Si aparece evidencia de expansión del tumor, se aconsejan procedimientos alternativos.
El beneficio terapéutico de una reducción en los niveles de la hormona de crecimiento (HC) y la normalización de la concentración del factor de crecimiento 1 tipo insulina (IGF-1) en pacientes femeninas acromegálicas podría potencialmente restaurar la fertilidad. Las pacientes femeninas con potencial fértil deben ser aconsejadas sobre el uso de un anticonceptivo adecuado si es necesario durante el tratamiento con octreótida (ver Restricciones de uso durante el embarazo y la lactancia).
La función de la tiroides debe monitorearse en pacientes que reciben tratamiento prolongado con octreótida.
Acontecimientos cardiovasculares: Se han señalado casos poco frecuentes de bradicardia, por lo que puede ser necesario reducir la dosis de medicamentos como betabloqueadores, bloqueadores de los canales de calcio o fármacos que controlan el equilibrio hidroelectrolítico.
Eventos adversos biliares: Se ha reportado el desarrollo de cálculos biliares en 15 a 30% de los pacientes que reciban tratamiento a largo plazo de Sandostatina® S.C. La prevalencia en la población general (de 40 a 60 años de edad) es de un 5 a 20%. La exposición a largo plazo a SANDOSTATINA® LAR® de pacientes con acromegalia o con tumores gastro-entero-pancreáticos sugiere que el tratamiento con SANDOSTATINA® LAR® no incrementa la formación de cálculos biliares, en comparación con el tratamiento S.C. Se recomienda, sin embargo, el examen con ultrasonido de la vesícula biliar antes y aproximadamente cada 6 meses durante el tratamiento con SANDOSTATINA® LAR®. Si se presentan cálculos biliares, éstos son generalmente asintomáticos; los cálculos sintomáticos deben tratarse ya sea mediante terapia de disolución con ácidos biliares o eliminarse mediante cirugía.
Metabolismo de la glucosa: Como SANDOSTATINA® LAR® inhibe la liberación de la hormona del crecimiento, glucagón e insulina, puede afectar la regulación de la glucosa así como la tolerancia a la glucosa posprandial. Se ha señalado que, en algunos pacientes tratados con Sandostatina® S.C., la administración crónica puede provocar un estado de hiperglucemia persistente.
En pacientes con diabetes mellitus Tipo I concomitante, es probable que SANDOSTATINA® LAR® afecte la regulación de la glucosa y los requerimientos de insulina pueden reducirse. En los no diabéticos y los diabéticos tipo II con reservas de insulina parcialmente intactas, la administración de Sandostatina® S.C. puede resultar en incrementos en la glucemia posprandial. Por lo tanto, se recomienda monitorear la tolerancia a la glucosa y el tratamiento antidiabético.
En pacientes con insulinomas, dado que la inhibición de la insulina es menos potente y de menor duración que la inhibición de la secreción de HC y de glucagón, la octreótida puede intensificar y prolongar la hipoglucemia. Estos pacientes deben ser monitoreados estrechamente.
Nutrición: La octreótida puede alterar la absorción de lípidos de la dieta en algunos pacientes.
Algunos pacientes tratados con la octreótida han presentado reducciones de las concentraciones de vitamina B12 y resultados anormales de las pruebas de Schilling. En los pacientes en tratamiento con SANDOSTATINA® LAR® con antecedentes de carencia de vitamina B12, se recomienda monitorear las concentraciones de esta vitamina.
Efectos sobre la capacidad para conducir y utilizar máquinas: Se carece de información sobre los efectos de SANDOSTATINA® LAR® en la capacidad para conducir y utilizar máquinas.
Incompatibilidades: Como SANDOSTATINA® LAR® en microesferas para inyectables se presenta en un envase monodosis y no debe diluirse con otros productos, no se han generado datos de compatibilidad con otros productos.
DOSIS Y VÍA DE ADMINISTRACIÓN:
Vía de administración: Inyección intramuscular profunda (región glútea).
Dosis: SANDOSTATINA® LAR® sólo debe administrarse mediante inyección profunda en los glúteos. El sitio de inyección debe alternarse entre el músculo glúteo derecho e izquierdo.
Acromegalia:
Para pacientes que están adecuadamente controlados con Sandostatina® S.C., se recomienda iniciar el tratamiento con la administración de 20 mg de SANDOSTATINA® LAR® en intervalos de 4 semanas durante 3 meses. El tratamiento con SANDOSTATINA® LAR® puede iniciarse el día posterior a la última dosis de Sandostatina® S.C. El ajuste subsecuente de la dosis debe basarse en las concentraciones séricas de somatotropina (hormona de crecimiento; HC) y somatomedina C (factor de crecimiento insulínico tipo 1; IGF-1), así como en los síntomas clínicos.
Para pacientes en quienes, dentro de un periodo de 3 meses, los síntomas clínicos y los parámetros bioquímicos (HC; IGF-1) no están completamente controlados (concentraciones de HC todavía superiores a 2.5 µg/L), la dosis puede incrementarse a 30 mg cada 4 semanas.
Para pacientes cuyas concentraciones de HC son constantemente inferiores a 1 µg/L, cuyas concentraciones séricas del IGF-1 están normalizadas y en quienes la mayoría de los signos y síntomas reversibles de la acromegalia han desaparecido después de 3 meses de tratamiento con 20 mg, la dosis de SANDOSTATINA® LAR® puede reducirse a 10 mg cada 4 semanas. Sin embargo, particularmente en este grupo de pacientes, se recomienda vigilar estrechamente las concentraciones séricas de HC y IGF-1, así como los signos y síntomas durante el tratamiento con dosis bajas de SANDOSTATINA® LAR®.
Para pacientes con una dosis estable de SANDOSTATINA® LAR®, debe hacerse una valoración de HC y de IGF-1 cada 6 meses.
Para pacientes en quienes la cirugía o la radioterapia es inapropiada o inefectiva, o en el periodo intermedio hasta que la radioterapia se hace completamente efectiva: Se recomienda un periodo corto de dosis de prueba de administración S.C. de Sandostatina® para valorar la respuesta y la tolerabilidad sistémica de la octreótida antes de iniciar el tratamiento con SANDOSTATINA® LAR® según lo descrito anteriormente.
Tumores endocrinos gastro-entero-pancreáticos:
Tratamiento de pacientes con síntomas asociados con los tumores neuroendocrinos gastro-entero-pancreáticos funcionales.
Para pacientes en quienes los síntomas están adecuadamente controlados con Sandostatina® S.C., se recomienda iniciar el tratamiento con la administración de 20 mg de SANDOSTATINA® LAR® en intervalos de 4 semanas.
El tratamiento con Sandostatina® S.C. debe continuarse con la dosis previamente eficaz durante 2 semanas después de la primera inyección de SANDOSTATINA® LAR®.
Para pacientes que no han sido previamente tratados con Sandostatina® S.C., se recomienda iniciar con la administración de Sandostatina® S.C. en una dosis de 0.1 mg tres veces diarias durante un corto periodo (aproximadamente 2 semanas) para evaluar la respuesta y la tolerabilidad sistémica de la octreótida antes de iniciar el tratamiento con SANDOSTATINA® LAR®, según lo descrito anteriormente.
Para pacientes en quienes los síntomas y los indicadores biológicos están bien controlados después de 3 meses de tratamiento, la dosis puede reducirse a 10 mg de SANDOSTATINA® LAR® cada 4 semanas.
Para pacientes en quienes los síntomas sólo están controlados parcialmente después de 3 meses de tratamiento, la dosis puede incrementarse a 30 mg de SANDOSTATINA® LAR® cada 4 semanas.
Para los días en que los síntomas asociados con los tumores gastro-entero-pancreáticos pueden incrementarse durante el tratamiento con SANDOSTATINA® LAR®, se recomienda la administración adicional de Sandostatina® S.C. en la dosis usada antes del tratamiento con SANDOSTATINA® LAR®. Esto puede ocurrir principalmente en los primeros 2 meses de tratamiento hasta que se alcanzan las concentraciones terapéuticas de octreótida.
Tratamiento de pacientes con tumores neuroendocrinos avanzados del intestino medio o sitio desconocido del tumor primario: La dosis recomendada de SANDOSTATINA® LAR® es de 30 mg administrado cada 4 semanas (ver Farmacocinética y farmacodinamia). El tratamiento con SANDOSTATINA® LAR® para el control del tumor debe continuarse en la ausencia del progreso del tumor.
Uso en pacientes con insuficiencia renal: La función renal deteriorada no afectó la exposición total (ABC) a octreótida cuando se administró S.C. SANDOSTATINA® LAR®. Por lo tanto, no es necesario ningún ajuste de dosis de SANDOSTATINA® LAR® en los pacientes con insuficiencia renal.
Uso en pacientes con insuficiencia hepática: En un estudio con Sandostatina® administrada S.C. e I.V. se demostró que la capacidad de eliminación puede reducirse en pacientes con cirrosis hepática, pero no en pacientes con enfermedad del hígado graso. Debido a la amplia ventana terapéutica de la octreótida, no es necesario ningún ajuste de dosis de SANDOSTATINA® LAR® en pacientes con cirrosis hepática.
Uso en ancianos: En un estudio con Sandostatina® S.C., no fue necesario ningún ajuste de dosis en pacientes ? 65 años. Por lo tanto, no es necesario ningún ajuste de dosis en este grupo de pacientes con SANDOSTATINA® LAR®.
Uso en niños: Es limitada la experiencia sobre el uso de SANDOSTATINA® LAR® en niños.
MANIFESTACIONES Y MANEJO DE LA SOBREDOSIFICACIÓN O INGESTA ACCIDENTAL: Se ha reportado un número limitado de sobredosis accidentales con SANDOSTATINA® LAR®. Las dosis estuvieron en un rango de 100 mg a 163 mg/mes de SANDOSTATINA® LAR®. El único evento adverso reportado fueron bochornos.
Se ha reportado que los pacientes con cáncer han recibido dosis de SANDOSTATINA® LAR® hasta de 60 mg/mes y hasta 90 mg/2 semanas. Estas dosis fueron en general bien toleradas; sin embargo, se han reportado los siguientes eventos adversos: orina frecuente, fatiga, depresión, ansiedad y falta de concentración.
El tratamiento de la sobredosis debe ser sintomático.
PRESENTACIONES: Caja con 1 frasco ámpula con 10, 20 o 30 mg, una jeringa prellenada con 2.5 ml de diluyente y 2 agujas.
RECOMENDACIONES SOBRE ALMACENAMIENTO: Conservar en refrigeración entre 2 y 8°C. No se congele.
Protéjase de la luz.
SANDOSTATINA® LAR® puede permanecer a una temperatura inferior a 25°C el día de la inyección. No obstante, la suspensión debe prepararse justo antes de la inyección intramuscular.
LEYENDAS DE PROTECCIÓN:
Consérvese en refrigeración entre 2 y 8ºC. No se congele. Protéjase de la luz. SANDOSTATINA® LAR® puede permanecer a una temperatura inferior a 25°C el día de la inyección. No obstante, la suspensión debe prepararse justo antes de la inyección intramuscular. Léase instructivo anexo. No se deje al alcance de los niños. Su venta requiere receta médica. No se use en el embarazo y lactancia. Hecha la mezcla, adminístrese de inmediato y deséchese el sobrante. No se administre si el cierre ha sido violado. No se administre si la solución no es transparente, si contiene partículas en suspensión o sedimentos. Literatura exclusiva para el médico.
NOVARTIS FARMACÉUTICA, S. A. de C. V.
La Candelaria No. 186, Local A
Col. Atlántida, C.P. 04370
Deleg. Coyoacán, D.F., México
Reg. Núm. 418M99, SSA IV
103501415D0019
BPI: 20 May 2010
NPI: 10 ene 13
T.N.: Not applicable

