
NUCALA
MEPOLIZUMAB
Solución inyectable
1 Caja, 1 Frasco(s) ámpula con liofilizado, 100 mg
1 Caja, 3 Frasco(s) ámpula con liofilizado, 100 mg
Para visualizar el contenido completo de la IPPA (información para prescribir amplia), deberá iniciar sesión.
FORMA FARMACÉUTICA Y FORMULACIÓN:
Solución:
• Polvo liofilizado:
Fórmula:
El frasco ámpula con polvo liofilizado contiene:
Mepolizumab 100 mg
Excipiente cbp
Diluyente recomendado: Agua estéril para uso inyectable, en un volumen de 1.2 mL para permitir un volumen extraíble de 1 mL después de la reconstitución.
Después de la reconstitución, cada mL de solución contiene 100 mg de mepolizumab
• Fórmula Líquida:
Fórmula:
Cada pluma precargada (auto-inyector) o jeringa precargada (jeringa de seguridad) contiene:
Mepolizumab 100 mg
Vehículo cbp 1 mL
Anticuerpo monoclonal humanizado IgG1 de origen ADN recombinante expresado en células de ovario de Hámster Chino (CHO)
INDICACIONES TERAPÉUTICAS:
• Asma Eosinofílica Grave Refractaria:
NUCALA está indicado como un tratamiento complementario de mantenimiento en pacientes adultos y adolescentes mayores de 12 años de edad, con asma eosinofílica grave refractaria. (Véase sección de estudios clínicos, en la sección de Farmacocinética).
Limitaciones de uso:
NUCALA no está indicado para el alivio del broncoespasmo agudo o estado asmático.
• Rinosinusitis crónica con pólipos nasales (RSCcPN):
NUCALA está indicado como tratamiento de mantenimiento complementario en pacientes adultos mayores de 18 años con rinosinusitis crónica con pólipos nasales (RSCcPN) inadecuadamente controlada.
• Granulomatosis eosinofílica con poliangeítis (EGPA):
NUCALA está indicado como tratamiento en pacientes adultos mayores de 18 años con granulomatosis eosinofílica con poliangeitis (EGPA).
• Síndrome hipereosinofílico (HES):
NUCALA está indicado para el tratamiento del síndrome hipereosinofílico (HES) en pacientes adultos y adolescentes mayores de 12 años de edad.
FARMACOCINÉTICA Y FARMACODINAMIA:
Código ATC:
Grupo farmacoterapéutico: Medicamentos para el tratamiento de enfermedades obstructivas de las vías aéreas, otros medicamentos sistémicos de enfermedades obstructivas de las vías aéreas
R03DX09
Mecanismo de acción:
NUCALA es un anticuerpo monoclonal humanizado (IgG1, kappa), que se dirige a interleucina humana 5 (IL-5) con alta afinidad y especificidad. IL-5 es la principal citosina responsable del crecimiento y diferenciación, reclutamiento, activación y supervivencia de los eosinófilos. NUCALA se une a IL - 5 con una constante de disociación de 100 pM. NUCALA inhibe la bioactividad de IL-5 al bloquear la unión de IL-5 a la cadena alfa del complejo receptor- IL-5 expresado en la superficie celular del eosinófilo. La inflamación es un importante componente de la patogénesis del asma, EGPA, HES y RSCcPN. En la inflamación están involucrados múltiples tipos celulares (por ejemplo, mastocitos, eosinófilos, neutrófilos, macrófagos, linfocitos) y mediadores (por ejemplo, histamina, eicosanoides, leucotrienos, citosinas). NUCALA inhibe la señalización IL-5 y se reduce la producción y supervivencia de eosinófilos, sin embargo, el mecanismo de la acción de mepolizumab no se ha establecido definitivamente en asma, EGPA, HES y RSCcPN.
Farmacodinamia:
La respuesta farmacodinámica (reducción de eosinófilos en sangre) después de dosis repetidas de mepolizumab administrado de manera subcutánea o intravenosa se evaluó en pacientes con asma y niveles de eosinófilos en sangre mayores a 200 células / μL. Los pacientes recibieron 1 de 4 tratamientos de mepolizumab (administrados cada 28 días para un total de 3 dosis): 12.5 mg SC, 125 mg SC, 250 mg SC o 75 mg IV. Sesenta y seis (66) de los 70 pacientes aleatorizados terminaron el estudio.
En estudios clínicos, se observó de forma consistente la reducción de eosinófilos en la sangre después del tratamiento con NUCALA. La magnitud y duración de esta reducción fue dependiente de la dosis.
Se observó una reducción en los eosinófilos en sangre en todos los grupos de tratamiento por el Día 3 (48 horas después de dosificarse). En el Día 84 (4 semanas después de la última dosis), la reducción de la media geométrica observada a partir del valor basal en eosinófilos en sangre fue del 64 %, 78 %, 84 % y 90 % en los grupos de tratamiento de 12.5 mg SC, 75 mg IV, 125 mg SC y 250 mg SC, respectivamente. Las dosis SC predichas por el modelo proporcionando el 50% y 90% de la reducción máxima de eosinófilos en sangre en el Día 84 se estimó que es 11 y 99 mg, respectivamente. Estos resultados, junto con los datos de eficacia clínica del estudio de exacerbación en sujetos adultos y adolescentes con asma grave variando la dosis (Estudio 1) apoyaron la evaluación de 75 mg de mepolizumab IV y 100 mg SC en estudios confirmatorios en estudios de asma grave.
Después de una dosis de 100 mg administrada por vía subcutánea cada 4 semanas durante 32 semanas en sujetos adultos y adolescentes con asma grave, los eosinófilos en sangre se redujeron a un recuento de la media geométrica de 40 células/μL. Esto corresponde a una reducción en la media geométrica de 84% en comparación con placebo.
En pacientes con RSCcPN, después de una dosis de 100 mg administrada por vía subcutánea cada 4 semanas durante 52 semanas, los eosinófilos en sangre fueron reducidos hasta un recuento promedio de 60 células/μL, que corresponde a una reducción promedio de 83% en comparación con el placebo. Esta magnitud de reducción se observó dentro de las 4 semanas de tratamiento y se mantuvo durante el periodo de tratamiento. (Consulte estudios Clínicos).
Para adultos con EGPA, después de la administración por vía subcutánea de mepolizumab 300 mg cada 4 semanas durante 52 semanas, los eosinófilos en sangre se redujeron a un recuento promedio de 38 células/μL. El promedio se redujo un 83% en comparación con el placebo, y esta magnitud de reducción se observó dentro de las 4 semanas de tratamiento (consulte Estudios clínicos).
Para adultos y adolescentes con HES, después de la administración subcutánea de mepolizumab 300 mg cada 4 semanas durante 32 semanas, los eosinófilos sanguíneos se redujeron a un promedio de 70 células / μL. Hubo una reducción promedio del 92% en comparación con el placebo (consulte Estudios clínicos).
Farmacocinética:
Después de la dosis subcutánea en sujetos con asma moderada/grave, mepolizumab exhibió una farmacocinética aproximadamente proporcional a la dosis a lo largo de un rango de dosis de 12.5 mg a 250 mg. Las propiedades farmacocinéticas de mepolizumab observadas en pacientes con RSCcPN, EGPA o HES, fue similar a las propiedades farmacocinéticas observadas en pacientes con asma grave. La administración subcutánea de 300 mg de NUCALA tuvo aproximadamente tres veces la exposición sistémica de 100 mg de mepolizumab.
Absorción:
Después de la administración subcutánea a sujetos sanos o pacientes con asma, mepolizumab se absorbió lentamente con una mediana de tiempo para alcanzar la concentración plasmática máxima (Tmáx) que varía de 4 a 8 días.
Después de una sola administración subcutánea en el abdomen, muslo o brazo de sujetos sanos, la biodisponibilidad absoluta de mepolizumab fue 64%, 71% y 75%, respectivamente. En pacientes con asma, la biodisponibilidad absoluta de mepolizumab administrado por vía subcutánea en el brazo fue de 74-80%. Después de la administración subcutánea repetida cada 4 semanas, existe aproximadamente una acumulación del doble en estado estable.
Distribución:
Después de una sola administración intravenosa de mepolizumab a pacientes con asma, la media del volumen de distribución fue de 55 a 85 mL/kg.
Metabolismo:
Mepolizumab es un anticuerpo monoclonal humanizado IgG1 degradado por enzimas proteolíticas que se distribuyen ampliamente en el cuerpo y no se restringen al tejido hepático.
Eliminación:
Después de una sola administración intravenosa a pacientes con asma, la media de la depuración sistémica (CL) varió de 1.9 a 3.3 mL/día/kg, con una vida media de aproximadamente 20 días. Después de la administración subcutánea de mepolizumab, la media de semivida terminal (t1/2) varió de 16 a 22 días. En el análisis farmacocinético de la población se estimó una depuración sistémica de mepolizumab de 3.1 mL/día/kg.
Poblaciones Especiales de Pacientes:
La farmacocinética poblacional de mepolizumab se analizó para valorar los efectos de las características demográficas. Los resultados de este análisis son limitados, y sugieren que no se requieren ajustes en las dosis por raza o género.
Pacientes ancianos (65 años o mayores):
No se han realizado estudios formales en pacientes geriátricos. Sin embargo, en el análisis farmacocinético poblacional no hubo un efecto de la edad en la farmacocinética de mepolizumab.
Deterioro renal:
No se han realizado estudios formales para investigar el deterioro renal sobre la farmacocinética de mepolizumab. Con base en los análisis de farmacocinética de la población, no se requiere ajuste de la dosis en pacientes con valores de depuración de creatinina entre 50-80 mL/min. Se cuenta con datos disponibles limitados con valores de depuración de creatinina <50 mL/min.
Deterioro hepático:
No se han realizado estudios formales para investigar el deterioro hepático sobre la farmacocinética de mepolizumab. Dado que mepolizumab se degrada por enzimas proteolíticas ampliamente distribuidas, no restringidas al tejido hepático, los cambios en la función hepática tienen pocas probabilidades de tener algún efecto sobre la eliminación de mepolizumab.
Interacciones medicamentosas:
No se han realizado estudios formales de interacción de fármacos con mepolizumab. En los análisis de farmacocinéticos de la población de los estudios fase 3, no hubo evidencia de un efecto de fármacos de molécula pequeña coadministrados comúnmente en la exposición de mepolizumab.
Las enzimas de citocromo P450, bombas de eflujo y los mecanismos de unión a proteína no están involucrados en la depuración de mepolizumab. Niveles aumentados de citosinas pro-inflamatorias (por ejemplo, IL-6), vía interacción con sus receptores análogos en hepatocitos, han mostrado reprimir la formación de enzimas CYP450 y transportadores de fármaco, sin embargo, la elevación de marcadores pro-inflamatorios en asma grave es mínima y no hay evidencia de expresión de alfa receptor IL-5 sobre hepatocitos. El potencial para interacciones medicamentosas con mepolizumab es por lo tanto considerada como baja.
Estudios clínicos:
Asma grave:
La eficacia de NUCALA en el tratamiento de un grupo objetivo de pacientes con asma eosinofílica refractaria grave fue evaluada en 3 estudios clínicos aleatorizados, doble ciego, de grupo paralelo de entre 24-52 semanas de duración, en pacientes de 12 años de edad y mayores. Estos pacientes permanecieron sin control (al menos dos exacerbaciones graves en los 12 meses previos) sobre su atención estándar vigente, incluyendo al menos dosis altas de corticosteroides inhalados (CSI) más un tratamiento de mantenimiento adicional, o fueron dependientes de corticosteroides sistémicos. Tratamientos de mantenimiento adicionales incluyeron agonistas beta2-adrenérgicos de acción prolongada (LABA), modificadores de leucotrienos, antagonistas muscarínicos de acción prolongada (LAMA), teofilina, y corticosteroides orales (CSO).
Los dos estudios de exacerbaciones MEA112997 y MEA115588 inscribieron un total de 1192 pacientes, 60% el sexo femenino, con una media de edad de 49 años (rango 12-82). La proporción de pacientes en mantenimiento CSO fue 31% y 24%, respectivamente. Se requirió que los pacientes tuvieran un historial de dos o más exacerbaciones de asma grave que requirieran tratamiento corticosteroide oral o sistémico en los últimos 12 meses y función pulmonar reducida en línea base (FEV1 < 80% pre-broncodilatador) en adultos y <90% en adolescentes. La media de número de exacerbaciones en el año previo fue 3.6 y la media de FEV1 predicho pre-broncodilatador fue 60%. Los pacientes continuaron recibiendo sus medicinas de asma existentes durante los estudios.
Para el estudio de reducción de corticosteroides orales MEA115575, un total de 135 pacientes fueron inscritos (55% fueron del sexo femenino con una media de edad de 50 años), quienes estaban siendo tratados diariamente con CSO (5-35 mg por día), y dosis altas de CSI, más un medicamento de mantenimiento adicional.
Estudio de eficacia de rango de dosis MEA112997 (DREAM):
En MEA112997, un estudio aleatorizado, con doble ciego, controlado con placebo, de grupo paralelo multicéntrico de 52 semanas de duración en 616 pacientes con asma eosinofílico refractario grave, NUCALA redujo significativamente las exacerbaciones del asma clínicamente significativas (definidas como empeoramiento del asma que requirió el uso de corticosteroides orales/sistémicos y/o hospitalización y/o visitas al departamento de emergencia) cuando se administró en dosis de 75 mg, 250 mg o 750 mg intravenosamente en comparación con placebo (ver Tabla 1).
Tabla 1: Frecuencia de exacerbaciones clínicamente significativas en la semana 52 en la población con intención de tratar
|
Mepolizumab intravenoso |
Placebo |
|||
|
75 mg n=153 |
250 n=152 |
750 mg n=156 |
n=155 |
|
|
Tasa de exacerbación/año |
1.24 |
1.46 |
1.15 |
2.40 |
|
Reducción por ciento |
48% |
39% |
52% |
|
|
Relación de tasa (IC del 95%) |
0.52 (0.39, 0.69) |
0.6 (0.46, 0.81) |
0.48 (0.36, 0.64) |
|
|
Valor-p |
< 0.001 |
< 0.001 |
< 0.001 |
- |
Estudio de reducción de exacerbación (MEA115588):
MEA115588 fue un estudio aleatorizado, con doble ciego, controlado con placebo, de grupo paralelo, multi-céntrico que evaluó la eficacia y seguridad de NUCALA como terapia agregada en 576 pacientes con asma eosinofílico refractario grave, definida como un recuento de eosinófilos sanguíneos periféricos mayores que o iguales a 150 células/μL en iniciación de tratamiento o mayores que o iguales a 300 células/μL dentro de los 12 meses anteriores.
Los pacientes recibieron 100 mg de mepolizumab administrado subcutáneamente, 75 mg de mepolizumab administrado intravenosamente o tratamiento de placebo una vez cada 4 semanas durante 32 semanas. El criterio de valoración primario fue la frecuencia de exacerbaciones clínicamente significativas de asma y las reducciones para ambos ramos de tratamiento de mepolizumab comparados con placebo fueron estadísticamente significativas (p < 0.001). La tabla 2 proporciona los resultados de los criterios de valoración primarios y secundarios de MEA115588.
Tabla 2: Resultados de los criterios de valoración primarios y secundarios en la semana 32 en la población con intención de tratamiento (MEA115588)
|
NUCALA 100 mg (subcutáneo) N=194 |
Placebo N=191 |
|
|
Criterio de valoración primario |
||
|
Frecuencia de exacerbaciones clínicamente significativas |
||
|
Tasa de exacerbación por año |
0.83 |
1.74 |
|
Reducción porcentual Relación de tasa (IC del 95%) |
53% 0.47 (0.35, 0.64) |
- |
|
Valor-p |
< 0.001 |
|
|
Criterios de valoración secundarios |
||
|
Frecuencia de exacerbaciones que requirieron hospitalizaciones/visitas a sala de emergencias |
||
|
Tasa de exacerbación por año |
0.08 |
0.20 |
|
Reducción porcentual Relación de tasa (IC del 95%) |
61% 0.39 (0.18, 0.83) |
- |
|
Valor-p |
0.015 |
|
|
Frecuencia de exacerbaciones que requirieron hospitalización |
||
|
Tasa de exacerbaciones por año |
0.03 |
0.10 |
|
Reducción porcentual Relación de tasa (IC el 95%) |
69% 0.31 (0.11, 0.91) |
- |
|
Valor-p |
0.034 |
|
|
FEV1 Pre-broncodilatador (mL) en semana 32 |
||
|
Media de cambo de Línea base (EE) |
183 (31) |
86 (31.4) |
|
Diferencia (mepolizumab frente a placebo) |
98 |
|
|
IC del 95% |
11, 184 |
|
|
Valor-p |
0.028 |
|
|
Cuestionario Respiratorio de St. George (CRSG) en semana 32 |
||
|
Media de cambo de Línea base (EE) |
-16.0 (1.1) |
-9.0 (1.16) |
|
Diferencia (mepolizumab frente a placebo) |
-7.0 |
|
|
IC del 95% |
(-10.2, -3.8) |
|
|
Valor-p |
<0.001 |
|
Reducción de tasa de exacerbación mediante conteo de eosinófilos sanguíneos:
La Tabla 3 muestra los resultados de un análisis combinado de los dos estudios de exacerbaciones (MEA112997 y MEA115588) mediante conteo de eosinófilos sanguíneos en línea base. La tasa de exacerbaciones en el ramo de placebo aumentó con conteo de eosinófilos sanguíneos en línea base en incremento. La tasa de reducción con mepolizumab fue mayor en pacientes con conteos eosinófilos sanguíneos más altos.
Tabla 3: Análisis combinado de la tasa de exacerbaciones clínicamente significativas mediante conteo de eosinófilos sanguíneos en línea base en pacientes con asma eosinofílico refractario grave
|
Mepolizumab 75 mg IV/ 100 mg SC N=538 |
Placebo N=346 |
|
|
MEA112997+MEA115588 |
||
|
< 150 células/μL |
||
|
N |
123 |
66 |
|
Tasa de exacerbación por año |
1.16 |
1.73 |
|
Mepolizumab frente a placebo |
||
|
Relación de tasa (IC del 95%) |
0.67 (0.46, 0.98) |
--- |
|
150 a < 300 células/μL |
||
|
N |
139 |
86 |
|
Tasa de exacerbación por año |
1.01 |
1.41 |
|
Mepolizumab frente a placebo |
||
|
Relación de tasa (IC del 95%) |
0.72 (0.47, 1.10) |
--- |
|
300 a < 500 células/μL |
||
|
N |
109 |
76 |
|
Tasa de exacerbación por año |
1.02 |
1.64 |
|
Mepolizumab frente a placebo |
||
|
Relación de tasa (IC del 95%) |
0.62 (0.41, 0.93) |
--- |
|
≥ 500 células/μL |
||
|
N |
162 |
116 |
|
Tasa de exacerbación por año |
0.67 |
2.49 |
|
Mepolizumab frente a placebo |
||
|
Relación de tasa (IC del 95%) |
0.27 (0.19, 0.37) |
--- |
Estudio de reducción de corticosteroides orales (MEA115575):
MEA115575 evaluó el efecto de NUCALA 100 mg administrado subcutáneamente sobre la reducción de requerimiento para corticosteroides orales (CSO) de mantenimiento mientras se mantenía control de asma en pacientes con asma eosinofílico refractario grave. Los pacientes tuvieron un conteo de eosinófilos sanguíneos de ≥ 150/μL en línea base o un conteo de eosinófilos sanguíneos de ≥ 300/μL en los 12 meses previos a exploración. Se administró tratamiento con mepolizumab o placebo a los pacientes una vez cada 4 semanas a lo largo del período de tratamiento. Los pacientes continuaron recibiendo sus medicamentos para asma existentes durante el estudio con la excepción de sus dosis de CSO que fueron reducidas cada 4 semanas durante la fase de reducción de CSO (Semanas 4-20), siempre que el control de asma fuera mantenido.
Un total de 135 pacientes fueron reclutados: media de edad fue 50 años, 55% fueron del sexo femenino y 48% habían recibido terapia de esteroides orales por al menos 5 años. La media en línea base de dosis equivalente a prednisona fue aproximadamente 13 mg por día.
El criterio de valoración primario fue el porcentaje de reducción de dosis CSO diaria (semanas 20-24), mientras se mantenía control de asma en comparación con pacientes tratados con placebo (consultar tabla 4).
Tabla 4: Resultados de los criterios de valoración primario y secundarios en la población de interacción de tratamiento MEA115575
|
Población IDT |
||
|
Mepolizumab 100 mg (subcutáneo) N=69 |
Placebo N=66 |
|
|
Criterio de valoración primario |
||
|
Reducción porcentual en CSO desde la línea base en las semanas 20-24 (%) |
||
|
90% - 100% 75% - < 90% 50% - < 75% < 0% - < 50% Sin disminución en CSO/falta de control de asma/retiro del tratamiento |
16 (23%) 12 (17%) 9 (13%) 7 (10%) 25 (36%) |
7 (11%) 5 (8%) 10 (15%) 7 (11%) 37 (56%) |
|
Razón de momios (IC del 95%) |
2.39 (1.25, 4.56) |
|
|
Valor-p |
0.008 |
|
|
Criterios de valoración secundarios |
||
|
Reducción en la dosis diaria de CSO (%) |
||
|
Reducción de al menos 50% |
37 (54%) |
22 (33%) |
|
Relación de probabilidad (CI del 95%) |
2.26 (1.10, 4.65) |
|
|
Valor-p |
0.027 |
|
|
Reducción en la dosis diaria de CSO (%) |
||
|
A ≤ 5 mg/día |
37 (54%) |
21 (32%) |
|
Relación de probabilidad (CI del 95%) |
2.45 (1.12, 5.37) |
|
|
Valor-p |
0.025 |
|
|
Reducción en la dosis de CSO diaria |
||
|
A 0 mg/día |
10 (14%) |
5 (8%) |
|
Relación de probabilidad (CI del 95%) |
1.67 (0.49, 5.75) |
|
|
Valor-p |
0.414 |
|
|
Mediana de la reducción porcentual en la dosis diaria de CSO |
||
|
Mediana % de reducción en desde línea base (IC del 95%) |
50.0 (20.0, 75.0) |
0.0 (-20.0, 33.3) |
|
Mediana de diferencia (IC del 95%) |
-30.0 (-66.7, 0.0) |
|
|
Valor-p |
0.007 |
|
Además, se midió la calidad de vida relacionada con la salud usando SGQR. A la semana 24 hubo una mejoría estadísticamente significativa en la media de la calificación SGRQ para NUCALA en comparación con placebo: -5.8 (CI de 95%: -10.6,-1.0; P=0.019). A la Semana 24, la proporción de los sujetos con una disminución clínicamente significativa en la calificación SGRQ (definida como una disminución de al menos 4 unidades del inicio) fue mayor para NUCALA (58%, 40/69) en comparación con placebo (41%, 27/66).
El perfil de eficacia a largo plazo de NUCALA en pacientes con asma grave (n=998) tratados durante una mediana de 2.8 años (intervalo de 4 semanas a 4.5 años) en los estudios de extensión abiertos MEA115666, MEA115661 y 201312 fue consistente en general con los 3 estudios controlados con placebo.
Población pediátrica:
Hubo 25 adolescentes 13 niñas y 12 niños, 9 con edad de 12-14 años y 16 con edad de 15- 17 años inscritos en el estudio MEA115588. De los 25 pacientes totales: 9 recibieron placebo; 9 recibieron mepolizumab 75 mg intravenosamente, y 7 recibieron 100 mg subcutáneamente. La misma proporción de pacientes (3/9) que recibieron placebo y mepolizumab intravenosamente reportaron exacerbaciones clínicamente significativas; no se reportaron exacerbaciones en aquellos que recibieron mepolizumab subcutáneamente.
Rinosinusitis crónica con pólipos nasales (RSCcPN):
El estudio 205687 fue un estudio aleatorizado, doble ciego, controlado con placebo, de 52 semanas que evaluó a 407 pacientes de 18 años y mayores con RSCcPN.
Los pacientes inscritos en el estudio debían contar con una puntuación de EVA (escala visual análoga) de síntomas de obstrucción nasal >5 de una puntuación máxima de 10, una puntuación general de EVA de síntomas >7 de una puntuación máxima de 10 y una puntuación endoscópica de pólipos nasales (PN) bilaterales ≥ 5 de una puntuación máxima de 8 (con una puntuación mínima de 2 en cada cavidad nasal). Los pacientes también debían tener antecedentes de al menos una cirugía previa por pólipos nasales en los últimos 10 años.
Los pacientes recibieron una dosis de 100 mg de NUCALA, o placebo, administrada por vía subcutánea una vez cada 4 semanas, además de la terapia de base con corticosteroides intranasales.
Las características demográficas y basales de los pacientes del estudio 205687 se proporcionan en la Tabla 5 a continuación:
Tabla 5. Características demográficas y basales en RSCcPN
|
N = 407 |
|
|
Edad (años) de los pacientes, media (SD) |
49 (13) |
|
Mujer, n (%) |
143 (35) |
|
Raza blanca, n (%) |
379 (93) |
|
Duración (años) de RSCcPN, media (SD) |
11.4 (8.39) |
|
Pacientes con ≥ 1 cirugía previa, n (%) |
407 (100) |
|
Pacientes con ≥ 3 cirugías previas, n (%) |
124 (30) |
|
Uso de OCS para PN (≥ 1 ciclo) en los últimos 12 meses, n (%) |
197 (48) |
|
Puntuación endoscópica de PN totalb c, media (SD), puntuación máxima = 8 |
5.5 (1.29) |
|
Puntuación de EVA de obstrucción nasala d, media (SD), puntuación máxima = 10 |
9.0 (0.83) |
|
Puntuación general de EVA de síntomasa d, media (SD), puntuación máxima = 10 |
9.1 (0.74) |
|
Puntuación total SNOT-22e, media (SD), rango 0-110 |
64.1 (18.32) |
|
Puntuación de EVA de pérdida del olfatoa,d, media (SD), puntuación máxima = 10 |
9.7 (0.72) |
|
Asma, n (%) |
289 (71) |
|
EREA, n (%) |
108 (27) |
|
Recuento medio geométrico de eosinófilos en la basal, células/mcL (CI de 95%) |
390 (360, 420) |
RSCcPN = rinosinusitis crónica con pólipos nasales, SD = desviación estándar, OCS = corticosteroide oral, PN = pólipos nasales, EVA = escala visual análoga, SNOT-22 = Prueba de desenlace sinonasal, EREA = enfermedad respiratoria exacerbada por la aspirina.
a Las puntuaciones más altas indican una mayor gravedad de la enfermedad.
b Calificada por evaluadores cegados independientes.
c La puntuación endoscópica de PN es la suma de las puntuaciones de ambas fosas nasales (escala de 0-8), en la que cada fosa nasal se calificó (0 = sin pólipos; 1 = pólipos pequeños en el meato medio que no llegan por debajo del borde inferior del cornete medio; 2 = pólipos que llegan por debajo del borde inferior del cornete medio; 3 = pólipos grandes que llegan al borde inferior del cornete inferior o pólipos mediales a cornete medio; 4 = pólipos grandes que causan congestión/obstrucción casi completa del meato inferior).
d Recopilado diariamente por los pacientes en una escala de 0 a 10 (0 = ninguno; 10 = tan malo como usted pueda imaginar).
e SNOT-22 es una herramienta de evaluación de la calidad de vida relacionada con la salud e incluyó 22 elementos en 6 dominios de síntomas e impacto asociado con RSCcPN (nasal, no nasal, oído/facial, sueño, fatiga, consecuencias emocionales). Las puntuaciones más altas indican una peor calidad de vida relacionada con la salud.
Los criterios de valoración coprimarios fueron el cambio desde la basal en la puntuación total de PN endoscópica en la semana 52 y el cambio desde la basal en la puntuación media de EVA de obstrucción nasal durante las semanas 49-52.
Los pacientes que recibieron NUCALA tuvieron mejorías (disminuciones) significativamente mayores en la puntuación endoscópica de PN total en la Semana 52 y en la puntuación de EVA de obstrucción nasal durante las semanas 49-52 en comparación con el placebo (véase la Tabla 6).
Tabla 6. Análisis de los criterios de valoración coprimarios (población con intención de tratar)
|
Placebo (N=201) |
NUCALA 100 mg SC (N=206) |
|
|
Puntuación total endoscópica en la semana 52a |
||
|
Mediana de la puntuación en la basal (mín, máx) |
6.0 (0, 8) |
5.0 (2, 8) |
|
Mediana del cambio desde la basal |
0.0 |
-1.0 |
|
Valor pb |
<0.001 |
|
|
Diferencia ajustada del tratamiento en medianas (CI de 95%)c |
-0.73 (-1.11, -0.34) |
|
|
Mejoría ≥ 1 punto, n (%) |
57 (28) |
104 (50) |
|
Mejoría ≥ 2 puntos, n (%) |
26 (13) |
74 (36) |
|
Puntuación de EVA de obstrucción nasal (semanas 49 a 52)a |
||
|
Mediana de la puntuación en la basal (mín, máx) |
9.14 (5.31, 10.00) |
9.01 (6.54, 10.00) |
|
Mediana del cambio desde la basal |
-0.82 |
-4.41 |
|
Valor pb |
<0.001 |
|
|
Diferencia ajustada del tratamiento en medianas (CI de 95%)c |
-3.14 (-4.09, -2.18) |
|
|
Mejoría >1 punto, n (%) |
100 (50) |
146 (71) |
|
Mejoría ≥3 puntos, n (%)d |
73 (36) |
124 (60) |
a) A los sujetos con cirugía nasal/sinuplastia antes de la visita se les asignó su peor puntuación observada antes de la cirugía nasal/sinuplastia. A aquellos que se retiraron del estudio sin cirugía nasal/sinuplastia se les asignó su peor puntuación observada antes del retiro del estudio.
b) Con base en la prueba de suma de rangos de Wilcoxon.
c) Regresión cuantílica con covariables de grupo de tratamiento, región geográfica, puntuación basal y recuento basal de eosinófilos en sangre log(e).
d) Se identificó una mejoría de tres puntos en la EVA de obstrucción nasal como un cambio significativo intrapaciente en esta evaluación.
Todos los criterios de valoración secundarios fueron estadísticamente significativos y respaldan los criterios de valoración coprimarios. El criterio de valoración secundario clave fue el tiempo hasta la primera cirugía de PN hasta la Semana 52. Los datos de los otros criterios de valoración secundarios se presentan en la Tabla 7.
Tiempo hasta la primera cirugía de PN:
Durante el periodo de tratamiento de 52 semanas, los pacientes del grupo de NUCALA tuvieron una menor probabilidad de someterse a una cirugía de PN que los pacientes del grupo de placebo (la cirugía se definió como cualquier procedimiento que involucrara instrumentos y que diera lugar a una incisión y extirpación de tejido [polipectomía] en la cavidad nasal).
En la Semana 52, 18 pacientes (9%) del grupo de NUCALA se habían sometido a cirugía de PN en comparación con 46 pacientes (23%) del grupo de placebo.
Los pacientes que recibieron NUCALA tuvieron un aumento en el tiempo hasta la primera cirugía de PN en comparación con el placebo. El riesgo de cirugía durante el período de tratamiento fue significativamente menor en un 57% en los pacientes tratados con NUCALA en comparación con el placebo (cociente de riesgo: 0.43; CI de 95% 0.25; 0.76; p = 0,003 no ajustado/ajustado), un análisis post-hoc mostró una reducción del 61% en las probabilidades de cirugía (OR: 0.39; CI de 95%: 0.21; 0.72; p= 0.003.
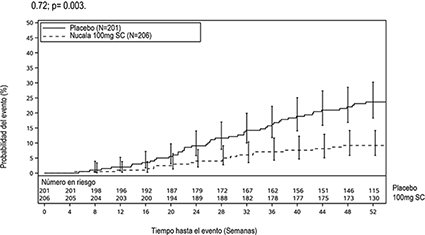
Figura 1: Curva de Kaplan Meier en el tiempo hasta la primera cirugía de pólipos nasales
Tabla 7. Resultados de otros criterios de valoración secundarios en la población con intención de tratar
|
Placebo (N=201) |
NUCALA (N=206) |
|
|
Puntuación general de EVA (Semanas 49-52) a |
||
|
Mediana de la puntuación en la basal (mín, máx) |
9.20 (7.21, 10.00) |
9.12 (7.17, 10.00) |
|
Mediana del cambio desde la basal |
-0.90 |
-4.48 |
|
Valor p no ajustado/ajustadob,c |
<0.001/0.003 |
|
|
Diferencia ajustada del tratamiento en medianas (CI de 95%)d |
-3.18 (-4.10, -2.26) |
|
|
Mejoría ≥ 2.5 puntos (%) |
40 |
64 |
|
Puntuación total SNOT-22 en la Semana 52a, g |
||
|
n |
198 |
205 |
|
Mediana de la puntuación en la basal (mín, máx) |
64.0 (19, 110) |
64.0 (17, 105) |
|
Mediana del cambio desde la basal |
-14.0 |
-30.0 |
|
Valor p no ajustado/ajustadob,c |
< 0.001/0.003 |
|
|
Diferencia ajustada del tratamiento en medianas (CI de 95%) d |
-16.49 (-23.57, -9.42) |
|
|
Mejoría ≥ 28 puntos (%)g |
32 |
54 |
|
Pacientes que requirieron esteroides sistémicos para pólipos nasales hasta la Semana 52 |
||
|
Número de pacientes con ≥1 ciclo |
74 (37) |
52 (25) |
|
Cociente de riesgo respecto al placebo (CI de 95%)e |
0.58 (0.36, 0.92) |
|
|
Valor p no ajustado/ajustado c, e |
0.020/0.020 |
|
|
Puntuación compuesta de EVA – Síntomas nasales (Semanas 49-52)a,f |
||
|
Mediana de puntuación en la basal (mín, máx) |
9.18 (6.03, 10.00) |
9.11 (4.91, 10.00) |
|
Mediana del cambio desde la basal |
-0.89 |
-3.96 |
|
Valor p no ajustado/ajustadob,c |
<0.001/0.020 |
|
|
Diferencia ajustada del tratamiento en medianas (CI de 95%)d |
-2.68 (-3.44, -1.91) |
|
|
Mejoría ≥ 2 puntos (%)h |
40 |
66 |
|
Puntuación de EVA de pérdida del olfato (Semanas 49-52)a |
||
|
Mediana de la puntuación en la basal (mín, máx) |
9.97 (6.69, 10.00) |
9.97 (0.94, 10.00) |
|
Mediana del cambio desde la basal |
0.00 |
-0.53 |
|
Valor p no ajustado/ajustadob,c |
<0.001/0.020 |
|
|
Diferencia ajustada del tratamiento en medianas (CI de 95%)d |
-0.37 (-0.65, -0.08) |
|
|
Mejoría ≥ 3 puntos (%)h |
19 |
36 |
a A los pacientes con cirugía nasal/sinuplastia antes de la visita se les asignó su peor puntuación observada antes de la cirugía nasal/sinuplastia. A aquéllos que se retiraron del estudio sin cirugía nasal/sinuplastia se les asignó su peor puntuación observada antes del retiro del estudio.
b Con base en la prueba de suma de rangos de Wilcoxon.
c Multiplicidad controlada mediante pruebas de criterios de valoración secundarios de siguiendo una jerarquía predefinida.
d Regresión cuantílica con covariables del grupo de tratamiento, región geográfica, puntuación basal y recuento basal de eosinófilos en sangre log(e).
e Análisis con un modelo de regresión logística con covariables del grupo de tratamiento, región geográfica, número de ciclos de OCS para PN en los últimos 12 meses (0, 1, >1 como ordinal), puntuación ENP total basal (lectura central), puntuación de EVA de obstrucción nasal basal y recuento basal de eosinófilos en sangre log(e).
f Puntuación compuesta de EVA de obstrucción nasal, secreción nasal, moco en garganta y pérdida del olfato.
g Se observó una mejoría en los 6 dominios de síntomas e impacto asociado con RSCcPN.
h Se identificó el umbral de mejoría de cada criterio de valoración como un cambio significativo intrapaciente en esta evaluación.
Criterios de valoración en pacientes con asma:
En 289 (71%) pacientes con asma comórbida, los análisis especificados previamente mostraron mejorías en los criterios de valoración coprimarios de manera consistente con aquellos observados en la población general en los pacientes que recibieron NUCALA 100 mg en comparación con el placebo. Asimismo, en estos pacientes, se observó una mayor mejoría respecto desde la basal en la Semana 52 en el control del asma evaluado con el Cuestionario de Control del Asma (ACQ-5) para NUCALA 100 mg en comparación con el placebo (mediana del cambio [Q1, Q3] de -0.80 [-2.20, 0.00] y 0.00 [- 1.10, 0.20], respectivamente).
Granulomatosis eosinofílica con poliangeítis (EGPA):
MEA115921 fue un estudio aleatorizado, doble ciego, controlado con placebo, de 52 semanas, en el que se evaluó a 136 pacientes de ≥ 18 años de edad con EGPA recidivante o refractaria, y que estaban bajo tratamiento estable con corticosteroides por vía oral (OCS; prednisolona/prednisona de ≥ 7.5 a ≤ 50 mg /día). Cincuenta y tres por ciento (n=72) también estaban bajo terapia inmunosupresora concomitante estable.
Los pacientes recibieron una dosis de 300 mg de NUCALA administrada por vía subcutánea una vez cada 4 semanas, además de su tratamiento previo con prednisolona/prednisona con o sin terapia inmunosupresora. El investigador ajustó la dosis de OCS a su discreción.
Los criterios de valoración coprimarios fueron la duración acumulada total de remisión, definida de acuerdo con la Valoración de la actividad en vasculitis de Birmingham (BVAS)=0 (vasculitis no activa) además de la dosis de ≤ 4 mg/día de prednisolona/prednisona, y la proporción de sujetos en remisión a las 36 y 48 semanas de tratamiento.
Remisión:
En comparación con placebo, los sujetos que recibían 300 mg de NUCALA lograron un tiempo acumulado en remisión significativamente mayor. Adicionalmente, en comparación con placebo, una proporción significativamente mayor de sujetos que recibían 300 mg de NUCALA lograron remisión a la Semana 36 y a la Semana 48. Tabla 8.
Table 8. Análisis de los criterios de valoración coprimarios (población con intención de tratar)
|
Número (%) de sujetos |
||
|
Placebo N=68 |
NUCALA 300 mg N=68 |
|
|
Duración acumulada de la remisión durante 52 semanas |
||
|
0 semanas |
55 (81) |
32 (47) |
|
> 0 a < 12 semanas |
8 (12) |
8 (12) |
|
12 a < 24 semanas |
3 (4) |
9 (13) |
|
24 a < 36 semanas |
0 |
10 (15) |
|
≥ 36 semanas |
2 (3) |
9 (13) |
|
Relación de probabilidades (NUCALA/placebo) |
5.91 |
|
|
95% CI |
--- |
2.68, 13.03 |
|
Valor de p |
--- |
< 0.001 |
|
Sujetos en remisión en las semanas 36 y 48 |
2 (3) |
22 (32) |
|
Relación de probabilidades (NUCALA/placebo) |
16.74 |
|
|
CI de 95% |
--- |
3.61, 77.56 |
|
Valor de p |
--- |
< 0.001 |
Una relación de probabilidades > 1 favorece a NUCALA
Los sujetos que recibían 300 mg de NUCALA lograron un tiempo acumulado en remisión significativamente mayor (p < 0.001), mientras que una proporción mayor de sujetos que recibían 300 mg de NUCALA estuvo en remisión tanto a la Semana 36 como a la Semana 48 (p < 0.001), en comparación con placebo, al utilizar la definición de remisión del criterio de valoración secundario de BVAS = 0 más prednisolona/prednisona ≤ 7.5 mg/día.
Recaída:
En comparación con placebo, el tiempo hasta la primera recaída (definido como deterioro relacionado con vasculitis, asma o síntomas sinunasales que requieren un incremento de la dosis de corticosteroides o terapia inmunosupresora, o bien, hospitalización), fue significativamente más largo para los sujetos que recibían 300 mg de NUCALA (p < 0.001). Asimismo, los sujetos que recibían NUCALA presentaron una reducción del 50% en el índice de recaída anual, en comparación con placebo 1.14 frente a 2.27, respectivamente.
Reducción de corticosteroides orales:
En comparación con placebo, los sujetos que recibían 300 mg de NUCALA tenían una dosis diaria promedio de corticosteroides orales menor durante las Semanas 48 a 52 (p < 0.001). En el grupo de 300 mg de NUCALA, 12 sujetos (18%) pudieron dejar gradualmente el tratamiento con OCS, en comparación con los 2 sujetos (3%) en el grupo de placebo.
Síndrome hipereosinofílico (HES):
El estudio 200622 fue un estudio aleatorio, doble ciego, controlado con placebo, de 32 semanas, que evaluó a 108 sujetos de ≥ 12 años de edad con HES. Los sujetos recibieron 300 mg de NUCALA o placebo administrados subcutáneamente una vez cada 4 semanas mientras continuaban con terapia estable para HES. De los 4 adolescentes reclutados, uno recibió 300 mg de NUCALA y 3 recibieron placebo por 32 semanas. La terapia estándar por HES podía incluir OCS y terapia inmunosupresora o citotóxica. Los sujetos que entraron en el estudio habían experimentado por lo menos dos exacerbaciones de HES en los últimos 12 meses y tenían un conteo de eosinófilos sanguíneos ≥ 1000 células/μL durante la revisión.
El criterio primario de valoración del estudio 200622 fue la proporción de sujetos que experimentaron una exacerbación de HES durante el periodo de tratamiento de 32 semanas. Una exacerbación de HES se definió como el empeoramiento de los síntomas y signos clínicos de HES o el incremento de los eosinófilos (en ≥ 2 ocasiones), dando como resultado la necesidad de aumentar el OCS o incrementar/agregar una terapia citotóxica o inmunosupresora por HES.
El análisis primario comparó sujetos que experimentaron una exacerbación de HES o se retiraron del estudio en los grupos de tratamiento con NUCALA o placebo. Durante el periodo de tratamiento de 32 semanas, 50% menos pacientes experimentaron una exacerbación de HES o se retiraron del estudio cuando fueron tratados con 300 mg de NUCALA en comparación con placebo; 28% versus 56%, respectivamente (o 0.28, CI del 95%: 0.12, 0.64) (ver Tabla 9)
Los criterios secundarios de valoración fueron el tiempo para la primera exacerbación de HES, la proporción de sujetos que experimentaron una exacerbación de HES durante la Semana 20 a la Semana 32, la tasa de exacerbaciones de HES y el cambio desde la basal en la severidad de la fatiga.
Todos los criterios secundarios de valoración fueron estadísticamente significativos y respaldaron el criterio primario de valoración (ver Tabla 9)
Table 9. Resultados del criterio primario de valoración/análisis en la población con Intención de Tratar (Estudio 200622)
|
NUCALA N = 54 |
Placebo N = 54 |
|
|
Proporción de sujetos que experimentaron una exacerbación de HES |
||
|
Sujetos con ≥ 1 exacerbación de HES o que se retiraron del estudio (%) |
15 (28) |
30 (56) |
|
Sujetos con ≥ 1 exacerbación de HES (%) |
14 (26) |
28 (52) |
|
Sujetos sin exacerbaciones de HES que se retiraron (%) |
1 (2) |
2 (4) |
|
Relación de probabilidades (CI del 95%) |
0.28 (0.12, 0.64) |
|
|
Valor de p de CMH |
0.002 |
|
CMH = Cochran-Mantel-Haenszel.
Tiempo para la primera exacerbación:
Los sujetos que recibieron 300 mg de NUCALA presentaron un incremento significativo en el tiempo para la primera exacerbación de HES en comparación con placebo. El riesgo de la primera exacerbación de HES durante el periodo de tratamiento fue 66% más bajo para los sujetos tratados con NUCALA, en comparación con placebo (Proporción de riesgos: 0.34; CI del 95% 0.18, 0.67; p = 0.002).
Figura 2: Curva de Kaplan Meier para el tiempo para la primera exacerbación de HES
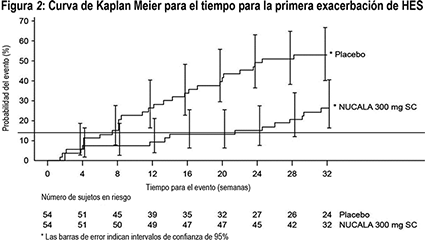
Tabla 10. Resultados de otros criterios secundarios de valoración en la población con intención de tratar (Estudio 200622).
|
NUCALA N = 54 |
Placebo N = 54 |
|
|
Exacerbaciones de HES durante la semana 20 y hasta la semana 32 inclusive |
||
|
Sujetos con ≥ 1 exacerbación de HES o que se retiraron del estudio (%) |
9 (17) |
19 (35) |
|
Relación de probabilidades (CI de 95%) |
0.33 (0.13, 0.85) |
|
|
Valor de p de CMH (sin ajustar/ajustado)a |
0.02/0.02 |
|
|
Tasa de exacerbaciones de HES |
||
|
Tasa media estimada/año |
0.50 |
1.46 |
|
Proporción de tasas (CI de 95%) |
0.34 (0.19, 0.63) |
|
|
Valor p de Wilcoxon (sin ajustar/ajustado)a |
0.002/0.02 |
|
|
Cambio desde la basal en la severidad de la fatiga con base en el Artículo 3 del Inventario de Fatiga Breve (BFI) (peor nivel de fatiga durante las últimas 24 horas) en la semana 32b |
||
|
Cambio mediano en el Artículo 3 del BFI |
-0.66 |
0.32 |
|
Valor de p en la comparación (NUCALA versus placebo) (sin ajustar/ajustado)a |
0.036/0.036 |
|
a Valores de p ajustados con base en la jerarquía previamente especificada de los criterios de valoración.
b Pacientes con datos faltantes incluidos con el peor valor observado.
CMH = Cochran-Mantel-Haenszel.
Extensión abierta de HES:
Los pacientes elegibles, incluidos 4 adolescentes que completaron el estudio 200622, continuaron en el estudio de extensión abierto 205203 de 20 semanas para investigar el perfil de seguridad a largo plazo y proporcionar datos adicionales sobre el beneficio clínico de NUCALA en pacientes con HES más allá de las 32 semanas.
Se mantuvo el efecto del tratamiento con NUCALA en la reducción de las exacerbaciones de HES observados durante el Estudio 200622 en los sujetos que continuaron el tratamiento con NUCALA en el estudio 205203, en el que el 94% (47/50) de los pacientes no experimentó una exacerbación. Durante la Semana 16 a 20, el 28% de todos los sujetos con una dosis media de OCS >0 mg/día (prednisona o equivalente) de la Semana 0 a 4 lograron una reducción de la dosis diaria media de OCS ≥ 50%.
CONTRAINDICACIONES:
NUCALA está contraindicado en pacientes con antecedentes de hipersensibilidad a mepolizumab o a cualquiera de los excipientes de la fórmula, y en menores de 12 años.
RESTRICCIONES DE USO DURANTE EL EMBARAZO Y LA LACTANCIA:
Embarazo:
Se desconoce el efecto de NUCALA sobre el embarazo humano. No se han observado efectos relacionados con el tratamiento sobre el desarrollo embrio-fetal o postnatal en estudios de animales.
NUCALA debe usarse en el embarazo sólo si el beneficio esperado para la madre justifica el riesgo potencial al feto.
Lactancia:
No se dispone de información sobre la presencia de mepolizumab en la leche materna. Sin embargo, mepolizumab se excretó en la leche de monos cynomolgus a concentraciones inferiores a 0.5% de lo detectado en plasma.
Debe tomarse una decisión sobre si descontinuar la lactancia o descontinuar NUCALA, considerando la importancia de la leche materna para el lactante y la importancia del medicamento para la madre.
REACCIONES SECUNDARIAS Y ADVERSAS:
Asma grave:
La seguridad de NUCALA se estudió en un programa de desarrollo clínico en adolescentes y adultos con asma eosinofílica grave, que incluyó tres estudios con asignación al azar, controlados con placebo, multicéntricos (n=1327). Los sujetos recibieron ya sea mepolizumab o placebo por vía subcutánea (SC) o intravenosa (IV) durante los estudios clínicos de 24 a 52 semanas de duración. Las reacciones adversas relacionadas con NUCALA 100 mg administrado por vía subcutánea (n=263) se presentan en la tabla que aparece más adelante. El perfil de seguridad de NUCALA en pacientes con asma grave (n=998) tratados durante una mediana de 2.8 años (intervalo de 4 semanas a 4.5 años) en estudios de extensión abiertos fue similar al observado en los estudios controlados con placebo.
La frecuencia de reacciones adversas se define usando la siguiente convención: muy común (≥1/10); común (≥1/100 a <1/10), no común (≥1/1,000 a <1/100) y rara (≥1/10,000 a <1/1,000).
|
Clase de sistema órgano |
Reacciones adversas |
Frecuencia |
|
Infecciones e infestaciones |
Faringitis Infección de vías respiratorias inferiores Infección de vías urinarias |
Común Común Común |
|
Trastornos del sistema nervioso |
Cefalea |
Muy Común |
|
Trastornos respiratorios, torácicos y mediastínicos |
Congestión nasal |
Común |
|
Trastornos gastrointestinales |
Dolor abdominal superior |
Común |
|
Trastornos cutáneos y de tejido subcutáneo |
Eccema |
Común |
|
Trastornos musculoesqueléticos y de tejido conjuntivo |
Lumbalgia |
Común |
|
Trastornos generales y alteraciones del sitio de administración |
Pirexia Reacciones en el sitio de inyección* |
Común Común |
* Los síntomas más frecuentes relacionados con inyecciones subcutáneas incluyen: dolor, eritema, hinchazón, prurito y sensación quemante.
RSCcPN:
En un estudio aleatorio, doble ciego, controlado con placebo de 52 semanas en sujetos con RSCcPN (NUCALA 100 mg n = 206, placebo n = 201), no se identificaron reacciones adversas adicionales a las reportadas para los estudios de asma grave.
EGPA:
En un estudio doble ciego controlado con placebo en sujetos con EGPA (NUCALA 300 mg n = 68, placebo n = 68) no se identificaron reacciones adversas adicionales a las reportadas para los estudios de asma grave.
HES:
En un estudio aleatorio, doble ciego, controlado con placebo de 32 semanas en sujetos con HES (300 mg de NUCALA n = 54, placebo n = 54), no se identificaron reacciones adversas adicionales a las reportadas para los estudios de asma grave. El perfil de seguridad de NUCALA en pacientes con HES (n = 102) registrados en un estudio de extensión abierto de 20 semanas fue similar al perfil de seguridad de los pacientes del estudio pivotal controlado con placebo.
Datos Post-comercialización:
|
Clase de sistema órgano |
Reacciones adversas |
Frecuencia |
|
Trastornos del sistema inmunológico |
Reacciones de hipersensibilidad incluyendo anafilaxia |
Rara |
PRECAUCIONES EN RELACIÓN CON EFECTOS DE CARCINOGÉNESIS, MUTAGÉNESIS, TERATOGÉNESIS Y SOBRE LA FERTILIDAD:
Dado que mepolizumab es un anticuerpo monoclonal, no se han realizado estudios de genotoxicidad o carcinogenicidad. No se han realizado estudios animales a largo plazo para evaluar el potencial carcinogénico de mepolizumab. La literatura publicada usando modelos animales sugiere que la IL - 5 y eosinófilos son parte de una reacción inflamatoria temprana en el sitio de génesis tumoral y pueden promover el rechazo del tumor. Sin embargo, otros reportes indican que la infiltración de eosinófilos en los tumores puede promover el crecimiento del tumor. Por lo tanto, el riesgo de neoplasia en humanos derivado de un anticuerpo para IL - 5 tal como mepolizumab se desconoce.
Toxicología reproductiva:
Fertilidad:
La fertilidad en hombres y mujeres no fue afectada ya que no hubo hallazgos histopatológicos adversos en los órganos reproductivos de monos cynomolgus tratados con mepolizumab por 6 meses en dosis IV hasta 100 mg/ kg una vez cada 4 semanas (aproximadamente 70 veces la MRHD en una base de ABC). No se observó ninguna alteración de la fertilidad en un estudio de toxicología de fertilidad y reproducción general en ratones CD-1 realizados con un anticuerpo análogo que inhibe IL-5 en ratones en una dosis IV de 50 mg / kg una vez por semana. Este estudio no incluyó una camada o una valoración funcional de F1.
No hay datos de fertilidad en humanos. Los estudios en animales no mostraron efectos adversos propios del tratamiento anti-IL5 en la fertilidad.
Embarazo:
En monos, mepolizumab no ejerció efecto alguno sobre el embarazo o en el desarrollo embrionario/fetal y posnatal (lo que incluyó la función inmunitaria) de los descendientes. No se realizaron exploraciones en busca de malformaciones internas o esqueléticas. Los datos en monos cinomólogos demuestran que mepolizumab cruza la placenta. Las concentraciones de mepolizumab fueron aproximadamente 2.4 veces mayores en lactantes que en las madres durante varios meses posparto y no afectaron el sistema inmunitario de los lactantes.
Toxicología y farmacología animal:
Los datos no clínicos no revelaron riesgos específicos para humanos con base en los estudios convencionales de farmacología de seguridad o estudios de toxicidad de dosis repetida en monos. La administración intravenosa y subcutánea a monos se relacionó con una reducción en los eosinófilos periféricos y pulmonares, sin datos toxicológicos.
Los eosinófilos se han relacionado con respuestas del sistema inmunitario a ciertas infecciones parasitarias. Estudios realizados en ratones tratados con anticuerpos anti-IL-5 o genéticamente deficientes en IL-5 o eosinófilos no han mostrado una alteración en la capacidad para eliminar infecciones parasitarias.
INTERACCIONES MEDICAMENTOSAS Y DE OTRO GÉNERO:
No se han realizado estudios formales de interacciones con mepolizumab.
Enzimas de citocromo P450, bombas de eflujo y mecanismos de unión a proteína no están involucrados en la depuración de mepolizumab. Niveles aumentados de citosinas pro- inflamatorias (por ejemplo, IL-6), vía interacción con sus recetores análogos en hepatocitos, han mostrado reprimir la formación de enzimas CYP450 y transportadores de fármaco, sin embargo, elevación de marcadores pro-inflamatorios en asma grave es mínima y no hay evidencia de expresión de alfa receptor IL-5 sobre hepatocitos. El potencial para interacciones medicamentosas con mepolizumab es por lo tanto considerada como baja.
Incompatibilidades:
No mezclar la solución reconstituida para inyección con otros productos farmacológicos.
ALTERACIONES EN LOS RESULTADOS DE PRUEBAS DE LABORATORIO:
No hay datos.
PRECAUCIONES GENERALES:
NUCALA no está indicado para uso intravenoso y solo debe administrarse por vía subcutánea.
La prescripción de mepolizumab debe ser por médicos con experiencia en el diagnóstico y tratamiento de asma eosinofílica, EGPA, HES y RsCcPN.
Los eventos adversos o las exacerbaciones relacionados con asma pueden ocurrir durante el tratamiento con NUCALA.
Síntomas Agudos de Asma o de Enfermedad Deteriorante:
NUCALA no debe ser usado para tratar síntomas agudos de asma o exacerbaciones agudas. No usar NUCALA para tratar broncoespasmo agudo o estado asmático. Los pacientes deben buscar consejo médico si su asma continúa sin controlarse o empeora después de iniciar el tratamiento con NUCALA.
Reducción en la Dosis de Corticoesteroides:
La descontinuación abrupta de los corticosteroides después de iniciar el tratamiento con NUCALA no se recomienda. Las reducciones en las dosis de corticosteroides, si se requieren, deben ser graduales y realizarse bajo la supervisión de un médico.
Hipersensibilidad y Reacciones de Administración:
Las reacciones sistémicas agudas y retrasadas, lo que incluye reacciones de hipersensibilidad (p. ej., anafilaxia, urticaria, angioedema, exantema, broncoespasmo, hipotensión) han ocurrido después de la administración de NUCALA. Estas reacciones por lo general ocurren en un lapso de horas de la administración, pero en algunos casos tienen un inicio retrasado (es decir, días).
Infecciones parasitarias:
Los eosinófilos pueden participar en la respuesta inmunológica a algunas infecciones por helmintos. Los pacientes con infecciones preexistentes por helmintos se excluyeron de participar en el programa clínico. Se desconoce si NUCALA influenciará una respuesta del paciente contra las infecciones parasitarias. Los pacientes con infecciones preexistentes por helmintos deben tratarse por la infección antes del tratamiento con mepolizumab. Si los pacientes se infectan mientras reciben tratamiento con mepolizumab y no responden al tratamiento contra helmintos, debe considerarse la descontinuación temporal de NUCALA.
Inmunogenicidad:
En pacientes adultos y adolescentes con asma grave que reciben NUCALA 100 mg, 15/260 (6%) se presentaron anticuerpos anti-mepolizumab detectables. Se detectaron anticuerpos neutralizantes en solo 1 paciente con asma recibiendo NUCALA 100 mg. Los anticuerpos anti-mepolizumab aumentaron ligeramente (aproximadamente un 20%) a depuración del mepolizumab. No hubo evidencia de una correlación entre los títulos de anticuerpos anti-mepolizumab y el cambio en el nivel de eosinófilos. Se desconoce la relevancia clínica de la presencia de anticuerpos anti-mepolizumab.
En pacientes con RSCcPN que recibieron 100 mg de NUCALA, 6/196 (3%) tenían anticuerpos anti- mepolizumab detectables. No se detectaron anticuerpos neutralizantes en ningún paciente con RSCcPN.
En pacientes con EGPA que recibieron 300 mg de NUCALA, 1/68 (< 2%) tenían anticuerpos anti-mepolizumab detectables. No se detectaron anticuerpos neutralizantes en ningún paciente con EGPA.
En pacientes adultos y adolescentes con HES que recibieron 300 mg de NUCALA, 1/53 (2%) tuvo anticuerpos anti-mepolizumab detectables. No se detectaron anticuerpos neutralizantes en ningún paciente con HES.
La frecuencia informada de anticuerpos anti-mepolizumab puede subestimar la frecuencia real debido a la menor sensibilidad del ensayo en presencia de una alta concentración de fármaco. Los datos reflejan el porcentaje de los pacientes cuyos resultados de las pruebas fueron positivos para anticuerpos contra mepolizumab en ensayos específicos. La incidencia observada de positividad de anticuerpos en un ensayo depende en gran medida de varios factores, incluidos sensibilidad y especificidad del ensayo, metodología del ensayo, manejo de la muestra, momento de la recolección de la muestra, medicamentos concomitantes y enfermedad subyacente.
Uso Pediátrico:
El uso pediátrico en pacientes pediátricos menores a 12 años no ha sido establecido. Un total de 28 adolescentes de edad de 12 a 17 años con asma fueron inscritos en los estudios fase 3. De estos, 25 fueron inscritos en el estudio de exacerbación de 32 semanas (Estudio 2) y tenían una edad media de 14.8 años. Los pacientes tenían un historial de 2 o más exacerbaciones en el año anterior a pesar del uso regular de corticosteroides inhalados de dosis alta más un controlador (es) adicional con o sin corticosteroides orales y tenían eosinófilos en sangre mayores a o iguales a 150 células/mcL en el examen o mayor a o igual a 300 células/mcL en los 12 meses previos a la inscripción. [Véase Estudios Clínicos en la sección de Farmacocinética]. Los pacientes tuvieron una reducción en la tasa de exacerbaciones que tendió a favor de mepolizumab. De los 19 adolescentes que recibieron mepolizumab, 9 recibieron NUCALA y la depuración media aparente en estos pacientes fue del 35% menos que los adultos. El perfil de evento adverso en adolescentes fue generalmente similar a la población general en los estudios fase 3.
Efectos sobre la Capacidad para Conducir y Usar Maquinaria Pesada:
No se han realizado estudios para investigar el efecto de mepolizumab sobre el desempeño en la capacidad para conducir o para operar maquinaria pesada. No se anticiparía un efecto deletéreo sobre estas actividades a partir del perfil farmacológico de reacciones adversas de NUCALA.
DOSIS Y VÍA DE ADMINISTRACIÓN:
Vía de administración: Subcutánea.
NUCALA sólo debe administrarse como inyección subcutánea (p. ej., parte superior del brazo, muslo o abdomen) (Véase Uso y manejo).
NUCALA es un tratamiento complementario de mantenimiento de adición en pacientes de 12 años de edad o mayores con asma eosinofílica grave refractaria. (Véase sección de estudios clínicos en la sección Farmacocinética).
La prescripción y administración de NUCALA debe ser por médicos de experiencia en el diagnóstico y tratamiento del asma eosinofílica grave refractaria, Rinosinusitis crónica con pólipos nasales (RSCcPN), Granulomatosis eosinofílica con poliangeitis (EGPA) y Síndrome hipereosinofílico (HES).
Mepolizumab está diseñado para un tratamiento a largo plazo. La necesidad de continuar el tratamiento debe ser considerado por lo menos anualmente según lo determinado por la valoración del médico de acuerdo a la gravedad de la enfermedad y el nivel de control de las exacerbaciones.
NUCALA debe ser reconstituido y administrado por un profesional de la salud. En línea con la práctica clínica, se recomienda el monitoreo de pacientes después de la administración de agentes biológicos (véase sección Precauciones generales).
USO Y MANEJO:
Fórmula liofilizada:
• Presentación frasco ámpula con liofilizado para reconstitución:
Instrucciones para reconstitución y administración:
1. Reconstituir NUCALA en el frasco ámpula con 1.2 mL de agua estéril para uso inyectable, utilizando preferentemente una jeringa de 2 o 3 mL y una aguja 21 - G. La solución reconstituida contendrá una concentración de 100 mg/mL de mepolizumab.
2. Dirigir el flujo de agua estéril para uso inyectable en sentido vertical hacia el centro del polvo liofilizado. Permitir que el frasco ámpula repose a temperatura ambiente durante la reconstitución, haciéndolo girar con cuidado por 10 segundos con movimientos circulares a intervalos de 15 segundos hasta que el polvo se haya disuelto.
Nota: No agitar la solución reconstituida durante el procedimiento ya que esto puede llevar a producir espuma o precipitación. La reconstitución suele completarse en un lapso de 5 minutos después de que se ha agregado el agua estéril para uso inyectable, pero puede tomar tiempo adicional.
Luego de la reconstitución con 1.2 mL de agua estéril para uso inyectable, el volumen final de la solución es 1.44 mL que incluye la contribución de volumen del polvo liofilizado, proporcionando una concentración objetivo de 100 mg/mL (144 mg/1.44 mL = 100 mg/mL).
Los 144 mg de mepolizumab reconstituidos con 1.2 mL de agua inyectable proporcionan una concentración de mepolizumab garantizada de 100 mg/mL y un volumen extraíble de 1.0 mL para cada frasco ámpula.
3. Si se utiliza un dispositivo de reconstitución mecánica (agitador orbital) para reconstituir NUCALA, girar a 450 rpm por no más de 10 minutos. Alternativamente, girar a 1 000 rpm por no más de 5 minutos es aceptable.
4. Inspeccionar de forma visual la solución reconstituida en cuanto a la presencia de partículas y claridad. La solución debe ser clara a opalescente, e incolora a amarillo pálido o café pálido, sin partículas visibles. Sin embargo, se espera que se formen pequeñas burbujas de aire y son aceptables. Si hay partículas presentes en la solución o si ésta parece estar turbia o lechosa, la solución no debe usarse.
5. Si la solución reconstituida no se usa inmediatamente, debe:
• Almacenar a menos de 30°C.
• No congelar, y
• Desechar si no se usa dentro de las siguientes 8 horas después de la reconstitución.
• No mezclarse con otros medicamentos.
Instrucciones para la administración:
1. Para administración subcutánea, usar preferiblemente una jeringa de polipropileno de 1 mL equipada con una aguja de 21 a 27 G x 0.5 pulgadas (13 mm).
2. Justo antes de la administración, remover 1 mL de NUCALA reconstituido de un frasco ámpula. No agitar la solución reconstituida durante el procedimiento ya que esto puede producir espuma o precipitación del producto.
3. Administrar la inyección de 1 mL (equivalente a 100 mg de mepolizumab) subcutáneamente en la parte superior del brazo, muslo o abdomen.
Si se requiere más de un frasco ámpula para administrar la dosis prescrita, repetir los pasos 1 al 3. Se recomienda que los sitios individuales de inyección estén separados por lo menos 5 cm.
Fórmula líquida:
• Presentación pluma precargada (auto-inyector) o jeringa precargada.
Preparación:
1. Tenga listo lo que va a necesitar:
• Encuentre una superficie cómoda, bien iluminada y limpia. Asegúrese de tener a la mano:
• La pluma/jeringa precargada de NUCALA.
• Una toallita con alcohol (no incluido).
• Una almohadilla de gasa o una bola de algodón (no incluido).
No realizar la inyección si no tiene todo lo anterior.
Vea las instrucciones para la administración de la pluma precargada (auto-inyector) o jeringa precargada (jeringa de seguridad) en el instructivo del producto.
NOTA: NUCALA puede ser autoadministrado por el paciente o administrado por un cuidador si el profesional médico determina que es apropiado y el paciente o cuidador están capacitados en técnicas de inyección.
Poblaciones:
Asma Eosinofílica Grave:
Adultos y Adolescentes (12 años en adelante):
La dosis recomendada es de 100 mg de NUCALA administrado por inyección subcutánea (SC) una vez cada 4 semanas.
RSCcPN:
Adultos:
La dosis recomendada es de 100 mg de NUCALA administrados mediante inyección subcutánea (SC) una vez cada 4 semanas.
EGPA:
Adultos:
La dosis recomendada es 300 mg (3 inyecciones de 100 mg) administrado por inyección subcutánea (SC) una vez cada 4 semanas. Los sitios de inyección deben estar separados por lo menos 5 cm (véase Uso y Manejo).
HES:
Adultos y adolescentes (12 años en adelante):
La dosis recomendada es de 300 mg (3 inyecciones de 100mg) de NUCALA administrados por inyección subcutánea (SC) una vez cada 4 semanas. Los sitios de inyección deben estar separados por lo menos 5 cm (véase Uso y Manejo).
Ancianos (65 años o mayores):
No se requiere ajustar la dosis en pacientes de 65 años de edad o mayores (véase sección Farmacocinética – Poblaciones especiales de pacientes).
Deterioro renal:
Es poco probable que se requieran ajustes de la dosis en pacientes con deterioro renal (véase sección Farmacocinética – Poblaciones especiales de pacientes).
Deterioro hepático:
Es poco probable que se requieran ajustes de la dosis en pacientes con deterioro hepático (véase sección Farmacocinética – Poblaciones especiales de pacientes).
MANIFESTACIONES Y MANEJO DE LA SOBREDOSIFICACIÓN O INGESTA ACCIDENTAL:
No hay experiencia clínica con la sobredosis de NUCALA.
Se administraron dosis únicas de hasta 1500 mg de mepolizumab por vía intravenosa en un estudio clínico a pacientes con enfermedad eosinofílica sin evidencia de toxicidades relacionadas con la dosis.
Tratamiento:
No hay un tratamiento específico para una sobredosis con mepolizumab. En caso de sobredosis, hay que proporcionar al paciente cuidados de apoyo con la vigilancia apropiada según se requiera.
El manejo ulterior debe ser según se indique en clínica o como lo recomiende el centro nacional de control de intoxicaciones, en los lugares en que esté disponible.
PRESENTACIONES:
Caja de cartón con 1 o 3 frasco(s) ámpula con 100 mg de polvo liofilizado e instructivo anexo.
Caja de cartón con 1 o 3 plumas pre-cargadas (autoinyector) o con 1 o 3 jeringas pre-cargadas (jeringa de seguridad). Todas con instructivo anexo.
RECOMENDACIONES SOBRE ALMACENAMIENTO:
Precauciones Especiales de Almacenamiento:
Frasco ámpula con liofilizado sin abrir:
No congelar. Proteger de la luz. Almacenar en el empaque original hasta su uso.
Solución reconstituida:
Después de la reconstitución con agua estéril para uso inyectable, el producto permanece estable hasta por 8 horas cuando se almacena a menos de 30°C.
No congelar.
Durante la administración, no se requiere la protección de la luz.
• Pluma precargada (auto-inyector) o jeringa precargada (jeringa de seguridad):
Almacenar en refrigeración (2-8ºC).
No congelar. Proteger de la luz. Almacenar en el empaque original hasta su uso.
Si es necesario, el producto se puede conservar fuera del refrigerador hasta por 7 días a temperatura ambiente (hasta 30°C), en su empaque original sin abrir, protegido de la luz. Desechar si se dejó fuera del refrigerador por más de 7 días.
La pluma precargada y la jeringa precargada deben administrarse dentro de 8 horas una vez que se abra el empaque. Desechar si no se administra dentro de 8 horas.
Deséchese inmediatamente después de su uso.
LEYENDAS DE PROTECCIÓN:
Literatura exclusiva para médicos. La prescripción y administración de NUCALA debe ser por médicos con experiencia en el diagnóstico y tratamiento del Asma eosinofílica grave refractaria, Rinosinusitis Crónica con Pólipos Nasales (RSCcPN), Granulomatosis Eosinofílica con Poliangeitis (EGPA) y Síndrome Hipereosinofílico (HES). Su venta requiere receta médica. No se deje al alcance de los niños. Este medicamento no está destinado para uso en niños menores a 12 años de edad, para el tratamiento de Asma Eosinofílica Grave o Síndrome Hipereosinofílico. El uso de este medicamento solo está destinado para adultos en el tratamiento de la Rinosinusitis Crónica con Pólipos Nasales o Granulomatosis Eosinofílica con Poliangeitis. NUCALA no debe mezclarse con otros medicamentos. En caso de embarazo y lactancia, consulte a su médico. No se administre si la solución no es transparente, o si contiene partículas en suspensión o sedimentos. No se administre si el cierre ha sido violado.
Reporte las sospechas de reacción adversa a los correos: farmacovigilancia@cofepris.gob.mx y
farmacovigilancia.mx@gsk.com
Titular del Registro:
Glaxo Wellcome, S.A.
Avda. Extremadura, 3, Pol. Ind. Allendeduero,
Aranda de Duero, 09400 Burgos, España.
Representante Legal:
GLAXOSMITHKLINE MÉXICO, S.A. de C.V.
Autopista México-Querétaro Km. 41.5 Edif. TR9,
Interior 5-C, Ex. Hacienda San Miguel,
C.P. 54715, Cuautitlán Izcalli, México, México.
Reg. Núm. 243M2017 SSA IV
Liofilizado GDS 14/IPI 14,
Líquido GDS14/IPI05 – 06-ago-20
Actualización: 26-Noviembre-2021
Puede contener leyendas relacionadas a la propiedad y/o licenciamiento de la Denominación Distintiva.


