
MOZOBIL
PLERIXAFOR
Solución
1 Caja , 1 Frasco(s) ámpula , 24/1.2 mg/ml
FORMA FARMACÉUTICA Y FORMULACIÓN:
Cada frasco ámpula contiene:
Plerixafor 24 mg
Vehículo cbp 1.2 mL
INDICACIONES TERAPÉUTICAS:
Adulto: MOZOBIL® es un movilizador de células madre hematopoyéticas. Está indicado en combinación con el factor de estimulación de colonias de granulocitos (por sus siglas en inglés G-CSF) para movilizar células madre progenitoras hematopoyéticas a la sangre periférica para su recolección y subsecuente trasplante autólogo en pacientes con linfoma no-Hodgkin (LNH) y mieloma múltiple (MM).
Pediátrico (1 año a menores de 18 años): MOZOBIL® está indicado en combinación con G-CSF para movilizar células madre hematopoyéticas a la sangre periférica para su recolección y subsecuente trasplante autólogo en niños con linfoma·o tumores sólidos malignos y:
• Bajo recuento de células madre circulantes en el día previsto de la recolección después de la movilización con G-CSF (con o sin quimioterapia), o
• Que previamente no lograron recolectar suficientes células madre hematopoyéticas.
FARMACOCINÉTICA Y FARMACODINAMIA:
Farmacología:
Mecanismo de acción: MOZOBIL® es un antagonista selectivo del receptor de quimiocina CXCR4 y bloquea la unión de su ligando afín, el factor derivado de células estromales 1α (por sus siglas en inglés SDF-1α), también conocida como CXCL12. SDF-1α y CXCR4 se conocen por desempeñar funciones regulatorias clave en la distribución y dirección ded CMH (células madre hematopoyéticas) humanas al compartimiento medular óseo. Las células madre expresan CXCR4 y se sabe que migran a la médula ósea a través de un efecto quimioatrayente de SDF-1α que se produce localmente por células estromales de la médula ósea. Una vez en la médula ósea, se postula que la CXCR4 de las célula madre puede actuar para ayudar a “anclar” estas células a la matriz ósea, ya sea directamente a través de SDF-1α o a través de la inducción de otras moléculas de adhesión. Se cree que la elevación de niveles de células progenitoras hematopoyéticas son resultado de una disrupción en la unión de CXCR4 a su ligando afín, lo que resulta en la aparición de células tanto maduras como pluripotentes en la circulación sistémica.
Las células CD34+ movilizadas por MOZOBIL® son funcionales y pueden injertarse, con capacidad de favorecer la repoblación a largo plazo.
Farmacodinamia: El valor del incremento en la sangre periférica del recuento de células CD34+ (células/mcL) por día de aféresis se evaluó en dos estudios clínicos controlados con placebo en pacientes con linfoma y MM (AMD3100- 3101 y AMD3100- 3102, respectivamente). El valor del incremento sobre el periodo de 24 horas del día previo a la primera aféresis hasta el momento antes de la primera aféresis se encuentra resumido en la Tabla 1. Durante ese periodo de 24 horas, la primera dosis de MOZOBIL® 0.24 mg/kg o placebo fue administrada de 10-11 horas antes de la aféresis.
Tabla 1: Valor del incremento en sangre periférica del recuento de células CD34+ después de la administración de MOZOBIL®
|
Estudio |
MOZOBIL® y G-CSF |
Placebo y G-CSF |
||
|
Mediana |
Media (DE) |
Mediana |
Media (DE) |
|
|
AMD3100-3101 |
5.0 |
6.1 (5.4) |
1.4 |
1.9 (1.5) |
|
AMD3100-3102 |
4.8 |
6.4 (6.8) |
1.7 |
2.4 (7.3) |
En los estudios de farmacodinamia en los voluntarios sanos de MOZOBIL®, el pico de movilización de células CD34+ se observó de 6 a 9 horas después de la administración. En estudios farmacodinamicos de MOZOBIL® en voluntarios sanos en conjunto con G-CSF, se observó una elevación sostenida en la sangre periférica en el conteo de CD34+ de 4 a 18 horas después de la administración de MOZOBIL®, con una respuesta pico entre 10 y 14 horas.
La eficacia y seguridad de MOZOBIL® se evaluaron en un estudio abierto, mullicéntrico y controlado en pacientes pediátricos con tumores sólidos (incluyendo neuroblastoma, sarcoma, sarcoma de Ewing) o linfoma. Cuarenta y cinco pacientes pediátricos (1 a < 18 años) fueron aleatorizados, 2:1, utilizando 0.24 mg/kg de MOZOBIL® más movilización estándar (G-CSF más o menos quimioterapia) versus control (movilización estándar sola). El análisis primario mostró que el 80% de los pacientes en el grupo de MOZOBIL® experimentaron al menos una duplicación del recuento de PB CD34+, observado desde la mañana del día anterior a la primera aféresis planificada hasta la mañana anterior a la aféresis, en comparación con el 28.6% de los pacientes en el grupo control (p = 0.0019).
La mediana del recuento de células PB CD34+ al inicio del estudio fue de 15 células/μL en el grupo de MOZOBIL® frente a 35 células/μL en el grupo de control. El aumento medio en los recuentos de células PB CD34+ desde el inicio hasta el día de la aféresis fue de 3.2 veces en el grupo de MOZOBIL® frente a 1.4 veces en el grupo control.
Farmacocinética: La farrnacocinética de plerixafor ha sido evaluada en linfoma y pacientes con MM a nivel de dosis clínicas de 0.24 mg/kg siguiendo el tratamiento previo con G-CSF (10 mcg/kg una vez al día por 4 días consecutivos).
Con el fin de comparar la farmacocinética y la farmacodinamia de plerixafor siguiendo una dosis basada en 0.24 mg/kg y fija (20 mg), se realizó un ensayo de seguimiento en pacientes con LNH (N=61) que fueron tratados con 0.24 mg/kg o 20 mg de plerixafor. El ensayo se realizó en pacientes que pesaban 70 kg o menos. La dosis fija de 20 mg mostró una exposición 1.43 veces mayor (AUCa-10 h) que la dosis de 0.24 mg/kg (Tabla 2). La dosis fija de 20 mg también mostró una tasa de respuesta numéricamente más alta (5.2% (60.0% frente a 54.8%] según los datos de laboratorio local y 11.7% [63.3% frente a 51.6%] según los datos del laboratorio central) para alcanzar el objetivo de ≥ 5 x 106 células CD34+/kg que la dosis basada en mg/kg. Sin embargo, el tiempo medio para alcanzar ≥ 5 x 106 células CD34+/kg fue de 3 días para ambos grupos de tratamiento, y el perfil de seguridad entre los grupos fue similar. Se seleccionó un peso corporal de 83 kg como punto de corte para la transición de los pacientes de la dosificación fija a la basada en el peso (83 kg x 0.24 mg = 19.92 mg/kg).
Tabla 2: Comparaciones de exposición sistémica (AUC0.10h) de regímenes fijos basados en el peso
|
Régimen |
Promedio geométrico AUc |
|
Fijo 20 mg (n = 30) |
3991.2 |
|
0.24 mg/kg (n = 31) |
2792.7 |
|
Ratio (90% CI) |
1.43 (1.32, 1.54) |
Absorción: Plerixafor se absorbe rápidamente después de la inyección SC con concentraciones pico en aproximadamente 30-60 minutos.
Distribución: Plerixafor se une moderadamente a las proteínas de plasma humano hasta 58%. El volumen aparente de distribución de plerixafor en los seres humanos es de 0.3 L/kg lo que demuestra que plerixafor se limita en gran medida al espacio del fluido extravascular, pero no se limita a él.
Metabolismo: Plerixafor No se metaboliza in vitro utilizando microsomas hepáticos humanos o hepatocitos primarios humanos y no exhibe actividad inhibitoria in vitro hacia la mayoría de las enzimas CYPA50 metabolizadoras de fármacos (1A2, 2A6, 2B6, 2C8, 2C9, 2C19, 2D6, 2E1, y 3A4/5). En los estudios in vitro con hepatocitos humanos, plerixafor no induce enzimas CYP1A2, CYP2B6, y CYP3A4. Estos descubrimientos sugieren que plerixafor tiene un potencial más bajo para su participación en las interacciones fármaco-fármaco dependientes de P450.
Eliminación: La principal vía de eliminación de plerixafor es la urinaria. Posterior a una dosis de 0.24 mg/kg en voluntarios sanos con función renal normal, aproximadamente el 70% de la dosis fue excretada en la orina como el fármaco original durante las primeras 24 horas después de la administración. La vida media en plasma es de 3-5 horas. MOZOBIL® no actuó como un sustrato o inhibidor de P-glicoproteína en un estudio in vitro con modelos de células MDCKII y MDCKII-MDR1.
Insuficiencia renal: Posterior a una dosis única de 0.24 mg/kg de MOZOBIL®, la depuración de plerixafor se redujo en pacientes con grados variables de disfunción renal y tuvo una correlación positiva con la depuración de creatinina (CrCI por sus siglas en inglés). Los valores promedio de AUCo.24 de plerixafor en pacientes con insuficiencia renal leve (CrCI 51-80 mL/min), moderada (CrCl 31-50 mL/min) y grave (CrCl < 30 mL/min) fueron 5410, 6780 y 6990 ng x hr/mL, respectivamente, siendo mayores que la exposición observada en pacientes sanos con función renal normal (5070 ng x hr/mL). La insuficiencia renal no tiene efecto sobre Cmax (Ver sección DOSIS Y VIA DE ADMINISTRACIÓN).
Sexo: Un análisis farmacocinético de la población no mostró ningún efecto con respecto al sexo en la farmacocinética de MOZOBIL®.
Edad: Un análisis farmacocinético de la población no mostró efecto con respecto a la edad en la farmacocinética de MOZOBIL®.
Población pediátrica: La farmacocinética de MOZOBIL® se evaluó a dosis de 0.16, 0.24 y 0.32 mg/kg en 27 pacientes pediátricos (2 a menores de 18 años) con tumores sólidos. La exposición a MOZOBIL® después de una sola dosis subcutánea mostró una proporcionalidad de dosis de 0.16 a 0.24 mg/kg en pacientes pediátricos, como en adultos. No se observó un aumento en la exposición (AUCo.gh) más allá de 0.24 mg/kg (la dosis para adultos), y no hubo un efecto clínicamente relevante de la edad en la exposición. La exposición observada en niños fue similar a la observada en adultos.
Estudios clínicos:
La eficacia y seguridad de MOZOBIL® en conjunto con G-CSF en pacientes con linfoma y MM fueron evaluadas en dos estudios controlados con placebo de Fase 3 (estudios AMD3100-3101 y AMD3100-3102). Los pacientes fueron aleatorizados para recibir ya sea MOZOBIL® de 0.24 mg/kg o placebo en cada tarde antes de la aféresis. Los pacientes recibieron dosis matutinas diarias de G-CSF 10 mcg/kg por 4 días previos a la primera dosis de MOZOBIL® o placebo y en cada mañana previa a la aféresis. El criterio de valoración primario fue la recolección de un número determinado de células CD34+/kg con un número dado de días de aféresis. Doscientos noventa y ocho (298) pacientes de LNH fueron incluidos en los análisis de eficacia primarios para el Estudio AMD3100-3101. La edad promedio fue de 55.1 años (29-75) y 57.5 años (22-75) en los grupos de MOZOBIL® y placebo, respectivamente, y 92.6% (276/298) de los pacientes eran caucásicos. Trescientos dos (302) de los pacientes con MM fueron incluidos en los análisis de eficacia primaria para el Estudio de AMD3100-3132. La edad promedio fue de 58.2 años (28-75) y 58.4 años (28-75) en los grupos de MOZOBIL® y placebo, respectivamente, y el 81.1% (245/302) de los pacientes eran caucásicos.
En AMD3100-3101, el 59.3% de pacientes de LNH fueron movilizados con MOZOBIL® y G-CSF alcanzó el criterio de valoración primario de colección de ≥ 5 X 106 células CD34+/kg de la sangre periférica en cuatro o menos sesiones de aféresis, comparado con el 19.6% de los pacientes que fueron movilizados con placebo y G-CSF (p < 0.001 ). Los resultados de la movilización de células CD34+ secundarias fueron consistentes con el punto final primario (tabla 3).
Tabla 3: Resultados de Eficacia en el estudio de AMD3100-3101- movilización de células CD34+ en Pacientes de LNH
|
Criterio de valoración de eficacia |
MOZOBIL® y G-CSF (n= 150) |
Placebo y G-CSF (n= 148) |
Valor Pª |
|
Pacientes que alcanzan ≥ 5 X 106 células/kg en ≤ 4 días de aféresis |
89 (59.3%) |
29 (19.6%) |
< 0.001 |
|
Pacientes que alcanzan ≥ 2 X 106 células/kg en ≤ 4 días de aféresis |
130 (86.7%) |
70 (47.3%) |
< 0.001 |
ª Valor-p calculado usando el método de cuadrados de Pearson.
La mediana del número de días a alcanzar el criterio de valoración primario de ≥ 5 x 106 CD34+ células/kg fue de 3 días para el grupo de MOZOBIL® y no evaluable para el grupo de placebo (figura 1).
Figura 1: Resultados de Eficacia del Estudio AMD3100-3101- días de aféresis requeridos para alcanzar ≥ 5 x 106 células/kg en pacientes de LNH
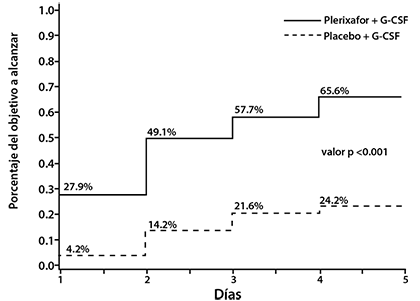
* Porcentajes determinados por el método de Kaplan Meier; valor p calculado con base en la prueba de rango logarítmico
En AMD3100-3102, el 71.6% de los pacientes con MM que fueron manejados con MOZOBIL® y G-CSF que alcanzaron el criterio primario de recolección de ≥ 6 X 106 células CD34+/kg de la sangre periférica en dos o menos sesiones de aféresis, comparado con el 34.4% de los pacientes que fueron manejados con placebo y G-CSF (p < 0.001). Los resultados secundarios de movilización de células CD34+ fueron consistentes con el criterio de valoración primario (tabla 4).
Tabla 4: Resultados de eficacia del Estudio AMD3100-3102 - movilización de células CD34+ en pacientes con MM
|
Criterio de valoración de eficacia |
MOZOBIL® y G-CSF (n= 148) |
Placebo y G-CSF (n= 154) |
Valor Pª |
|
Pacientes que alcanzan ≥ 6 X 106 células/kg en ≤ 2 días de aféresis |
106 (71.6%) |
53 (34.4%) |
< 0.001 |
|
Pacientes que alcanzan ≥ 6 X 106 células/kg en ≤ 4 días de aféresis |
112 (75.7%) |
79 (51.3%) |
< 0.001 |
|
Pacientes que alcanzan ≥ 2 X 106 células/kg en ≤ 4 días de aféresis |
141 (95.3%) |
136 (88.3%) |
0.031 |
ª valor p calculado utilizando la estadística Cochran-Mantel-Haenszel obstruida por recuentos de plaquetas del valor basal.
La mediana del número de días para alcanzar el punto final primario de ≥ 6x106 células CD34+ /kg fue de 1 día para el grupo de MOZOBIL ® y 4 días para el grupo placebo (figura 2).
Figura 2: Resultados de eficacia del Estudio AMD3100-3102 - días de aféresis requeridos para alcanzar ≥ 6 x 106 células/kg en pacientes con MM

* Porcentajes determinados por el método de Kaplan Meier; valor p calculado con base en la prueba de rango logarítmico establecido por cuentas de plaqueta del valor basal.
Para los pacientes trasplantados en los estudios de Fase 3, el tiempo de fijación de neutrófilos y de plaquetas y la durabilidad del injerto hasta 12 meses posteriores al implante fueron similares en los grupos MOZOBIL® y placebo. La mediana de tiempo para la fijación de neutrófilos fue de 10 días en AMD3100-3101 y 11 días en AMD3100-3102 (p = 0.360 y 0.690, respectivamente) y para la fijación de plaquetas fueron 20 días en AMD3100-3101 y 18 días en AMD3100-3102 (p = 0.630 y 0.180, de manera respectiva). No se observaron diferencias de durabilidad de la fijación a través de grupos de MOZOBIL® y placebo en AMD3100-3101 o AMD3100-3102.
La eficacia y la seguridad de MOZOBIL® en conjunto con G-CSF en linfoma y MM también fueron evaluadas en dos estudios de apoyo de Fase 2 (Estudios AMD3100-2101 y AMD3100-2106). En estos estudios, los pacientes con LNH enfermedad de Hodgkin, o MM recibieron 0.24 mg/kg de MOZOBIL® en la noche o mañana antes de la aféresis. Los pacientes recibieron dosis diarias en la mañana de 10 mcg/kg de G-CSF por 4 días antes de su primera dosis de MOZOBIL® y en cada mañana antes de la aféresis. La información de movilización e injerto para estos estudios fue similar a la información para los estudios de Fase 3.
CONTRAINDICACIONES: Hipersensibilidad al fármaco o a alguno de los componentes de la fórmula, durante el embarazo y lactancia y en personas con leucemia.
RESTRICCIONES DE USO DURANTE EL EMBARAZO Y LA LACTANCIA:
Embarazo: MOZOBIL® puede causar daño fetal cuando se le administra a mujeres embarazadas. Estudios en animales han mostrado teratogenicidad. No hay estudios adecuados o bien controlados en mujeres embarazadas usando MOZOBIL®.
Si este fármaco se usa durante el embarazo, o la paciente se embaraza mientras toma este fármaco, la paciente debe estar informada del riesgo potencial para el feto. Aconseje a las mujeres en edad fértil respecto a la utilización de métodos anticonceptivos eficaces durante el tratamiento.
SDF-1α y CXCR4 desempeñan una función esencial en el desarrollo embriofetal. Plerixafor ha mostrado causar un incremento de reabsorciones, disminución del peso fetal, retraso en el desarrollo del esqueleto y el aumento de anomalías fetales en ratas y conejos. Los modelos animales también sugieren una modulación de hematopoyesis, vascularización, y desarrollo del cerebelo por SDF-1α y CXCR4 a nivel fetal. Los niveles a los cuales no se observaron efectos adversos (NOAEL) de plerixafor en ratas y conejos (3 mg/kg/día y 0.6 mg/kg/día, respectivamente) son de aproximadamente 2.0 y 0.8 veces la dosis humana recomendada de 0.24 mg/kg/día (8.9 mcg/m2/día). No hay estudios adecuados y bien controlados en mujeres embarazadas que usan MOZOBIL®.
Lactancia: Se desconoce si MOZOBIL® se excreta a través de leche materna; pero en vista de que muchos medicamentos son excretados por esta vía, el médico debe decidir si se descontinúa plerixafor o se interrumpe la lactancia considerando la importancia que tiene el tratamiento para la madre.
REACCIONES SECUNDARIAS Y ADVERSAS: A continuación se describen las reacción secundarias y adversas identificados durante estudios clínicos y en la experiencia post-comercialización clasificándolas con base en la clasificación CIOMS: Muy común ≥ 10%; común ≥ 1 y < 10%; infrecuente ≥ 0.1 y < 1%; rara ≥ 0.01 y < 0.1%; muy rara < 0.01%, frecuencia desconocida.
Estudios clínicos: La información de seguridad para MOZOBIL® en conjunto con G-CSF en pacientes oncológicos se obtuvieron de dos estudios de Fase 3 controlados con placebo (301 pacientes) y 10 estudios de Fase 2 no controlados (242 pacientes). Los pacientes fueron tratados principalmente con dosis diarias de 0.24 mg/kg de la inyección SC. La exposición a MOZOBIL® en estos estudios fue en un rango entre 1 y 7 días consecutivos (mediana = 2 dias).
En los dos estudios de Fase 3 en pacientes con LNH y MM (AMD3100-3101 y AMD3100-3102, respectivamente), se trató un total de 301 pacientes en el grupo con MOZOBIL® y G-CSF y a 292 pacientes en el grupo placebo y GCSF.
Los pacientes recibieron dosis diariamente por la mañana de G-CSF 10 mcg/kg por 4 días previos a la primera dosis de MOZOBIL® o placebo y en cada mañana antes de la aféresis. Los eventos adversos que ocurrieron de forma más frecuente con MOZOBIL® que con placebo y se reportaron con relación en ≥ 1% de los pacientes que recibieron MOZOBIL® durante la movilización de CMH y aféresis antes de la quimioterapia/tratamiento ablativo en preparación para el trasplante se muestran en la tabla 5. De la quimioterapia/tratamiento ablativo en preparación para el trasplante, hasta 12 meses posteriores al trasplante, ninguna diferencia notable en la incidencia de los eventos adversos se observó a lo largo de los grupos de tratamiento.
La Tabla 5 enlista las reacciones adversas organizadas por frecuencia de acuerdo a la clasificación por órganos y sistemas (SOC) del Diccionario Médico para Actividades Regulatorias (MedDRA).
Tabla 5: Eventos Adversos que ocurren de manera más frecuente con MOZOBIL® que con placebo y que se consideran relacionadas a MOZOBIL® durante la movilización de CMH y estudios de aféresis en Fase 3
|
Trastornos psiquiátricos |
|
|
Común |
Insomnio |
|
Trastornos del sistema nervioso |
|
|
Común |
Cefalea, mareos |
|
Trastornos gastrointestinales |
|
|
Muy Común |
Diarrea, náuseas |
|
Común |
Flatulencia, dolor abdominal, vómitos, distensión abdominal, boca seca, malestar estomacal, estreñimiento, dispepsia, hipostesia oral |
|
Trastornos la piel y del tejido subcutáneo |
|
|
Común |
Hiperhidrosis, eritema |
|
Trastornos musculoesqueléticos y del tejido conjuntivo |
|
|
Común |
Artralgia, dolor musculoesquelético |
|
Trastornos generales y condiciones del sitio de administración |
|
|
Muy Común |
Reacciones en el área de la inyección |
|
Común |
Fatiga, malestar general |
Las reacciones adversas reportadas en pacientes oncológicos que recibieron MOZOBIL® en los estudios de Fase 3 controlados y estudios no controlados, incluyendo un estudio de Fase 2 de MOZOBIL® como monoterapia para la movilización de CMH, son similares. No se observaron diferencias notables en la incidencia de reacciones adversas en los pacientes oncológicos por la enfermedad, la edad o el sexo.
Reacciones alérgicas: En los estudios clínicos oncológicos de MOZOBIL®, menos del 1% de los pacientes experimentaron reacciones alérgicas leves o moderadas dentro de aproximadamente 30 minutos después de la administración de MOZOBIL®, incluida una o varias de las siguientes: urticaria (n = 2), hinchazón periorbitaria (n = 2), disnea (n = 1) o hipoxia (n = 1).
Infarto de miocardio: En estudios clínicos, siete de 679 pacientes oncológicos experimentaron infartos de miocardio después de la movilización de CMH con MOZOBIL® y G-CSF. Todos los hechos ocurrieron por lo menos 14 días después de la última administración de MOZOBIL®. Además, dos pacientes oncológicas de sexo femenino en el programa de uso compasivo experimentaron infartos de miocardio tras la movilización de CMH con MOZOBIL® y G-CSF. Uno de estos eventos ocurrió 4 días después de la última administración de MOZOBIL®. La falta de relación temporal en 8 de los 9 pacientes, junto con el perfil de riesgo de los pacientes con infarto de miocardio no sugiere que MOZOBIL® confiere un riesgo independiente para el infarto de miocardio en pacientes que también reciben G-CSF.
Reacciones vasovagales: En estudios clínicos de MOZOBIL® en pacientes oncológicos y voluntarios sanos, menos del 1% de los pacientes experimentaron reacciones vasovagales (hipotensión y/ o síncope ortostático) después de la administración subcutánea de dosis de ≤ 0,24 mg/kg de plerixafor. La mayoría de estos eventos se produjo dentro de 1 hora posterior a la administración de MOZOBIL®.
Trastornos gastrointestinales: En los estudios clínicos de MOZOBIL® con pacientes oncológicos, se han notificado casos raros de eventos gastrointestinales graves, como diarrea, náuseas, vómitos y dolor abdominal.
Parestesias: Las parestesias son comúnmente observadas en los pacientes oncológicos sometidos a trasplante autólogo después de múltiples intervenciones contra la enfermedad. En los estudios de Fase 3 controlados con placebo, la incidencia de parestesias fue del 20.6% y del 21.2% en los grupos de MOZOBIL® y placebo, respectivamente.
Hiperleucocitosis: En los estudios clínicos de Fase 3 se observaron recuentos leucocitarios de 100,000 células/mcl o mayores, el día anterior o algún día de aféresis, en el 7% de los pacientes que recibieron MOZOBIL® y en el 1% de los pacientes que recibieron placebo. No se observaron complicaciones o síntomas clínicos de leucostasis.
Población pediátrica: Fueron tratados treinta pacientes con 0.24 mg/kg de MOZOBIL® en un estudio abierto, mullicéntrico, controlado. (Ver sección FARMACOCINÉTICA Y FARMACODINAMIA).
En este estudio, 23 de 30 (76.7%) pacientes en el grupo de MOZOBIL® y 10 de 15 (66.7%) pacientes en el grupo de control experimentaron eventos adversos.
Los eventos adversos más comunes (> 10% de pacientes) para MOZOBIL® fueron anemia, disminución del recuento de plaquetas, rinitis, neutropenia febril, hipoalbuminemia, diarrea, vómitos y pirexia. Los eventos adversos más comunes (> 10% de pacientes) en el grupo control fueron hipokalemia, aumento de la alanin aminotransferasa, neutropenia febril, vómitos, disminución del recuento de plaquetas, fatiga, náuseas y anemia.
Ningún paciente suspendió el tratamiento del estudio debido a eventos adversos. No se identificaron nuevas situaciones preocupantes respecto a la seguridad en este estudio.
Experiencia post-comercialización: Además de las reacciones adversas que se reportaron en los estudios clínicos, las siguientes reacciones adversas se reportaron después de una experiencia posterior a la comercialización mundial con MOZOBIL®. Dado que estas reacciones se reportan de forma voluntaria desde una población de un tamaño incierto, no siempre es posible calcular de forma fiable su frecuencia o establecer una relación casual a la exposición del fármaco.
Trastornos de la sangre y del sistema linfático: esplenomegalia y ruptura esplénica (Ver sección PRECAUCIONES GENERALES).
Trastornos del sistema inmunológico: Reacciones anafilácticas, incluido el shock anafiláctico (Ver sección PRECAUCIONES GENERALES).
Trastornos psiquiátricos: Sueños anormales y pesadillas (referido de experiencia posterior a la comercialización y estudios de Fase 3).
PRECAUCIONES EN RELACIÓN CON EFECTOS DE CARCINOGÉNESIS, MUTAGÉNESIS, TERATOGÉNESIS Y SOBRE LA FERTILIDAD:
Carcinogénesis: No se han realizado estudios de carcinogénesis con MOZOBIL®.
Mutagénesis: MOZOBIL® no fue genotóxico en un ensayo de mutación bacteriana in vitro (Prueba de Ames en Salmonella), una prueba de aberraciones cromosómicas usando células de ovario de hámster chino y una prueba in vivo de micronúcleos en la médula ósea de una rata.
Fertilidad: Los efectos potenciales de MOZOBIL® sobre la fertilidad masculina y desarrollo posterior al nacimiento no han sido evaluados en estudios no clínicos. En estudios realizados para medir la distribución de 14C-plerixafor, no hubo evidencia de acumulación en los testículos. La estadificación de la espermatogénesis medida en un estudio de toxicidad en una dosis repetida por 28 días en ratas, no reveló anomalías que pudieran estar relacionadas con MOZOBIL®. No se observó evidencia histopatológica de toxicidad en los órganos reproductivos masculinos o femeninos en estudios de toxicidad de dosis repetida.
No se observaron efectos adversos en un estudio de investigación de fertilidad femenina en ratas, aunque las concentraciones de plerixafor en los ovarios fueron detectables hasta los últimos días de la convivencia.
Toxicología:
Dosis única: Inyecciones únicas IV o SC de plerixafor en ratas y ratones indujeron la aparición rápido (< 2 horas) de efectos neuromusculares de tipo sedantes (hipoactividad), disnea, postración ventral o lateral y/o espasmos transitoria pero severa. La recuperación completa de la mayoría de los signos se produjo dentro de las 4 horas siguientes a la administración de MOZOBIL®. En los ratones, se observaron muertes posteriores a las dosis de ≥ 14 mg/kg de SC y ≥ 5 El nivel al cual no se observaron efectos adversos (NOAEL, por sus siglas en inglés) fue de 2 mg/kg y < 2 mg/kg IV. En las ratas, se observaron muertes posteriores a las dosis de ≥ 40 mg/kg se y ≥ 5 mg/kg IV.
Dosis repetida: En los estudios de dosis repetidas en ratas y perros con una o dos dosis diarias, la inyección se de MOZOBIL® trajo signos clínicos negativos similares a los que se vieron en los estudios de dosis única en ratas y ratones. El inicio de estos signos clínicos ocurrieron entre 15 min y 1 h después de la inyección de plerixafor SC; sin embargo, a diferencia de los estudios de dosis única, los signos generalmente no se observaron hasta después de haberse administrado aproximadamente 5 a 8 dosis diarias se de plerixafor a ratas o perros.
Los efectos adicionales de MOZOBIL® observados de forma consistente en estudios con animales a dosis repetidas incluyen leucocitosis y aumento de la excreción urinaria de calcio y magnesio en ratas y perros, y diarrea y taquicardia en perros. No se observaron cambios en la presión arterial o en trazos electrocardiográficos relacionados con plerixafor a las dosis toleradas. Se documentaron hallazgos histopatológicos de hematopoyesis extramedular en el hígado, el bazo y ocasionalmente en otros órganos de ratas y/o perros. Se observaron pesos de bazo ligeramente mayores en ratas. Estos hallazgos se consideraron como una extensión de la acción farmacológica de MOZOBIL® para movilizar células hematopoyéticas y/o leucocitos. Excepto por la irritación local del sitio de la inyección, no se observaron cambios histopatológicos toxicológicamente significativos. La toxicidad limitante de dosis en la dosificación de una vez y dos veces al día en los estudios en ratas y perros durante 28 días fue la mortalidad y los signos clínicos adversos graves de características neuromusculares observados en las primeras 1 y 2 horas posteriores a la dosis. La dosis máxima total diaria tolerada fue aproximadamente dos veces mayor en ratas y perros cuando se administró MOZOBIL® vía SC en un régimen de dos veces al día en comparación con un régimen de una dosis diaria. La dosis máxima tolerada en un programa de dosificación de una o dos veces diarias en ratas fue de 11.4 mg/kg/día y 12 mg/kg BID (24 mg/kg/dia), respectivamente. La dosis máxima tolerada en perros en un programa de dosificación de una a dos veces diarias fue de 4.0 mg/kg/día y 4.0 mg/kg BID (8.0 mg/kg/día), respectivamente.
Un tamiz in vitro de la actividad general del receptor, mostró que plerixafor, a una concentración (5 mcg/mL) varias veces mayor que el nivel sistémico máximo en humanos tiene afinidad moderada a fuerte de unión a un número de distintos receptores localizados predominantemente en terminaciones nerviosas presinápticas en el SNC y/o SNP (receptores de canal de calcio tipo N, canal de potasio SKCA, histamina H3, acetilcolina muscarinica M1 y M2, adrenérgicos alfa 18 y alfa 2c, neuropéptidos Y/Y1 y poliamina NMDA glutamato). Se desconoce la relevancia clínica de estos hallazgos.
Los resultados del estudio de búsqueda de rango de dosís en cerdos miniatura jóvenes y los estudios de determinación de rango y definitivos en ratas jóvenes fueron similares a los observados en ratones, ratas y perros adultos. Se observaron signos clínicos de postración lateral y malestar en cerdos miniatura a 8 mg/kg y se observó mortalidad a 12 mg/kg. Plerixafor produjo la esperada leucocitosis mediada farmacológicamente en cerdos y ratas. Los efectos sobre el peso de los órganos observados en ratas fueron considerados farmacológicos (timo) o una respuesta adaptativa (hematopoyesis extramedular en el hígado).
Los márgenes de dosis en el estudio de ratas jóvenes a la dosis máxima tolerada (DMT) de 90 mg/m2 (es decir, 15 mg/kg/día) son 7.6 veces en función del área de superficie corporal o 47 veces en función de la dosis en comparación con la dosis pediátrica clínica más alta de 11.8 mg/m2 (es decir, 0.32 mg/kg) en niños de 2 a 12 años de edad.
INTERACCIONES MEDICAMENTOSAS Y DE OTRO GÉNERO:
Fármaco-Fármaco: Basados en los estudios in vitro, el Plerixafor no es un sustrato, inhibidor o inductor de isoenzimas del citocromo P450 humano. No se han realizado estudios formales de interacción medicamentosa. Plerixafor no actuó como sustrato o inhibidor de la P-glicoproteína en un estudio in vitro.
En estudios clínicos de pacientes con LNH la adición de rituximab a un régimen de movilización de MOZOBIL® y G-CSF no afectó la seguridad del paciente ni el rendimiento de células CD34+.
Fármaco-Alimento: MOZOBIL® se administra por vía parenteral, y las interacciones con alimentos y bebidas se consideran poco probables.
Incompatibilidades farmacéuticas: En ausencia de estudios de compatibilidad, MOZOBIL® no debe mezclarse con otros medicamentos en la misma inyección.
ALTERACIONES EN LOS RESULTADOS DE PRUEBAS DE LABORATORIO: No se ha demostrado que MOZOBIL® interfiera con ninguna prueba de laboratorio clínico de rutina.
PRECAUCIONES GENERALES:
Movilización de células tumorales en pacientes con leucemia:
En un programa de uso compasivo, MOZOBIL® y G-CSF han sido administrados a pacientes con leucemia mieloide aguda y leucemia de células plasmáticas. En algunas instancias, estos pacientes experimentaron un incremento en el número de células de leucemia circulantes. Con el propósito de la movilización de CMH, MOZOBIL® puede causar movilización de células de leucemia y contaminación subsecuente del producto de la aféresis. Por lo tanto, MOZOBIL® no está destinado para la movilización y cosecha de CMH en pacientes con leucemia.
Efectos hematológicos:
Leucocitosis: La administración de MOZOBIL® en conjunto con G-CSF aumenta la circulación de leucocitos así como las poblaciones de CMH. Los recuentos de leucocitos deben ser monitoreados durante el uso de MOZOBIL®. En pacientes con recuentos de neutrófilos en sangre periférica superiores a 50,000 células/mcL, la administración de MOZOBIL® debe hacerse bajo un análisis clínico cuidadoso.
Trombocitopenia: La trombocitopenia es una complicación conocida de la aféresis y se ha observado en pacientes que reciben MOZOBIL®. El recuento de plaquetas debe controlarse en todos los pacientes que reciben MOZOBIL® que después son sometidos a aféresis.
Potencial para la movilización de células tumorales en el linfama y pacientes con mieloma múltiple: Cuando MOZOBIL® se emplea en conjunto con G-CSF para la movilización CMH en pacientes con linfoma o MM, las células tumorales pueden ser liberadas de la médula y posteriormente recolectadas en el producto de leucoféresis. El efecto de la reinfusión potencial de las células tumorales no ha sido bien estudiado. En estudios clinicos de pacientes con linfoma no Hodgkin (LNH) y mieloma múltiple (MM), la movilización de las células tumorales no ha sido observada con MOZOBIL®.
Reacciones alérgicas: En los estudios clínicos oncológicos de MOZOBIL®, menos del 1% de los pacientes experimentaron reacciones alérgicas leves o moderadas dentro de aproximadamente 30 minutos después de la administración de MOZOBIL®, incluyendo una o más de las siguientes: urticaria, inflamación periorbitaria, disnea o hipoxia. En general, los síntomas respondieron a los tratamientos (por ejemplo, antihistamínicos, corticosteroides, hidratación u oxígeno suplementario) o se resolvieron de manera espontánea. Los casos de reacciones anafilácticas, incluido el shock anafiláctico, se han registrado en la experiencia posterior a la comercialización en todo el mundo. Deben tomarse las precauciones adecuadas debido al potencial para estas reacciones.
Reacciones vasovagales: Pueden ocurrir reacciones vasovagales, hipotensión ortostática y/o síncope después de las inyecciones. En estudios clínicos de MOZOBIL® en oncología y con voluntarios sanos, menos del 1% de los sujetos experimentaron reacciones vasovagales (hipotensión ortostática y/o síncope) después de la administración de dosis de MOZOBIL® ≤ 0.24 mg/kg. La mayoría de estos eventos ocurrieron dentro de 1 hora de la administración de MOZOBIL®. Deben tomarse precauciones apropiadas debido al potencial riesgo de estas reacciones.
Efecto potencial en el bazo: Se han reportado casos de agrandamiento y/o ruptura esplénica después de la administración de MOZOBIL® en conjunto con el factor de crecimiento G-CSF. En pacientes que reciben MOZOBIL® en conjunto con G-CSF reportan dolor abdominal superior izquierdo y/o escapular o dolor de hombros, debe evaluarse la integridad esplénica.
Se ha observado elevación del peso absoluto y relativo del bazo asociado a hematopoyesis extramedular posterior a la administración prolongada de plerixafor SC diario (2 a 4 semanas) en ratas, a dosis aproximadamente 4 veces mayores a la dosis recomendada en humanos. El efecto de MOZOBIL® en el tamaño del bazo en pacientes no ha sido evaluado de forma especifica en estudios clínicos.
Deterioro renal: En pacientes con deterioro renal moderado o severo (CrCI ≤ 50 mL/min) se debe reducir MOZOBIL® en un tercio, para recibir una dosis de 0.16 mg/kg (ver sección DOSIS Y VÍA DE ADMINISTRACIÓN).
Prolongación de QT/QTc: No existe evidencia de un efecto de prolongación de QT/QTc de MOZOBIL® en dosis únicas por encima de 0.40 mg/kg. En un estudio aleatorizado, bajo condiciones doble ciego, cruzado, 48 pacientes sanos recibieron una dosis única subcutánea de MOZOBIL® de (0.24 mg/kg y 0.40 mg/kg) y placebo.
Las concentraciones pico para 0.40 mg/kg de MOZOBIL® fueron aproximadamente 1.8 veces mayores que la concentración pico después de la dosis subcutánea única de 0.24 mg/kg.
Monitoreo de leucocitos y plaquetas: El recuento de leucocitos y de plaquetas debe monitorearse durante el uso de MOZOBIL® y aféresis.
DOSIS Y VÍA DE ADMINISTRACIÓN:
Dosis recomendada y vía de administración: La dosis recomendada diariamente de MOZOBIL® por inyección subcutánea (SC) es:
Adulto:
• Dosis fija de 20 mg o 0.24 mg/kg de peso corporal para pacientes que pesan ≤ 83 kg por inyección (Ver sección FARMACOCINÉTICA Y FARMACODINAMIA).
• 0.24 mg/kg de peso corporal para pacientes que pesan > 83 kg.
Pediátrico (de 1 año a menores de 18 años):
• 0.24 mg/kg de peso corporal (Ver sección FARMACOCINÉTICA Y FARMACODINAMIA).
MOZOBIL® deberá ser administrado de 6 a 11 horas antes del inicio de cada aféresis siguiendo 4 días de tratamiento con G-CSF. MOZOBIL® debe ser administrado por una enfermera, médico u otro profesional del cuidado de la salud.
MOZOBIL® ha sido utilizado frecuentemente por 2 a 4 días consecutivos. Ha sido usado por más de 7 días consecutivos en el entorno clínico.
Debe usarse el peso corporal actual del paciente para calcular el volumen de MOZOBIL® a ser administrado.
Cada frasco contiene 1.2 ml de 20 mg/ml de solución, y el volumen que debe ser administrado a los pacientes debe calcularse de acuerdo a la siguiente ecuación:
|
0.012 X el peso actual del pacientes (en kg) |
= |
dosis a administrar (en mL) |
En los estudios clínicos, las dosis de MOZOBIL® han sido calculadas basadas en el peso corporal actual en pacientes que rebasan el peso corporal ideal hasta un 175%. No se han investigado la dosis y el tratamiento de MOZOBIL® en pacientes que rebasan el peso corporal ideal por más de 175%.
Basado en el aumento de la exposición con el aumento del peso corporal, la dosis de MOZOBIL® no debe exceder a 40 mg/día.
El peso que se usa para calcular el volumen de MOZOBIL® debe ser obtenido dentro de una semana de la primera dosis de MOZOBIL®.
Medicamentos concomitantes recomendados: En los estudios clínicos pivótales que soportan el uso de MOZOBIL®, todos los pacientes recibieron dosis diarias en la mañana de factores estimulantes de colonias de granulocitos (G-CSF, por sus siglas en inglés) de 10 mcg/kg por 4 dias antes de la primera dosis de MOZOBIL® y en cada mañana antes de la aféresis. (Ver sección ESTUDIOS CLiNICOS).
Lineamientos para la modificación de dosis: En pacientes con insuficiencia renal moderada y grave (depuración de creatinina (CrCI) ≤ 50 mL/min) se debe reducir MOZOBIL® en un tercio para recibir una dosis de 0.16 mg/kg. Se espera exposición sistémica similar si la dosis es reducida un tercio en los pacientes con discapacidad renal moderada y grave comparada con los pacientes con función renal normal. La información clínica con este ajuste de dosis en pacientes con daño renal es limitada.
De acuerdo al aumento de la exposición con el aumento del peso del cuerpo, la dosis de MOZOBIL® no debe exceder 27 mg/dia si la CrCI < 50 mL/min.
La siguiente fórmula (Cockroft-Gault) puede ser usada para calcular la CrCI:
Hombres: Depuración de creatinina (mL/min)=
|
Peso (kg) x (140 - edad en años) |
|
72 x creatinina sérica (mg/dL) |
Mujeres: Depuración de creatinina (mL/min)= Multiplicar el valor de la fórmula anaerior x 0.85
No existe información suficiente para hacer recomendaciones de dosis en pacientes con hemodiálisis.
Poblaciones específicas:
Uso pediátrico: La eficacia y seguridad de MOZOBIL® en pacientes pediátricos (1 año a menores de 18 años) fueron estudiados en estudios abiertos, multicéntricos, clínicos controlados (Ver secciones REACCIONES SECUNDARIAS Y ADVERSAS Y FARMACOCINÉTICA Y FARMACODINAMIA).
Uso geriátrico: Del número total de sujetos en los dos estudios clinicos controlados de MOZOBIL®, 24% tenían 65 años o más.
No se observaron diferencias notables en la incidencia de reacciones adversas a medicamentos en pacientes mayores y más jóvenes.
Ya que MOZOBIL® se excreta principalmente por el riñón, no son necesarias modificaciones a la dosis en pacientes mayores con función renal normal. En general se debe tener cuidado en la selección de la dosis para pacientes mayores debido a la mayor frecuencia de función renal disminuida con edad avanzada. Se recomienda el ajuste de dosis en los pacientes ancianos con CrCl < 50 mL/min (Ver sección DOSIS Y VIA DE ADMINISTRACIÓN Y FARMACOLOGÍA).
MANIFESTACIONES Y MANEJO DE LA SOBREDOSIFICACIÓN O INGESTA ACCIDENTAL: Basados en datos limitados a dosis por arriba de la dosis recomendada de 0.24 mg/kg SC y hasta 0.48 mg/kg, la frecuencia de los trastornos gastrointestinales, reacciones vagales, hipotensión ortostática y/o síncope puede ser mayor. Los síntomas generalmente respondieron a los tratamientos (p. ej. antihistamínicos, corticoesteroides, hidratación u oxígeno suplementario), pero debido al potencial de estas reacciones, se deben tomar las precauciones apropiadas.
PRESENTACIÓN:
Caja con frasco ámpula con 1.2 mL e instructivo anexo.
RECOMENDACIONES SOBRE ALMACENAMIENTO:
Consérvese a no más de 25ºC.
LEYENDAS DE PROTECCIÓN:
Literatura exclusiva para el médico. Su venta requiere receta médica. No se deje al alcance de los niños. No se administre si el cierre ha sido violado. No se administre si la solución no es transparente, si contiene partículas en suspensión o sedimentos. Para usar una sola vez. Si no se administra todo el producto, deséchese el sobrante. No se use durante el embarazo o lactancia. Este medicamento deberá ser administrado únicamente por médicos especialistas en antineoplásicos o con experiencia en el tratamiento de la leucemia linfoblástica. MOZOBIL® no debe ser diluido ni mezclado con ningún otro medicamento. MOZOBIL® no debe considerarse como intercambiable con otro plerixafor, debido a que no se han establecido pruebas de bioequivalencia para los medicamentos huérfanos, por lo que implica un riesgo en términos de seguridad y/o eficacia para el paciente. No deberán mezclarse dos o más medicamentos con el mismo principio activo de diferenfe fabricante.
Reporte de sospechas de reacciones adversas al correo:
farmacovigilancia@cofepris.gob.mx
SANOFI-AVENTIS DE MÉXICO, S.A. de C.V.
Acueducto del alto Lerma No. 2,
Zona industrial Ocoyoacac,
C.P. 52740 Ocoyoacac, México, México.
193300EL870112


