METALYSE - Solución inyectable
Sustancia(s):
- Tenecteplasa
Presentaciones:
- 1 Caja, 1 Frasco(s) ámpula con liofilizado, 25 mg,
- 1 Caja, 1 Frasco(s) ámpula con liofilizado, 10 mL, 50 mg
- 1 Caja, 1 Frasco(s) ámpula con liofilizado, 8 mL, 40 mg
FORMA FARMACÉUTICA Y FORMULACIÓN:
El frasco ámpula con polvo liofilizado contiene:
METALYSE® 25 mg:
Tenecteplasa (5,000 U)
Excipiente cs
METALYSE® 40 mg:
Tenecteplasa (8,000 U)
Excipiente cs
METALYSE® 50 mg:
Tenecteplasa (10,000 U)
Excipiente cs
La jeringa prellenada con diluyente contiene:
METALYSE® 25 mg:
No se incluye jeringa con diluyente*
METALYSE® 40 mg:
Agua estéril 8 mL
Para uso inyectable
METALYSE® 50 mg:
Agua estéril 10 mL
Para uso inyectable
* Reconstituir con 5 ml de agua estéril para uso inyectable
Activador de plasminógeno tisular de origen ADN recombinante expresado en células de ovario de hámster chino (CHO). La solución reconstituida contiene 1,000 unidades (5 mg) de tenecteplasa por mL
La potencia de tenecteplasa se expresa en unidades (U) usando un estándar de referencia que es específico para tenecteplasa y no es comparable con las unidades utilizadas para otros agentes trombolíticos
INDICACIONES TERAPÉUTICAS:
METALYSE® 25 mg: está indicado en pacientes adultos para el tratamiento trombolítico del accidente cerebrovascular (ACV) isquémico agudo en las 4.5 horas siguientes al último momento en el que el paciente se encontraba bien y tras haber descartado hemorragia intracraneal.
METALYSE® 40 y 50 mg: está indicado en el tratamiento trombolítico del infarto agudo al miocardio (IAM). El tratamiento debe ser iniciado tan pronto sea posible después del inicio de los síntomas.
FARMACOCINÉTICA Y FARMACODINAMIA:
Código ATC: B01AD11.
Grupo farmacoterapéutico: Agentes antitrombóticos; Enzimas.
Farmacocinetica:
Absorción y distribución:
Tenecteplasa es una proteína recombinante administrada por vía intravenosa que activa el plasminógeno. Después de una administración en bolo IV de 30 mg de tenecteplasa en pacientes con infarto agudo de miocardio, la concentración plasmática inicialmente estimada fue 6.45 ± 3.60 μg/mL (media ± DE). La fase de distribución representa del 31% ± 22% al 69% ± 15% (media ± DE) del ABC total después de la administración de rangos de dosis de entre 5 y 50 mg.
Se obtuvieron datos sobre distribución tisular en estudios con tenecteplasa radiomarcada en ratas. El órgano principal al que se distribuyó la tenecteplasa fue el hígado. No se sabe si, y en qué medida, la tenecteplasa se une a las proteínas plasmáticas en seres humanos. El tiempo de residencia medio (TRM) en el organismo es de aproximadamente 1 hora y la media (± DE) del volumen de distribución en estado de equilibrio dinámico (Vss) fue de entre 6.3 ± 2 L y 15 ± 7 L.
Metabolismo:
La tenecteplasa se elimina de la circulación mediante la unión a receptores específicos en el hígado, seguido de un catabolismo con formación de pequeños péptidos. La unión a los receptores hepáticos, no obstante, es reducida en comparación con el tPA natural, lo que se traduce en una semivida prolongada.
Eliminación:
Después de la inyección de un bolo intravenoso único de tenecteplasa en pacientes con infarto agudo de miocardio, el antígeno tenecteplasa evidencia una eliminación bifásica del plasma. En el rango de dosis terapéutica, no se observa dependencia de la dosis en la depuración de tenecteplasa. La semivida dominante inicial es de 24 ± 5.5 (media ± DE) min, la cual es 5 veces más prolongada que la de tPA natural. La semivida terminal es 129 ± 87 min, y la depuración plasmática es 119 ± 49 mL/min.
El incremento del peso corporal tuvo relación con un incremento moderado de la depuración de tenecteplasa, y el aumento de edad tuvo correlación con una ligera reducción de la depuración. Las mujeres, por lo general, presentan una depuración menor que los hombres, pero ello puede explicarse por el peso corporal, que usualmente es menor en las mujeres.
Linealidad/no linealidad:
El análisis de la linealidad de la dosis en base al ABC sugirió que la tenecteplasa exhibe una farmacocinética no lineal en el rango de dosis estudiado, es decir, de 5 a 50 mg.
Poblaciones especiales:
Deterioro de la función renal y de la función hepática:
Dado que la eliminación de tenecteplasa se realiza por vía hepática, no se prevé que la disfunción renal afecte la farmacocinética de METALYSE®. Datos obtenidos en animales también lo confirman. Sin embargo, el efecto de la disfunción renal y hepática sobre la farmacocinética de tenecteplasa en los seres humanos no se ha investigado específicamente.
Farmacodinamia:
Mecanismo de acción:
La tenecteplasa es un activador del plasminógeno específico de fibrina recombinante, derivado a partir del t-PA (activador de plasminógeno) natural mediante modificaciones en tres sitios de la estructura proteica. Se une al componente fibrina del trombo (coágulo sanguíneo) y convierte selectivamente el plasminógeno unido al trombo en plasmina, la cual degrada la matriz de fibrina del trombo. La tenecteplasa posee una mayor especificidad por la fibrina y una mayor resistencia a la inactivación por inhibidor endógeno (PAI-1), en comparación con el t-PA natural.
Efectos farmacodinámicos:
Después de la administración de tenecteplasa, se ha observado un consumo de α2-antiplasmina dependiente de la dosis (el inhibidor de la plasmina de fase fluida), con el consecuente aumento en el nivel de producción de plasmina sistémica. Esta observación es compatible con el efecto deseado de la activación del plasminógeno. En estudios comparativos, se observó una reducción del fibrinógeno de menos del 15% y una reducción del plasminógeno de menos del 25% en sujetos tratados con la dosis máxima de tenecteplasa (10,000 U, correspondientes a 50 mg), en tanto que la alteplasa ocasionó una disminución de aproximadamente un 50% en los niveles de fibrinógeno y plasminógeno. No se detectó ninguna formación de anticuerpos clínicamente relevante a los 30 días.
Estudios clínicos:
METALYSE® 40 y 50 mg:
Para el tratamiento trombolítico del infarto agudo de miocardio:
Los datos de permeabilidad de estudios angiográficos de fase I y II sugieren que la tenecteplasa, administrada como bolo intravenoso único, es eficaz en la disolución de los coágulos sanguíneos de la arteria relacionada con el infarto en los sujetos con IAM, de forma dependiente de la dosis.
Estudio ASSENT 2:
Un estudio de mortalidad a gran escala (ASSENT 2) realizado en aproximadamente 17,000 pacientes demostró que la tenecteplasa es terapéuticamente equivalente a la alteplasa en términos de la reducción de la mortalidad (6.2% para ambos tratamientos a los 30 días) y que el uso de tenecteplasa está asociado con una incidencia significativamente más baja de hemorragias no intracraneales (26.4% vs. 28.9%, p=0.0003). La reducción del riesgo de sangrado es probable que esté relacionada con la mayor especificidad por la fibrina de la tenecteplasa y con el hecho de que su régimen está adaptado al peso del paciente.
Esto se traduce en una necesidad significativamente menor de transfusiones (4.3% vs. 5.5%, p=0.0002). La hemorragia intracraneal se produjo con una incidencia de 0.93% vs. 0.94% para la tenecteplasa y la alteplasa, respectivamente. En los 475 pacientes tratados más de 6 horas después de producido el evento, se observaron diferencias numéricas a favor de la tenecteplasa en lo que respecta a la tasa de mortalidad a los 30 días (4.3% vs. 9.6%), accidente cerebrovascular (0.4% vs. 3.3%) y HIC (0 % vs. 1.7%).
Estudio ASSENT 3:
El estudio ASSENT 3 estuvo destinado a optimizar el tratamiento antitrombótico concomitante de tenecteplasa, tanto en términos de una mejoría de las tasas de permeabilidad temprana como del mantenimiento de la reperfusión, fundamentalmente a fin de superar el efecto procoagulante paradójico debido a la liberación de la trombina atrapada que se produce cuando tiene lugar la lisis del coágulo. Se compararon tres regímenes antitrombóticos concomitantes diferentes en 6,095 pacientes: dosis completa de tenecteplasa + heparina no fraccionada (HNF) vs. dosis completa de tenecteplasa + heparina (enoxaparina) de bajo peso molecular (BPM) vs. media dosis de tenecteplasa + heparina no fraccionada + dosis completa de abciximab.
La HNF se utilizó de acuerdo con las recomendaciones de las guías de la AHA/ACC, las cuales están basadas en el peso corporal adaptado a un régimen de dosis baja, de la siguiente manera: un bolo único IV de 60 UI/kg (máximo 4,000 UI), inmediatamente seguido de una infusión intravenosa de 12 UI/kg/h (máximo 1,000 UI/h) durante las primeras 3 horas, y a partir de ese momento, de acuerdo con el monitoreo del TTPa, durante un máximo de 48 horas para mantener el TTPa en valores de entre 50 y 70 segundos.
Las tasas de mortalidad a los 30 días fueron del 6.0%, 5.4% y 6.6%, respectivamente; las tasas de hemorragias graves durante la internación hospitalaria (excepto HIC) del 2.16%, 3.04% y 4.32%, respectivamente; y las tasas de hemorragia intracraneal del 0.93%, 0.88% y 0.94%, respectivamente.
En el estudio ASSENT 3, el régimen recomendado por la ACC/AHA de heparina no fraccionada en dosis bajas ajustado por peso corporal, administrado de forma concomitante con tenecteplasa, evidenció una incidencia menor de sangrado sistémico, pero tasas similares de HIC en comparación con los regímenes posológicos de heparina no fraccionada más agresivos utilizados en el estudio ASSENT 2, sin pérdida de eficacia.
Estudio ASSENT 3 plus:
El estudio ASSENT 3 PLUS, un estudio satélite (complementario) del estudio ASSENT 3, fue diseñado para investigar el uso en etapa prehospitalaria. La eficacia y la seguridad del régimen de dosis completa de tenecteplasa + heparina no fraccionada vs. el régimen de dosis completa de tenecteplasa + heparina (enoxaparina) de bajo peso molecular (BPM) se evaluaron en 1,639 pacientes.
El diseño del estudio y la posología terapéutica utilizados son idénticos a los del estudio ASSENT 3. La terapia de reperfusión prehospitalaria con tenecteplasa y HNF o enoxaparina permitió el tratamiento dentro de las 2 horas del inicio de los síntomas en >50% de los pacientes con infarto de miocardio con elevación del segmento ST (IMEST).
En los estudios ASSENT 3 y ASSENT 3 PLUS, el tratamiento concomitante con enoxaparina tanto en etapa prehospitalaria como hospitalaria redujo la incidencia de complicaciones isquémicas en comparación con el tratamiento concomitante con HNF: la incidencia del criterio de valoración compuesto de eficacia a los 30 días (muerte, reinfarto, o isquemia refractaria) fue, respectivamente, de 11.4% vs. 15.4% en el estudio ASSENT 3, y de 14.2% vs. 17.4% en el estudio ASSENT 3 PLUS. No obstante, en el contexto del uso prehospitalario, la administración de tenecteplasa con enoxaparina en la dosis utilizada estuvo asociada con un aumento del riesgo de sangrado grave y de HIC en pacientes de más de 75 años.
Los datos de permeabilidad coronaria y los pocos datos de resultados clínicos disponibles indican que los pacientes con IAM han sido tratados con éxito más de 6 horas después de la aparición de los síntomas.
Estudio ASSENT 4 PCI:
El estudio ASSENT-4 PCI fue diseñado para demostrar si, en 4,000 pacientes con infarto de miocardio extenso, el pretratamiento con la dosis completa de tenecteplasa y un bolo único concomitante de hasta 4,000 UI de heparina no fraccionada, administrado antes de la intervención coronaria percutánea (ICP) primaria, a realizarse dentro de los 60 y 180 minutos subsiguientes, redunda en mejores resultados que la ICP primaria sola. Este estudio se interrumpió prematuramente, con 1,667 pacientes aleatorizados, debido a una mortalidad numéricamente superior en el grupo con ICP facilitada que recibió tenecteplasa. La incidencia del criterio de valoración primario, un compuesto de muerte o shock cardiogénico o insuficiencia cardiaca congestiva dentro de los 90 días, fue significativamente superior en el grupo que recibió el régimen exploratorio de tenecteplasa inmediatamente seguido de ICP: 18.6% (151/810) frente a 13.4% (110/819) en el grupo de ICP sola, p=0.0045. Esta diferencia significativa entre los grupos en lo que respecta al criterio de valoración primario a los 90 días ya estaba presente en la internación hospitalaria y a los 30 días. Numéricamente, todos los componentes del criterio de valoración compuesto clínico estuvieron a favor del régimen de ICP sola: muerte: 6.7% vs. 4.9%, p=0.14; shock cardiogénico: 6.3% vs. 4.8%, p=0.19; insuficiencia cardiaca congestiva: 12.0% vs. 9.2%, p=0.06; respectivamente. Las incidencias de los criterios de valoración secundarios de reinfarto y nueva revascularización del vaso culpable fueron significativamente superiores en el grupo pretratado con tenecteplasa: reinfarto: 6.1% vs. 3.7%, p=0.0279; nueva revascularización del vaso culpable: 6.6% vs. 3.4%, p=0.0041.
Los siguientes eventos adversos fueron más frecuentes en el grupo tratado con tenecteplasa antes de la ICP: hemorragia intracraneal: 1% vs. 0%, p=0.0037; accidente cerebrovascular: 1.8% vs. 0%, p <0.0001; sangrados graves: 5.6% vs. 4.4%, p=0.3118; sangrados menores: 25.3% vs. 19.0%, p=0.0021; transfusiones de sangre: 6.2% vs. 4.2%, p=0.0873; cierre vascular abrupto: 1.9% vs. 0.1%, p=0.0001.
Estudio STREAM:
El estudio STREAM fue diseñado para evaluar la eficacia y la seguridad de una estrategia fármaco-invasiva, de un tratamiento fibrinolítico temprano con tenecteplasa y un tratamiento antiplaquetario y anticoagulante adicional seguido de una angiografía dentro de las 6 a 24 horas subsiguientes o intervención coronaria de rescate frente a una estrategia de ICP primaria estándar.
La población de estudio consistió en pacientes con infarto agudo de miocardio con elevación del segmento ST tratados dentro de las 3 horas siguientes al inicio de los síntomas que no podían ser sometidos a una ICP primaria dentro del lapso de una hora posterior al primer contacto médico.
Para este estudio exploratorio se planeó un tamaño de la muestra de aproximadamente 1,000 pacientes por grupo de tratamiento. Después de que 382 pacientes habían sido enrolados (19.5% de la población del estudio prevista), la dosis de la inyección en bolo de tenecteplasa se redujo a la mitad para los pacientes de ≥75 años, debido a una mayor incidencia de hemorragia intracraneal (HIC) en dicho subgrupo.
Se aleatorizaron 1,892 pacientes a través de un sistema interactivo de respuesta de voz. El criterio de valoración primario, un compuesto de muerte, shock cardiogénico, insuficiencia cardiaca congestiva o reinfarto a los 30 días se observó en el 12.4% (116/939) de la rama tratada con la estrategia fármaco-invasiva frente a un 14.3% (135/943) en la rama de ICP primaria (riesgo relativo: 0.86 (0.68-1.09).
Los componentes individuales del criterio de valoración compuesto primario para la estrategia fármaco-invasiva frente a la ICP primaria, respectivamente, se observaron con las siguientes frecuencias:
|
Estrategia fármaco-invasiva (n=944) |
ICP primaria (n=948) |
P |
|
|
Compuesto de muerte, shock, insuficiencia cardiaca congestiva, o reinfarto Mortalidad por todas las causas Shock cardiogénico Insuficiencia cardiaca congestiva Reinfarto |
116/939 (12.4%) 43/939 (4.6%) 41/939 (4.4%) 57/939 (6.1%) 23/938 (2.5%) |
135/943 (14.3%) 42/946 (4.4%) 56/944 (5.9%) 72/943 (7.6%) 21/944 (2.2%) |
0.21 0.88 0.13 0.18 0.74 |
|
Mortalidad cardiaca |
31/939 (3.3%) |
32/946 (3.4%) |
0.92 |
La incidencia observada de hemorragias no HIC, graves y menores, fueron similares en ambos grupos:
|
Estrategia fármaco-invasiva (n=944) |
ICP primaria (n=948) |
P |
|
|
Sangrado grave no HIC |
61/939 (6.5%) |
45/944 (4.8%) |
0.11 |
|
Sangrado menor no HIC |
205/939 (21.8%) |
191/944 (20.2%) |
0.40 |
Incidencia de accidentes cerebrovasculares de todo tipo y hemorragias intracraneales
|
Estrategia fármaco-invasiva (n=944) |
ICP primaria (n=948) |
P |
|
|
Total de accidentes cerebrovasculares (todos los tipos) |
15/939 (1.6%) |
5/946 (0.5%) |
0.03a |
|
Hemorragia intracraneal Hemorragia intracraneal después de la enmienda al protocolo con la mitad de la dosis en pacientes de ≥75 años: |
9/939 (0.96%) 4/747 (0.5%) |
2/946 (0.21%) 2/758 (0.3%) |
0.04b 0.45 |
a Las incidencias en ambos grupos son las esperadas en los pacientes con IMEST tratados con fibrinolíticos o ICP primaria (según lo observado en estudios clínicos previos).
b La incidencia en el grupo de la estrategia fármaco-invasiva es la esperada para la fibrinólisis con METALYSE (según lo observado en estudios clínicos previos).
Ninguna de las diferencias entre los grupos que se presentan en las tablas anteriores alcanza el umbral de significación estadística, con excepción de la incidencia de accidentes cerebrovasculares de todo tipo y HIC; no obstante, las incidencias en el grupo de la estrategia fármaco-invasiva fueron similares a las observadas en estudios clínicos previos.
Tras la reducción de la dosis de tenecteplasa a la mitad en los pacientes de ≥75 años, no hubo más hemorragias intracraneales (0 de 97 pacientes) (IC del 95% 0.0-3.7) vs. 8.1% (3 de 37 pacientes) (IC del 95% 1.7-21.9) antes de la reducción de la dosis. Los límites del intervalo de confianza de las incidencias observadas antes y después de la reducción de la dosis se superponen.
En pacientes de ≥75 años, las incidencias observadas del compuesto del criterio de valoración de eficacia primario para la estrategia fármaco-invasiva y la ICP primaria fueron las siguientes: antes de la reducción de la dosis 11/37 (29.7%) (IC del 95% 15.9-47.0) vs. 10/32 (31.3%) (IC del 95%: 16.1-50.0); después de la reducción de la dosis: 25/97 (25.8%) (IC del 95% 17.4-35.7) vs. 25/88 (24.8%) (IC del 95% 19.3-39.0). En ambos grupos, los límites del intervalo de confianza de las incidencias observadas antes y después de la reducción de la dosis se superponen.
METALYSE® 25 mg:
Para el tratamiento trombolítico del accidente cerebrovascular isquémico agudo:
Estudio ACT:
El estudio “Alteplase compared to Tenecteplase” (AcT, alteplasa comparada con tenecteplasa) fue diseñado como un estudio pragmático, basado en estudios de registro, prospectivo, aleatorizado, abierto y controlado de tenecteplasa intravenosa comparada con alteplasa intravenosa, que tuvo por objetivo demostrar que tenecteplasa es no inferior a alteplasa en pacientes con ACV isquémico agudo cuando se utiliza en las 4.5 horas siguiente al último momento en el que el paciente se encontraba bien y que son elegibles para recibir trombólisis intravenosa de acuerdo con las guías actuales. Se alcanzó el criterio de valoración primario del estudio, ya que se demostró una no inferioridad clínicamente relevante con tenecteplasa 0.25 mg/kg (máx. 25 mg) frente a alteplasa 0.9 mg/kg (máx. 90 mg): 296 (36.9%) de 802 pacientes en el grupo de tenecteplasa y 266 (34.8%) de 765 pacientes en el grupo de alteplasa tuvieron un puntaje de la escala Rankin modificada de 0-1 a los 90 a 120 días (diferencia de riesgo no ajustada 2.1% [IC del 95%: -2.6 a 6.9], lo que cumple con el umbral pre-especificado de no inferioridad de -5%).
Los criterios de valoración clave de seguridad fueron hemorragia intracerebral sintomática, angioedema orolingual y sangrado extracraneal que requiriera transfusión sanguínea, todos dentro de las 24 horas posteriores a la administración de agentes trombolíticos, y mortalidad por todas las causas a 90 días.
No se observaron diferencias significativas en la tasa de hemorragia intracerebral sintomática a 24 horas. Las tasas de hemorragia intracraneal definida por diagnóstico por imágenes (que se evaluó en forma ciega con respecto al estado de los síntomas y a la asignación del tratamiento) no mostraron diferencias entre los dos grupos, y las tasas definidas con base en los métodos de diagnóstico por imágenes de hematoma parenquimatoso tipo 2 (es decir, hematoma que corresponde a ≥30% del infarto con un efecto de masa evidente) fueron similares a las tasas observadas de hemorragia intracerebral sintomática en el ensayo. No se produjeron diferencias significativas en la tasa de mortalidad a 90 días del tratamiento. Los eventos de angioedema orolingual y de sangrado periférico que requirió transfusión de sangre fueron poco frecuentes y similares en ambos grupos (Ver tabla 1).
Tabla 1. Incidencia de los criterios de valoración clave de seguridad en los grupos de tenecteplasa y alteplasa
|
Grupo de tenecteplasa |
Grupo de alteplasa |
Diferencia de riesgo (IC del 95%) |
|
|
Hemorragia intracerebral sintomática a 24 horas |
27/800 (3.4%) |
24/763 (3.2%) |
0.2 (-1.5 a 2.0) |
|
Hemorragia intracraneal identificada por diagnóstico por imágenes |
154/800 (19.3%) |
157/763 (20.6%) |
-1.3 (-5.3 a 2.6) |
|
Hemorragia extracraneal que requiere transfusión sanguínea |
6/800 (0.8%) |
6/763 (0.8%) |
0.0 (-0.9 a 0.8) |
|
Muerte dentro de los 90días de la aleatorización (n=1.554) |
122/796 (15.3%) |
117/758 (15.4%) |
-0,1 (-3.7 a 3.5) |
|
Angioedema orolingual |
9/800 (1.1%) |
9/763 (1.2%) |
-0.1 (-1.1 a 1.0) |
|
Hematoma parenquimatoso tipo 2 (hematoma que corresponde a ≥30% del infarto con efecto de masa evidente) |
21/800 (2.6%) |
18/763 (2.4%) |
0.3 (-1.3 a 1.8) |
Estudio EXTEND-IA TNK:
El estudio EXTEND-IA TNK se diseñó con el objetivo de evaluar si tenecteplasa es no inferior a alteplasa en la reperfusión al momento de la angiografía inicial cuando se administra dentro de las 4.5 horas del inicio del ACV isquémico en pacientes en quienes se planifica un tratamiento endovascular.
Los pacientes con ACV isquémico que tenían oclusión de la carótida interna, la arteria basilar o la arteria cerebral media y que eran aptos para someterse a una trombectomía fueron aleatorizados a recibir tenecteplasa 0.25 mg/kg o alteplasa 0.9 mg/kg dentro de las 4.5 horas posteriores al inicio de los síntomas. Hubo 101 pacientes en cada grupo de tratamiento. El criterio de valoración primario fue la reperfusión de más del 50% del territorio isquémico afectado o una ausencia de trombo que pudiera recuperarse al momento de la evaluación angiográfica inicial. Se evaluó primero la no inferioridad de tenecteplasa, seguida por la superioridad. Los criterios de valoración secundarios incluyeron el puntaje de la escala Rankin modificada a 90 días.
El criterio de valoración primario ocurrió en 22% de los pacientes tratados con tenecteplasa frente al 10% de los pacientes tratados con alteplasa (diferencia en la incidencia del 12%, IC del 95%: 2, 21; razón de incidencia: 2.2; IC del 95%: 1.1, 4.4; p=0.002 para la no inferioridad; p=0.03 para la superioridad).
En un análisis ordinal del puntaje de la escala de Rankin modificada a los 90 días, los pacientes en el grupo de tenecteplasa tuvieron una mediana de puntaje de 2 (rango intercuartilo de 0 a 3), lo que indicó una función significativamente mejor que la mediana de puntaje de 3 (rango intercuartilo de 1 a 5) que se observó en los pacientes del grupo alteplasa (cociente de probabilidades común 1.7; IC del 95 %, 1.0 a 2.8; p=0.04). No se observó diferencia significativa en la incidencia de la recuperación con independencia funcional (puntaje de la escala Rankin modificada 0 a 2 o ausencia de cambios en la capacidad funcional registrada al inicio) a los 90 días, lo que ocurrió en 65 de 101 pacientes (64%) en el grupo de tenecteplasa y en 52 de 101 pacientes (51%) en el grupo de alteplasa (razón de incidencia ajustada: 1,2; IC del 95%, 1,0 a 1,5; p = 0.06; cociente de probabilidades ajustado, 1,8; IC del 95%, 1,0 a 3,4; p = 0.06).
La proporción de puntajes de 0 a 1 en la escala Rankin modificada a los 90 días fue del 51% para el grupo de tenecteplasa y de 43% para el grupo de alteplasa (p=0.23).
Eventos de hemorragia intracraneal sintomática (HICs) se observó en el 1% de los pacientes en cada grupo. Hubo un total de 10 muertes (10%) en el grupo de tenecteplasa y 18 (18%) en el grupo de alteplasa, una diferencia no significativa en el análisis de regresión logística pre-especificado. La mayoría de las muertes se relacionó con la progresión del ACV grave (9 en el grupo de tenecteplasa y 14 en el grupo de alteplasa). Tenecteplasa 0.25 mg/kg mostró un perfil de seguridad similar a alteplasa 0.9 mg/kg.
Estudio EXTEND-IA TNK, parte 2:
El estudio EXTEND-IA TNK se diseñó con el objetivo de determinar si la dosis de 0.4 mg/kg de tenecteplasa generaba una mejoría segura en la reperfusión previo a la trombectomía endovascular, en comparación con la dosis de 0.25 mg/kg en pacientes con ACV isquémico con oclusión de grandes vasos (OGV).
Pacientes adultos con ACV isquémico por oclusión de la carótida interna intracraneal, la arteria basilar o la arteria cerebral media dentro de las 4.5 horas posteriores al inicio de los síntomas fueron incluidos, utilizando los criterios de elegibilidad estándar para la trombólisis IV Los pacientes fueron asignados aleatoriamente en una proporción 1:1 para recibir tenecteplasa 0.25 mg/kg o 0.4 mg/kg. El criterio de valoración primario fue la reperfusión de más del 50% del territorio isquémico afectado antes de la trombectomía, evaluado a través del consenso de 2 neurorradiólogos ciegos al tratamiento asignado.
La cantidad de participantes con más del 50% de reperfusión del territorio vascular previamente ocluido fue 29 de 150 (19.3%) en el grupo de 0.4 mg/kg frente a 29 de 150 (19.3%) en el grupo que recibió 0.25 mg/kg (diferencia de riesgos no ajustada: 0.0% [IC del 95%: -8.9%, 8.9%]; cociente de riesgos ajustado, 1.03 [IC del 95 %: 0.66, 1.61]; p=0.89). No se observaron diferencias significativas en ninguno de los 4 criterios de valoración funcionales entre los grupos de 0.4 mg/kg y 0.25 mg/kg ni en las muertes por todas las causas (26 [17%] frente a 22 [15%], diferencia de riesgos no ajustada, 2.7% [IC del 95% -5.6%, 11.0%]).
Se observó HICs en 7 pacientes (4.7%) en el grupo de 0.40 mg/kg y en 2 pacientes (1.3%) en el grupo de 0.25 mg/kg (diferencia de riesgos no ajustada 3.3% [IC del 95 %, -0.5, 7.2]; cociente de riesgos [RR, risk ratio], 3.50 [IC del 95%: 0.74, 16.62]; p=0.12). Se produjeron 26 muertes (17%) en el grupo de tenecteplasa 0.40 mg/kg y 22 muertes (15%) en el grupo de tenecteplasa 0.25 mg/kg (RR ajustado, 1.27 [IC del 95% 0.77, 2.11]; p=0.35).
Datos de la vida real:
En varios estudios no intervencionistas se comparó tenecteplasa (0.25 mg/kg) frente a alteplasa (0.9 mg/kg) en pacientes con ACV isquémico agudo con o sin oclusión de grandes vasos (OGV) dentro de las 4.5 horas posteriores al inicio de los síntomas. En estos estudios observacionales se informaron estimaciones ajustadas (o pareamiento por puntaje de propensión) de un total de >2,900 pacientes con ACV isquémico agudo (de estudios con más de 100 pacientes tratados con tenecteplasa), y se informó un perfil de seguridad y eficacia favorable o similar consistente con tenecteplasa en comparación con alteplasa intravenosa. Los criterios de valoración analizados incluyeron los resultados funcionales (puntaje de la escala de Rankin modificada a 3 meses), mortalidad por todas las causas, hemorragia intracraneal y hemorragia intracraneal sintomática, tasas de angioedema, tiempo desde el ingreso hospitalario hasta el tratamiento, tiempo desde el ingreso hospitalario hasta el alta hospitalaria, tiempo desde la obtención de imágenes a la trombólisis, tiempo desde la trombólisis a la punción arterial, y tiempo desde el inicio de los síntomas hasta la administración del tratamiento.
CONTRAINDICACIONES:
METALYSE® está contraindicado en:
– Pacientes con hipersensibilidad conocida al principio activo, tenecteplasa, a la gentamicina (un residuo traza del proceso de fabricación) o a cualquiera de los excipientes de la fórmula.
– Situaciones asociadas con riesgo de sangrado como:
• Trastorno hemorrágico significativo actual o dentro de los últimos 6 meses, diátesis hemorrágica conocida.
• Cualquier antecedente de lesión del sistema nervioso central (p. ej., neoplasia, aneurisma, cirugía intracraneal o de columna).
• Hipertensión arterial severa no controlada.
• Disfunción hepática severa, incluyendo insuficiencia hepática, cirrosis, hipertensión portal (várices esofágicas) y hepatitis activa.
• Enfermedad gastrointestinal ulcerosa activa.
• Aneurisma arterial y/o malformación arteriovenosa conocida.
• Neoplasma con aumento del riesgo de sangrado.
• Endocarditis bacteriana, pericarditis.
• Pancreatitis aguda.
METALYSE® 40 y 50 mg:
• Accidente cerebrovascular hemorrágico o accidente cerebrovascular de origen desconocido producido en cualquier momento
• Pacientes que reciben tratamiento anticoagulante eficaz (p. ej., antagonistas de la vitamina K con INR >1.3) (véase la sección Precauciones generales, subsección “Sangrado”)
• Cirugía mayor, biopsia de un órgano parenquimatoso o traumatismo significativo dentro de los últimos 2 meses (esto incluye cualquier traumatismo asociado con el IAM actual), traumatismo de cabeza o cráneo reciente.
METALYSE® 25 mg: Para el tratamiento trombolítico del accidente cerebrovascular isquémico agudo.
• Accidente cerebrovascular isquémico agudo sin déficit neurológico incapacitante.
• Antecedentes, evidencia o sospecha de hemorragia intracraneal, incluida hemorragia subaracnoidea.
• Pacientes que reciben tratamiento anticoagulante efectivo (p. ej. INR >1.7) (Ver sección Precauciones generales, subsección “Hemorragia”).
RESTRICCIONES DE USO DURANTE EL EMBARAZO Y LA LACTANCIA:
Embarazo:
Existe una cantidad limitada de datos sobre el uso de METALYSE® en mujeres embarazadas.
En estudios no clínicos realizados con tenecteplasa, se ha observado sangrado con mortalidad secundaria en las hembras preñadas como consecuencia de la actividad farmacológica conocida del fármaco, y en unos pocos casos se produjo aborto y reabsorción del feto (efectos que sólo se han observado con la administración de dosis repetidas). La tenecteplasa no se considera teratogénica (Ver sección Precaución en relación con efectos de carcinogénesis, mutagénesis, teratogénesis y sobre la fertilidad).
El beneficio del tratamiento debe ser sopesado frente a los potenciales riesgos durante el embarazo.
Lactancia:
Se desconoce si la tenecteplasa es excretada en la leche en los seres humanos. Se debe tener precaución cuando METALYSE® se administra a mujeres lactantes y debe decidirse si interrumpir la lactancia las primeras 24 horas tras la administración de METALYSE®.
REACCIONES SECUNDARIAS Y ADVERSAS:
Al igual que con otros agentes trombolíticos, la hemorragia es la reacción adversa más común asociada con el uso de METALYSE®. La hemorragia se puede producir en cualquier sitio o cavidad del cuerpo, y puede dar lugar a situaciones potencialmente mortales, o puede provocar discapacidad permanente o la muerte.
El tipo de hemorragia asociada con la terapia trombolítica se puede dividir en dos grandes categorías:
• Sangrado superficial, normalmente de los sitios de punción.
• Sangrado interno en cualquier sitio o cavidad del cuerpo.
Con la hemorragia intracraneal se pueden presentar síntomas neurológicos asociados, tales como somnolencia, afasia, hemiparesia y convulsiones.
Excepto por la reacción adversa al medicamento (RAM) de arritmias por reperfusión para la indicación de infarto agudo de miocardio y la frecuencia de la RAM de hemorragia intracraneal para la indicación de accidente cerebrovascular isquémico agudo, se considera que el perfil de seguridad de METALYSE® para las indicaciones de accidente cerebrovascular isquémico agudo e infarto agudo de miocardio es similar en base a los resultados demostrados en ensayos clínicos aleatorizados académicos publicados y a los datos de la vida real (RWE, Real World Evidence).
Tabla 2. Categorías de frecuencias.
|
Muy frecuentes |
≥1/10 |
|
Frecuentes |
≥1/100 – <1/10 |
|
Poco frecuentes |
≥1/1,000 – <1/100 |
|
Raros |
≥1/10,000 – <1/1,000 |
|
Muy raros |
<1/10,000 |
|
Desconocidos |
No pueden estimarse a partir de los datos disponibles |
Reacciones adversas para las indicaciones de infarto agudo de miocardio y ACV isquémico agudo:
Tabla 3. Frecuencias de las Reacciones adversas.
|
MedDRA Clasificación por sistema y órgano |
Reacciones adversas a tenecteplasa según terminología de la CCDS |
Categorías de frecuencia según la Guía SmPC de la UE |
|
Trastornos del sistema inmunológico |
Reacción anafilactoide – Exantema – Urticaria – Broncoespasmo – Edema laríngeo |
Raros |
|
Trastornos oculares |
Hemorragia ocular |
Poco frecuentes |
|
Trastornos cardiacos |
Hemorragia del pericardio |
Raros |
|
Trastornos vasculares |
Hemorragia |
Muy frecuentes |
|
Embolia |
Raros |
|
|
Trastornos respiratorios, torácicos y mediastínicos |
Epistaxis |
Frecuentes |
|
Hemorragia pulmonar |
Raros |
|
|
Trastornos gastrointestinales |
Hemorragia gastrointestinal: – Hemorragia gástrica – Hemorragia de úlcera gástrica – Hemorragia rectal – Hematemesis – Melena – Hemorragia bucal |
Frecuentes |
|
Náuseas |
Desconocidos |
|
|
Vómitos |
Desconocidos |
|
|
Hemorragia retroperitoneal: - Hematoma retroperitoneal |
Poco frecuentes |
|
|
Trastornos de la piel y del tejido subcutáneo |
Equimosis |
Frecuentes |
|
Trastornos renales y urinarios |
Hemorragia urogenital: – Hematuria – Hemorragia de las vías urinarias |
Frecuentes |
|
Trastornos generales y afecciones del sitio de administración |
Hemorragia en el lugar de la inyección, hemorragia en el sitio de la punción |
Frecuentes |
|
Exploraciones complementarias |
Descenso de la presión arterial |
Raros |
|
Aumento de la temperatura corporal |
Desconocidos |
|
|
Lesiones, intoxicaciones y complicaciones relacionados a procedimientos |
Embolia grasa |
Desconocidos |
|
Procedimientos médicos y quirúrgicos |
Transfusión |
Desconocidos |
Reacciones adversas para la indicación de infarto agudo de miocardio:
Tabla 4. Frecuencias de las reacciones adversas (IAM únicamente).
|
MedDRA Clasificación |
Reacciones adversas a tenecteplasa según |
Categorías de frecuencia según la Guía SmPC de la UE |
|
Trastornos del sistema nervioso |
Hemorragia intracraneal: – Hemorragia cerebral – Hematoma cerebral – Accidente cerebrovascular hemorrágico – Transformación hemorrágica de accidente cerebrovascular – Hematoma intracraneal – Hemorragia subaracnoidea |
Poco frecuentes |
|
Trastornos cardiacos |
Arritmias de reperfusión: – Asistolia – Arritmia idioventricular acelerada – Arritmia – Extrasístoles – Fibrilación auricular – Bloqueo auriculoventricular de primer grado – Bloqueo auriculoventricular completo – Bradicardia – Taquicardia – Arritmia ventricular – Fibrilación ventricular – Taquicardia ventricular |
Poco frecuentes |
Reacciones adversas para la indicación de ACV isquémico agudo:
Tabla 5. Frecuencias de las reacciones adversas (ACV isquémico agudo).
|
MedDRA Clasificación por sistema y órgano |
Reacciones adversas a tenecteplasa según terminología de la CCDS |
Categorías de frecuencia según la Guía SmPC de la UE |
|
Trastornos del sistema nervioso |
Hemorragia intracraneal – Hemorragia cerebral – Hematoma cerebral – Accidente cerebrovascular hemorrágico – Transformación hemorrágica de accidente cerebrovascular – Hematoma intracraneal – Hemorragia subaracnoidea |
Muy frecuentes |
PRECAUCIONES EN RELACIÓN CON EFECTOS DE CARCINOGÉNESIS, MUTAGÉNESIS, TERATOGÉNESIS Y SOBRE LA FERTILIDAD:
Con respecto a la indicación y a la administración de dosis únicas en los seres humanos, los estudios de toxicidad reproductiva se limitaron al conejo, dado que es una especie sensible. La tenecteplasa no indujo teratogenia alguna. La administración de dosis repetidas provocó sangrado con mortalidad secundaria en las hembras preñadas. En unos pocos casos se produjeron abortos y reabsorción del feto. No se observaron efectos tras la administración de una dosis única de tenecteplasa.
Para esta clase de proteínas recombinantes no se espera mutagenia ni carcinogenia, con lo cual no fue necesario realizar estudios de genotoxicidad y carcinogenicidad.
No se observó irritación local de los vasos sanguíneos tras la administración intravenosa, intraarterial o paravenosa de la formulación final de tenecteplasa.
No hay datos clínicos ni estudios preclínicos disponibles sobre la fertilidad en relación con la tenecteplasa (METALYSE®).
INTERACCIONES MEDICAMENTOSAS Y DE OTRO GÉNERO:
No se ha realizado ningún estudio formal de interacción con METALYSE® y los productos medicinales administrados habitualmente a pacientes con IAM.
METALYSE® no es compatible con la solución de dextrosa.
No se debe de adicionar otro medicamento a la solución de METALYSE® o a la línea de infusión cuando se esté administrando éste.
Fármacos que afectan la coagulación/función plaquetaria:
METALYSE® 40 y 50 mg:
Los productos medicinales que afectan la coagulación o aquellos que alteran la función plaquetaria pueden aumentar el riesgo de sangrado antes, durante o después del tratamiento con METALYSE®. (Ver sección Contraindicaciones).
METALYSE® 25 mg: Los medicamentos que afectan la coagulación o aquellos que alteran la función plaquetaria pueden aumentar el riesgo de sangrado y por lo tanto, se deben evitar en las primeras 24 horas posteriores al tratamiento con METALYSE® del ACV isquémico agudo; (Ver sección Contraindicaciones).
Inhibidores de la ECA:
El tratamiento concomitante con inhibidores de la ECA puede aumentar el riesgo de sufrir una reacción de hipersensibilidad (Ver sección Precauciones Generales).
Otras especialidades farmacéuticas:
METALYSE® 40 y 50 mg:
El análisis de datos de más de 12,000 pacientes tratados durante estudios de fases I, II y III no reveló interacciones clínicamente relevantes con especialidades farmacéuticas que se utilizan comúnmente en pacientes con IAM y que se usan de forma concomitante con METALYSE®.
METALYSE® 25 mg:
Ensayos aleatorizados académicos publicados con más de 2,000 pacientes tratados con tenecteplasa no mostraron interacciones clínicamente relevantes con otras especialidades farmacéuticas que se utilizan comúnmente en pacientes con ACV isquémico agudo.
ALTERACIONES EN LOS RESULTADOS DE PRUEBAS DE LABORATORIO:
Los resultados de las pruebas de laboratorio relacionadas con los tiempos de sangrado y de coagulación pueden sufrir modificación durante el periodo de aplicación de METALYSE® tal como sucede con la terapia trombolítica y fibrinolítica en general.
La administración de dosis única intravenosa en ratas, conejos y perros resultó en alteraciones dosis-dependientes y reversibles de los parámetros de coagulación con hemorragia local en el sitio de la inyección, que fue atribuido como consecuencia del efecto farmacodinámico de la tenecteplasa. Estudios de toxicidad con dosis múltiple en ratas y perros confirmaron las observaciones mencionadas previamente, pero la duración del estudio estuvo limitada a dos semanas por formación de anticuerpos a la proteína humana tenecteplasa, que resultó en anafilaxia.
Datos farmacológicos de seguridad en monos Cynomolgus reveló reducción de la tensión arterial seguida de cambios transitorios en el electrocardiograma, pero esto ocurrió en exposiciones que fueron considerablemente mayores a la exposición clínica.
PRECAUCIONES GENERALES:
Se debe elegir cuidadosamente la presentación adecuada del producto tenecteplasa que se encuentre en línea con la indicación. Se prevé que METALYSE® 25 mg se utilice únicamente en casos de accidente cerebrovascular isquémico agudo y que METALYSE® 40 mg o 50 mg se utilicen únicamente en casos de infarto agudo de miocardio.
METALYSE® debe ser recetado por médicos con experiencia en el uso de tratamiento trombolítico y con instalaciones para monitorizar su uso. Esto no excluye el uso prehospitalario de METALYSE®. Al igual que con otros trombolíticos, se recomienda que cuando se administre METALYSE® se encuentre disponible medicación y equipamiento de resucitación en todo momento.
METALYSE® 25 mg: El tratamiento debe ser realizado bajo la responsabilidad de un médico capacitado y con experiencia en cuidados neurológicos. Para verificar la indicación a tratar, las medidas de diagnóstico remoto pueden considerarse adecuadas (Ver sección Dosis y vía de administración, Tratamiento trombolítico del accidente cerebrovascular isquémico agudo).
Trazabilidad:
A fin de mejorar la trazabilidad de los productos de origen biotecnológico, el nombre comercial y el número de lote del producto administrado deben registrarse claramente en la historia clínica del paciente.
Hemorragia:
La complicación más frecuente observada durante el tratamiento con METALYSE® es el sangrado. El uso concomitante de otros principios activos que afecten la coagulación o la función plaquetaria (p. ej., la heparina) puede contribuir al sangrado (Ver sección Contraindicaciones).
Dado que la fibrina es lisada durante el tratamiento con METALYSE®, se puede presentar sangrado en los sitios de punción recientes. Por lo tanto, la terapia trombolítica requiere de una cuidadosa atención de todos los posibles sitios de sangrado (lo que incluye los lugares donde se haya realizado inserción de catéteres, punción arterial o venosa, disección o punción por aguja). Durante el tratamiento con METALYSE® se debe evitar el uso de catéteres rígidos e inyecciones intramusculares, así como también toda manipulación del paciente que no sea imprescindible.
En el caso de que se produzca un sangrado serio, en particular hemorragia cerebral, debe interrumpirse de inmediato la administración concomitante de heparina. Debe considerarse la administración de protamina si se ha administrado heparina dentro de las 4 horas anteriores al inicio del sangrado. En un número reducido de casos, es posible que el paciente no responda a estas medidas conservadoras; en dicha instancia, puede estar indicado el uso criterioso de productos de transfusión. Se debe considerar la transfusión de crioprecipitado, plasma fresco congelado y plaquetas, repitiendo las evaluaciones clínicas y de laboratorio luego de cada administración. Para la infusión de crioprecipitado es deseable un nivel de fibrinógeno objetivo de 1g/L. También se deben tomar en consideración los agentes antifibrinolíticos.
El uso del tratamiento con METALYSE® debe ser evaluado cuidadosamente a fin de asegurar que los potenciales riesgos de sangrado estén debidamente compensados por los beneficios esperados en las siguientes situaciones:
• Peso corporal bajo <50 kg.
• Pacientes que reciben tratamiento con anticoagulantes orales; puede considerarse el uso de METALYSE® cuando las pruebas pertinentes muestren que no existe una actividad anticoagulante clínicamente relevante.
METALYSE® 40 y 50 mg:
• Reanimación cardiopulmonar prolongada (>2 minutos) o traumática, o masaje cardiaco.
• Inyección intramuscular reciente o traumatismos menores recientes, como biopsias, punción de vasos mayores.
• Antecedentes de accidente cerebrovascular o ataque isquémico transitorio (AIT).
• Presión arterial sistólica >160 mmHg, (Ver sección Contraindicaciones)
• Sangrado gastrointestinal o urogenital reciente (en los últimos 10 días).
• Edad avanzada, es decir, pacientes de 75 años o más.
METALYSE® 25 mg:
• Inyección intramuscular reciente o traumatismos menores recientes, como biopsias, punción de vasos mayores, masaje cardiaco para reanimación.
Las hemorragias intracerebrales representan el evento adverso más frecuente (observadas en hasta aproximadamente 19% de los pacientes). Sin embargo, esto no ha producido un aumento en la morbilidad o mortalidad generales.
El riesgo de hemorragia intracraneal en pacientes con accidente cerebrovascular isquémico agudo puede aumentar con el uso de METALYSE®. Esto aplica específicamente a los siguientes casos:
• Todas las situaciones que involucran un alto riesgo de hemorragia, con inclusión de las enumeradas en la sección VI. Contraindicaciones;
• Demora en el inicio del tratamiento;
• Los pacientes pretratados con ácido acetilsalicílico (AAS) pueden tener un mayor riesgo de hemorragia intracerebral, particularmente si se demora el tratamiento con METALYSE®;
• En comparación con los pacientes más jóvenes, los pacientes de edad avanzada (más de 80 años) pueden tener resultados ligeramente peores independientemente del tratamiento y un riesgo incrementado de hemorragia intracerebral cuando son tratados con trombólisis. En general, la relación riesgo-beneficio de la trombólisis en los pacientes de edad avanzada sigue siendo positiva. En los pacientes con accidente cerebrovascular isquémico agudo (ACV isquémico agudo), la trombólisis debe evaluarse en función de la relación riesgo-beneficio en cada caso.
• El tratamiento no debe iniciarse más de 4.5 horas después de la aparición de los síntomas, dado que en este caso la relación riesgo-beneficio es desfavorable, principalmente debido a lo siguiente:
° Los efectos positivos del tratamiento disminuyen con el tiempo;
° Particularmente en los pacientes que recibieron tratamiento previo con AAS, la tasa de mortalidad se incrementa;
° Existe un mayor riesgo de hemorragia sintomática.
Hipersensibilidad:
METALYSE® 40 y 50 mg:
No se ha observado formación sostenida de anticuerpos a la molécula de tenecteplasa tras el tratamiento. Sin embargo, no existe experiencia sistemática sobre la readministración de METALYSE®.
Las reacciones anafilactoides asociadas con la administración de METALYSE® son eventos raros en términos de frecuencia, y pueden ser causadas por hipersensibilidad al principio activo tenecteplasa, a la gentamicina (residuo traza del proceso de fabricación) o a cualquiera de los excipientes. Si se produce una reacción anafilactoide, debe interrumpirse la inyección y se debe iniciar el tratamiento apropiado.
METALYSE® 25 mg:
Las reacciones de hipersensibilidad mediadas por el sistema inmunitario asociadas a la administración de METALYSE® pueden ser causadas por el principio activo tenecteplasa, la gentamicina (una traza residual del proceso de fabricación), o cualquiera de los excipientes (Ver sección Contraindicaciones).
No se ha observado formación sostenida de anticuerpos a la molécula de tenecteplasa tras el tratamiento. Sin embargo, no existe experiencia sobre la readministración de METALYSE®.
También existe un riesgo de que se produzcan reacciones de hipersensibilidad mediadas por un mecanismo no inmunitario.
El angioedema representa la reacción de hipersensibilidad más frecuentemente informada con METALYSE®. Este riesgo puede aumentar en la indicación de accidente cerebrovascular isquémico agudo y/o por el tratamiento concomitante con inhibidores de la enzima convertidora de angiotensina (ECA).
Se debe monitorear a los pacientes tratados con METALYSE® a fin de detectar casos de angioedema durante la administración y por hasta 24 horas después de ella.
En el caso de producirse una reacción de hipersensibilidad severa (p. ej., angioedema), debe iniciarse de inmediato el tratamiento adecuado, que puede incluir la intubación.
Tromboembolia:
El uso de METALYSE® puede aumentar el riesgo de eventos tromboembólicos en pacientes con trombos existentes, p. ej., trombo cardiaco izquierdo (estenosis mitral o fibrilación auricular, etc.).
Intervención coronaria:
Traslado a un establecimiento de intervención coronaria para la intervención coronaria percutánea (ICP) complementaria:
METALYSE® 40 y 50 mg: Los pacientes que reciban METALYSE® como tratamiento de reperfusión coronaria primaria deben ser trasladados sin demora a un establecimiento que cuente con los recursos y el equipamiento necesario para realizar angiografías e intervenciones coronarias dentro de un plazo de 6 a 24 horas o antes si estuviera medicamente indicado (Ver sección Farmacocinética y Farmacodinamia).
Intervención coronaria percutánea (ICP) primaria:
Si está programada una ICP primaria de acuerdo con las guías actuales de tratamiento pertinentes, METALYSE® no se debe administrar como se administró en el estudio ASSENT-4 PCI (Ver sección Farmacocinética y Farmacodinamia).
Arritmias:
La trombólisis coronaria puede dar lugar a arritmias asociadas con la reperfusión.
Las arritmias por reperfusión pueden provocar un paro cardiaco, pueden ser potencialmente mortales y pueden requerir el uso de terapias antiarrítmicas convencionales.
Antagonistas de la glucoproteína IIb/IIIa:
El uso concomitante de antagonistas de la GPIIb/IIIa aumenta el riesgo de sangrado.
Control de la presión arterial:
METALYSE® 25 mg:
Se deben tratar rápida y agresivamente los casos de presión arterial (PA) sistólica > 180 mmHg, PA diastólica > 105 mmHg o hipertensión arterial no controlada con el fin de minimizar las demoras hasta el inicio de la trombólisis. Es necesario controlar la PA durante hasta 24 horas posteriores al tratamiento con tenecteplasa. Se recomienda tratamiento antihipertensivo intravenoso si la PA sistólica es > 180 mmHg o la PA diastólica es > 105 mmHg.
Grupos especiales de pacientes con relación beneficio-riesgo reducida:
La relación riesgo-beneficio beneficio-riesgo del tratamiento trombolítico se considera menos favorable en pacientes que han experimentado un accidente cerebrovascular (ACV) previo o en quienes tienen diabetes no controlada, aunque se considera que continúa siendo positiva en estos pacientes.
Se debe considerar cuidadosamente la relación riesgo-beneficio, beneficio-riesgo de la administración de METALYSE® en pacientes con ACV isquémico agudo que presentan las siguientes condiciones:
• Síntomas que hayan mejorado rápidamente.
• Infartos extensos (p. ej., NIHSS >25).
• Convulsiones al inicio del ACV.
• Antecedentes recientes de ACV previo, o traumatismos de cabeza o columna vertebral graves, o cirugía mayor (como cirugía cardiaca, torácica, abdominal u ortopédica).
• Tiempo de tromboplastina parcial activado (TTPa) elevado al momento de la presentación.
• Recuento de plaquetas menor a 100,000/mm3.
• Hipertensión resistente al tratamiento antihipertensivo hiperagudo agresivo, que no permite que se alcance o se mantenga una presión arterial objetivo inferior a 180/105 mmHg.
• Glucemia <2.7 mmol/L o >22.2 mmol/L.
En los pacientes con accidente cerebrovascular, la probabilidad de un resultado favorable disminuye cuanto mayor es el tiempo transcurrido desde el último momento en el que el paciente se encontraba bien hasta el tratamiento trombolítico, con el aumento de la edad, el aumento de la gravedad del accidente cerebrovascular y el aumento de los niveles de glucemia al momento del ingreso, mientras que la probabilidad de discapacidad severa y muerte o sangrado intracraneal sintomático aumenta, independientemente del tratamiento.
Edema cerebral:
La reperfusión del área afectada por la isquemia puede inducir edema cerebral en la zona infartada.
Población pediátrica:
No se cuenta con datos de seguridad y eficacia en niños menores de 18 años de edad para METALYSE®. Por lo tanto, no se recomienda el uso de METALYSE® en niños menores de 18 años de edad.
Excipientes:
METALYSE® contiene polisorbato 20.
Este medicamento contiene 3.2 mg de polisorbato 20 en cada vial de 40 mg, y 4.0 mg de polisorbato 20 en cada vial de 50 mg. Los polisorbatos pueden causar reacciones alérgicas.
DOSIS Y VÍA DE ADMINISTRACIÓN:
Vía de administración: Intravenosa.
La solución reconstituida debe administrarse por vía intravenosa y utilizarse inmediatamente después de su reconstitución. La dosis requerida se debe administrar como bolo intravenoso único a lo largo de 5 a 10 segundos.
METALYSE® 40 y 50 mg: para el tratamiento trombolítico del infarto agudo de miocardio.
METALYSE® se debe administrar tan pronto como sea posible después del inicio de los síntomas en función del peso corporal, con una dosis máxima de 10,000 unidades (50 mg de tenecteplasa). El volumen requerido para administrar la dosis correcta puede calcularse a partir del siguiente esquema (Ver Tabla 6).
Tabla 6. METALYSE 40 y 50 mg - Esquema para el cálculo del volumen para la administración de la dosis correcta.
|
Categoría del peso corporal de los pacientes (kg) |
Tenecteplasa (U) |
Tenecteplasa (mg) |
Volumen correspondiente de solución reconstituida (mL) |
|
<60 |
6,000 |
30 |
6 |
|
≥60 a <70 |
7,000 |
35 |
7 |
|
≥70 a <80 |
8,000 |
40 |
8 |
|
≥80 a <90 |
9,000 |
45 |
9 |
|
≥90 |
10,000 |
50 |
10 |
Tratamiento complementario:
Se recomienda tratamiento complementario antitrombótico de conformidad con las guías internacionales vigentes para el manejo de pacientes con infarto de miocardio con elevación del segmento ST.
Para información sobre intervención coronaria, (Ver sección Precauciones Generales).
METALYSE® 25 mg: Para el tratamiento trombolítico del accidente cerebrovascular isquémico agudo.
METALYSE® debe administrarse a la mayor brevedad posible en las 4.5 horas siguientes al último momento en el que el paciente se encontraba bien y tras haber descartado hemorragia intracraneal mediante técnicas de diagnóstico por imágenes adecuadas; (Ver sección VII. Precauciones Generales). El efecto del tratamiento depende del tiempo de administración; por lo tanto, cuanto antes se inicie el tratamiento, mayores son las probabilidades de un resultado favorable.
METALYSE® debe administrarse en base al peso corporal, con una dosis máxima única de 5,000 unidades (25 mg) de tenecteplasa. Se debe evaluar cuidadosamente la relación riesgo-beneficio del tratamiento con tenecteplasa en pacientes que pesan 50 kg o menos debido a que los datos disponibles son limitados.
El volumen requerido para administrar la dosis total correcta puede calcularse a partir del siguiente esquema (Ver Tabla 7):
Tabla 7. METALYSE 25 mg - Esquema para el cálculo del volumen para la administración de la dosis correcta.
|
Categoría del peso corporal de los pacientes (kg) |
Tenecteplasa (U) |
Tenecteplasa (mg) |
Volumen correspondiente de solución reconstituida (mL) |
|
<60 |
3,000 |
15.0 |
3.0 |
|
≥60 a <70 |
3,500 |
17.5 |
3.5 |
|
≥70 a <80 |
4,000 |
20.0 |
4.0 |
|
≥80 a <90 |
4,500 |
22.5 |
4.5 |
|
≥90 |
5,000 |
25.0 |
5.0 |
Tratamiento complementario:
No se ha evaluado de manera suficiente la seguridad y la eficacia de este régimen con la administración concomitante de heparina o antiagregantes plaquetarios tales como el ácido acetilsalicílico durante las primeras 24 horas posteriores al tratamiento con METALYSE®. Por lo tanto, debe evitarse la administración de heparina intravenosa o de antiagregantes plaquetarios tales como el ácido acetilsalicílico en las primeras 24 horas luego del tratamiento con METALYSE® debido al mayor riesgo de hemorragias.
Si se requiere heparina para otras indicaciones, la dosis no debe superar las 10,000 UI por día, administrada por vía subcutánea.
Instrucciones de uso:
METALYSE® 40 y 50 mg:
Instrucciones para su reconstitución:
|
|
Abrir el recipiente del adaptador. Revise que la tapa del frasco ámpula se encuentre cerrada. Retirar el tapón de la jeringa. Retirar la tapa de plástico del frasco ámpula. |
|
|
Enroscar firmemente la jeringa prellenada en el adaptador para lograr un cierre hermético. |
|
|
Penetrar el tapón del frasco ámpula a la mitad con la punta del adaptador. |
|
|
Adicionar el agua estéril para uso inyectable presionando el émbolo de la jeringa lentamente hacia abajo para evitar formación de espuma. |
|
|
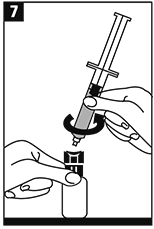
Reconstituir girando suavemente el frasco ámpula. |
|
|
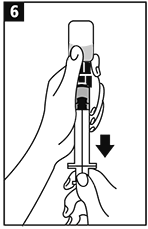
Invertir el frasco ámpula/jeringa y transferir el volumen apropiado de la solución a la jeringa de acuerdo con las instrucciones de dosificación. |
|
|
Retirar la jeringa del adaptador del frasco ámpula. La solución ahora está lista para la inyección IV en bolo. |
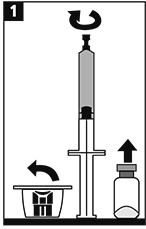
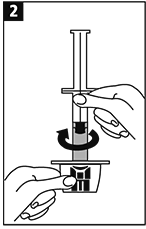

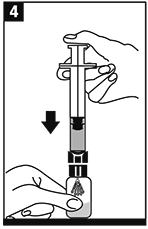

METALYSE® se debe reconstituir agregando el volumen completo de agua estéril para uso inyectable de la jeringa precargada al frasco ámpula que contiene el polvo liofilizado para inyección.
1. El volumen requerido para administrar la dosis correcta se debe calcular a partir de la tabla (Ver sección Dosis y vía de administración. Tabla 3).
2. Revise que la tapa del frasco ámpula esté aún intacta.
3. Retire la tapa del frasco ámpula.
4. Retire el tapón de la jeringa, luego enrosque inmediatamente la jeringa prellenada en el adaptador del frasco y penetre la tapa del frasco ámpula en el centro con la punta del adaptador del mismo.
5. Agregue el agua estéril para uso inyectable al frasco ámpula empujando el émbolo de la jeringa lentamente hacia abajo para evitar hacer espuma.
6. Reconstituir girándolo suavemente.
7. La preparación reconstituida es una solución transparente entre incolora y amarillo pálido. Solo se debe administrar una solución sin partículas.
8. Inmediatamente antes de la administración de la solución, invierta el frasco ámpula con la jeringa aún ligada, de tal manera que la jeringa esté debajo del frasco ámpula.
9. Saque el volumen apropiado de solución reconstituida de METALYSE® a la jeringa, basado en el peso del paciente.
10. Desconecte la jeringa del frasco ámpula.
11. METALYSE® puede utilizarse en una vía intravenosa preexistente, que solamente se haya utilizado para la administración de solución de cloruro de sodio al 0.9%. METALYSE® no se debe mezclar con otros fármacos, ni en el mismo vial de infusión ni en la misma vía venosa (ni siquiera con heparina).
12. METALYSE® debe ser administrado al paciente por vía intravenosa en aproximadamente 5 a 10 segundos. No se debe utilizar una vía que contenga dextrosa.
13. Se debe hacer un lavado de la vía después de la inyección de METALYSE para una correcta administración.
14. Toda solución no utilizada debe desecharse.
15. Alternativamente, la reconstitución se puede realizar con aguja en lugar del adaptador del vial incluido.
METALYSE® 25 mg:
Instrucciones para su reconstitución:
METALYSE® se debe reconstituir agregando el volumen adecuado de agua estéril para uso inyectable al frasco ámpula que contiene el polvo liofilizado para la solución inyectable por medio de una aguja y una jeringa (no se proporcionan en esta presentación).
1. Retire la tapa del frasco ámpula.
2. Llene una jeringa con 5 mL de agua estéril para uso inyectable y penetre el tapón del frasco ámpula en el centro con la aguja.
3. Agregue toda el agua estéril para uso inyectable al vial presionando suavemente el émbolo de la jeringa hacia abajo para evitar que se forme espuma.
4. Mantenga la jeringa unida al frasco ámpula y reconstituya la solución girando el frasco ámpula suavemente.
5. La preparación reconstituida es una solución de incolora a ligeramente amarilla y límpida. Solo se debe utilizar la solución si se observa límpida y sin partículas.
6. Inmediatamente antes de administrar la solución, invierta el frasco ámpula con la jeringa aún insertada, de modo tal que la jeringa quede debajo del frasco ámpula.
7. Transfiera el volumen adecuado de la solución reconstituida de METALYSE® a la jeringa en función del peso del paciente (Ver sección Dosis y vía de administración. Tabla 7)
8. Para la administración de METALYSE® puede utilizarse en una vía intravenosa preexistente, que solamente se haya utilizado para la administración de solución de cloruro de sodio al 0.9%. METALYSE® no se debe mezclar con otros fármacos, ni en el mismo vial de infusión ni en la misma vía venosa (ni siquiera con heparina)
9. METALYSE® debe ser administrado al paciente como única dosis por vía intravenosa en un lapso de entre 5 a 10 segundos. No se debe administrar utilizando una vía que contenta dextrosa dado que METALYSE® es incompatible con la solución de dextrosa.
10. Se debe hacer un lavado de la vía después de la inyección de METALYSE® para una administración adecuada.
11. Toda solución no utilizada debe descartarse.
MANIFESTACIONES Y MANEJO DE LA SOBREDOSIFICACIÓN O INGESTA ACCIDENTAL:
En caso de sobredosis, puede haber un mayor riesgo de sangrado. En caso de sangrado severo prolongado, se deberá considerar la terapia de reemplazo.
PRESENTACIONES:
Caja de cartón con un frasco ámpula con 25 mg (5,000 U) de polvo liofilizado e instructivo anexo.
Caja de cartón con un frasco ámpula con 40 mg (8,000 U) de polvo liofilizado, una jeringa prellenada con 8 mL de agua estéril para uso inyectable y un adaptador e instructivo anexo.
Caja de cartón con un frasco ámpula con 50 mg (10,000 U) de polvo liofilizado, una jeringa prellenada con 10 mL de agua estéril para uso inyectable y un adaptador e instructivo anexo.
RECOMENDACIONES SOBRE ALMACENAMIENTO:
Vida útil: 36 meses.
Consérvese a no más de 30 °C. Protéjase de la luz.
LEYENDAS DE PROTECCIÓN:
Literatura exclusiva para médicos. Su venta requiere receta médica. No se deje al alcance de los niños. No se use durante el embarazo o lactancia, ni en menores de 18 años. Solo deberá ser administrado por médicos especialistas. Hecha la mezcla, adminístrese de inmediato y deséchese el sobrante.
Reporte las sospechas de reacción adversa al correo: farmacovigilancia@cofepris.gob.mx y
farmacovigilancia.mex@boehringer-ingelheim.com
Titular del registro:
BOEHRINGER INGELHEIM PROMECO, S.A. de C.V.
Calle del Maíz No. 49, Col. Barrio Xaltocán,
C.P. 16090, Xochimilco, Ciudad de México, México.
Reg. Núm. 449M2001 SSA IV
®Marca Registrada