
LUCENTIS
RANIBIZUMAB
Solución
1 Caja , 1 Frasco vial , 2.3 Miligramos
1 Caja, 1 Jeringa(s) prellenada(s),
1 Caja, 1 Frasco(s) ámpula, 2.3/0.23 mg/ml, 10 mg/ml
1 Caja, 1 Frasco(s) ámpula, 2.3/0.23 mg/ml, 10 mg/ml
FORMA FARMACÉUTICA Y FORMULACIÓN:
El frasco ámpula contiene:
Ranibizumab 2.3 mg
Excipiente, cbp 0.23 mL
INDICACIONES TERAPÉUTICAS:
LUCENTIS® está indicado para el tratamiento de:
• La degeneración macular relacionada con la edad (DMRE) de tipo neovascular (húmeda).
• La pérdida de visión por edema macular diabético (EMD).
• La pérdida de visión por edema macular secundario a oclusión venosa retiniana, OVR (oclusión de rama venosa, ORVR u oclusión venosa central, OVC).
FARMACOCINÉTICA Y FARMACODINAMIA:
Grupo farmacoterapéutico: Agentes antineovascularizantes. Código ATC: S01LA04.
Propiedades farmacocinéticas:
Absorción: Tras la administración intravítrea mensual de LUCENTIS® a pacientes con DMRE neovascular, las concentraciones séricas de ranibizumab fueron generalmente bajas; las concentraciones máximas (Cmáx) resultaron usualmente inferiores a la concentración necesaria para inhibir la actividad biológica de VEGF en un 50% (entre 11 y 27 ng/mL, determinada en un ensayo de proliferación celular in vitro). La Cmáx fue proporcional a la dosis en la gama de dosis de 0.05 a 1.0 mg/ojo. Tras la administración intravítrea mensual de LUCENTIS® (0.5 mg/ojo), la Cmáx de ranibizumab en suero (alcanzada después de aproximadamente un día) por lo general varía entre 0.79 y 2.90 ng/mL, y la Cmin entre 0.07 y 0.49 ng/mL. Las concentraciones séricas de ranibizumab en los pacientes con EMD y OVR fueron similares a las observadas en los pacientes con DMRE neovascular.
Distribución y eliminación: Los análisis de farmacocinética poblacional y la desaparición de ranibizumab del suero en los pacientes tratados con la dosis de 0.5 mg indican que la vida media de eliminación vítrea de ranibizumab es de unos 9 días en promedio. La exposición sérica a ranibizumab es unas 90 000 veces menor que la exposición intravítrea al fármaco.
Poblaciones especiales:
Disfunción renal: No se han llevado a cabo estudios formales para examinar la farmacocinética de LUCENTIS® en pacientes con disfunción renal. En un análisis de farmacocinética poblacional de pacientes con DMRE neovascular el 68% de los pacientes (136 de 200) tenían disfunción renal (leve [50-80 mL/min] en el 46.5%, moderada [30-50 mL/min] en el 20%, y grave [< 30 mL/min] en el 1.5%). Entre los pacientes con OVR, el 48.2% (253 de 525) tenían disfunción renal (leve en el 36.4%, moderada en el 9.5%, y grave en el 2.3%). La depuración sistémica fue ligeramente inferior, sin llegar a ser clínicamente significativa.
Disfunción hepática: No se han llevado a cabo estudios formales para examinar la farmacocinética de LUCENTIS® en pacientes con disfunción hepática.
Propiedades farmacodinámicas:
Mecanismo de acción: El ranibizumab es un fragmento de anticuerpo monoclonal recombinado humanizado dirigido contra el factor de crecimiento endotelial vascular de tipo A (VEGF-A). Tiene gran afinidad por las isoformas del VEGF-A (por ejemplo, VEGF110, VEGF121 y VEGF165), por lo que impide que el VEGF-A se una a sus receptores VEGFR-1 y VEGFR-2.
La unión del VEGF-A a sus receptores promueve la proliferación de células endoteliales, la neovascularización y la hiperpermeabilidad vascular, y todo ello contribuye a la progresión de la forma neovascular de la DMRE, de la insuficiencia visual causada por el edema macular diabético y del edema macular causante de pérdida de visión en la diabetes y la oclusión venosa retiniana.
Estudios clínicos:
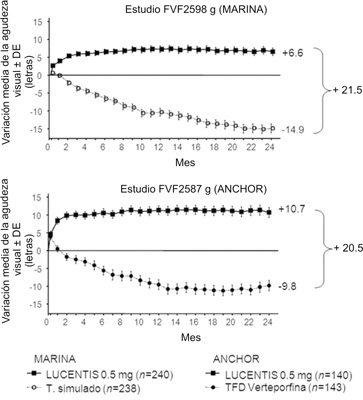
Tratamiento de la Degeneración Macular Relacionada con la Edad (DMRE): Se han evaluado la seguridad y la eficacia clínicas de LUCENTIS® en tres estudios controlados con tratamiento simulado** o fármaco activo, con aleatorización y doble enmascaramiento, en pacientes con DMRE neovascular. Participaron en estos estudios 1,323 pacientes en total (879 en el grupo que recibió fármaco activo y 444 en el grupo de control). En el estudio FVF2598g (MARINA) pacientes con neovascularización coroidea oculta sin componente clásico o mínimamente clásica recibieron inyecciones intravítreas mensuales de 0.3 mg o 0.5 mg de ranibizumab o inyecciones simuladas. En este estudio se incluyeron 716 pacientes en total (tratamiento simulado: 238; 0.3 mg de ranibizumab: 238; 0.5 mg de ranibizumab: 240). Se dispone de datos hasta el final del mes 24.
En el estudio FVF2587g (ANCHOR), los pacientes con lesiones de neovascularización coroidea de tipo predominantemente clásico recibieron alguno de los tratamientos siguientes: 1) terapia fotodinámica simulada e inyecciones intravítreas mensuales de 0.3 mg de ranibizumab; 2) terapia fotodinámica simulada e inyecciones intravítreas mensuales de 0.5 mg de ranibizumab; o 3) terapia fotodinámica activa con verteporfina e inyecciones intravítreas simuladas. Se administró terapia fotodinámica simulada o activa con verteporfina con la inyección de LUCENTIS® inicial y luego cada tres meses si la angiografía fluoresceínica indicaba persistencia o recurrencia de la hiperpermeabilidad vascular. En este estudio se incluyeron 423 pacientes en total (tratamiento simulado: 143; 0.3 mg de ranibizumab: 140; 0.5 mg de ranibizumab: 140). Se dispone de datos hasta el final del mes 24.
** El procedimiento de control consistente en la inyección simulada de LUCENTIS® supuso anestesiar el ojo igual que para la inyección intravítrea de LUCENTIS®. Luego se presionó la punta de una jeringuilla sin aguja contra la conjuntiva y se oprimió el émbolo.
Los resultados clave se resumen en las tablas 1 y 2 y en la figura 1:
Tabla 1. Resultados a los 12 meses y a los 24 meses. Estudio FVF2598g (MARINA).
|
Criterio de valoración |
Mes |
Tratamiento simulado (n = 238) |
LUCENTIS® 0.5 mg (n = 240) |
|
Pérdida de agudeza visual < 15 letras (%)a (conservación de la visión) |
Mes 12 |
62% |
95% |
|
Mes 24 |
53% |
90% |
|
|
Mejora de la agudeza visual ³ 15 letras (%)a |
Mes 12 |
5% |
34% |
|
Mes 24 |
4% |
33% |
|
|
Variación media de la agudeza visual (letras) (DE) a |
Mes 12 |
-10.5 (16.6) |
+7.2 (14.4) |
|
Mes 24 |
-14.9 (18.7) |
+6.6 (16.5) |
a p < 0.01.
Tabla 2. Resultados a los 12 meses en el estudio FVF2587g (ANCHOR)
|
Criterio de valoración |
Mes |
TFD con verteporfina (n = 143) |
LUCENTIS® 0.5 mg (n = 140) |
|
Pérdida de agudeza visual < 15 letras (%)a (conservación de la visión) |
Mes 12 |
64% |
96% |
|
Mes 24 |
66% |
90% |
|
|
Mejora de la agudeza visual ? 15 letras (%)a |
Mes 12 |
6% |
40% |
|
Mes 24 |
6% |
41% |
|
|
Variación media de la agudeza visual (letras) (DE)a |
Mes 12 |
-9.5 (16.4) |
+11.3 (14.6) |
|
Mes 24 |
-9.8 (17.6) |
+10.7 (16.5) |
a p < 0.01.
Figura 1. Variación media de la agudeza visual desde el inicio hasta el 24° mes en el estudio FVF2598g (MARINA) y hasta el 12° mes en el estudio FVF2587g (ANCHOR): Población ITT.
En los pacientes del grupo tratado con LUCENTIS®, la lesión de neovascularización coroidea mostró, en promedio, un crecimiento observable mínimo. En el duodécimo mes, la variación media del área total de la neovascularización coroidea era de 0.1-0.3 áreas papilares (DA) en los grupos tratados con LUCENTIS®, frente a 2.3-2.6 DA en los grupos de control.
Los resultados de ambos ensayos indicaron que el tratamiento con ranibizumab puede ser benéfico también en pacientes que perdieron ³ 15 letras de la agudeza visual con mayor corrección (AVMC) en el primer año de tratamiento.
En ambos estudios MARINA y ANCHOR, la mejora de la agudeza visual observada con LUCENTIS® 0.5 mg a los 12 meses fue acompañada por ciertos beneficios reportados por el paciente, medidos por los resultados del Cuestionario de Función visual del Instituto Nacional de los Ojos (VFQ-25). Las diferencias entre LUCENTIS® 0.5 mg y los dos grupos de control fueron evaluados con valores de p que van de 0.009 a < 0.0001.
El estudio FVF3192g (PIER) era un estudio de dos años con aleatorización, doble enmascaramiento y control con tratamiento simulado, concebido para evaluar la seguridad y la eficacia de LUCENTIS® en pacientes con DMRE neovascular (con o sin componente de neovascularización coroidea clásica). Los pacientes recibieron inyecciones intravítreas de 0.3 mg o 0.5 mg de LUCENTIS® o inyecciones simuladas una vez al mes durante tres meses consecutivos, seguidas de una dosis cada tres meses. Desde el decimocuarto mes, los pacientes sometidos a simulación del tratamiento fueron autorizados a recibir LUCENTIS®, y a partir del decimonoveno mes, fue posible realizar más tratamientos. Los pacientes tratados con LUCENTIS® en el estudio PIER recibieron un total de 10 tratamientos en promedio. El criterio principal de eficacia era la variación media de la agudeza visual a los 12 meses en comparación con la inicial (ver la figura 2). Tras un aumento inicial de la agudeza visual (con la administración de dosis mensuales), en promedio, los pacientes tratados una vez cada tres meses con LUCENTIS® perdieron agudeza visual y al cabo de 12 meses habían regresado a los valores basales. En el estudio PIER, casi todos los pacientes (90%) tratados con LUCENTIS® conservaban su agudeza visual en el duodécimo mes y este efecto se mantuvo en la mayoría de los pacientes tratados con LUCENTIS® (82%) en el mes 24. Los datos de un número limitado de pacientes que recibían tratamiento simulado y que fueron cambiados al tratamiento con LUCENTIS® sugieren que la iniciación temprana del tratamiento puede estar asociada con una mejor preservación de la agudeza visual.
El estudio FVF3689g (SAILOR) fue un ensayo de fase IIIb, a un ciego, multicéntrico de un año en sujetos tratados previamente con NVC secundaria a DMAE. El objetivo primario del estudio fue estimar la incidencia de efectos adversos oculares y no oculares graves, en sujetos tratados durante 12 meses. Dos mil trescientos setenta y ocho pacientes fueron colocados al azar en una proporción de 1:1 para recibir una inyección intravítrea de 0.3 mg o 0.5 mg de ranibizumab cada mes por tres meses consecutivos seguidos de un retratamiento cuando sea necesario, no más a menudo de una vez al mes.
En general, no se observó un desequilibrio entre los dos grupos de dosis en la frecuencia de los eventos adversos oculares y los no oculares. Hubo una tendencia estadísticamente significativa hacia una mayor incidencia de accidente cerebrovascular en el grupo de 0.5 mg en comparación con el grupo de 0.3 mg. Las CI´s respectivas del 95% para el rango en general fueron amplias (0.3% a 1.3% para el grupo de 0.3 mg frente a 0.7% a 2.0% para el grupo de 0.5 mg). El número de cambios fue pequeño en ambos grupos de dosis, y no es suficiente evidencia para concluir (o descartar) que hay una verdadera diferencia en las tasas de accidente cerebrovascular entre los grupos de tratamiento. La diferencia en las tasas de accidente cerebrovascular pueden ser mayor en pacientes con factores de riesgo conocidos, incluyendo antecedentes de accidente cerebrovascular previo y ataque isquémico transitorio.
Tratamiento de la deficiencia visual por Edema Macular Diabético (EMD): La seguridad y la eficacia de LUCENTIS® fue evaluada en dos estudios aleatorizados, doblemente enmascarados y controlados con simulación y activo de 12 meses de duración en pacientes con deficiencia visual debida al edema macular diabético. Un total de 496 pacientes, (336 con el medicamento, 160 con el control) fueron inscritos en los estudios, la mayoría tuvo diabetes de tipo II, mientras que 28 de ellos tuvieron diabetes de tipo I.
En el estudio D2301 (RESTORE), participaron 345 pacientes con pérdida de visión por edema macular a los que se administró de forma aleatoria ya sea una inyección intravítrea inicial de ranibizumab en monoterapia (0.5 mg) y fotocoagulación con láser simulada (n = 116), o bien ranibizumab (0.5 mg) y fotocoagulación con láser (n = 118), o bien una inyección simulada** y fotocoagulación con láser (n = 111). El tratamiento con ranibizumab comenzó con inyecciones intravítreas mensuales y continuó, hasta que se alcanzó la estabilidad de la agudeza visual y ésta se mantuvo por 3 evaluaciones mensuales consecutivas. El tratamiento se reanudó cuando se observó una disminución de la AVMC debido a la progresión del EMD. La fotocoagulación con láser se administró al inicio durante el primer día, al menos 30 minutos previamente a la inyección de ranibizumab, y posteriormente cuando se necesite basado en los criterios del ETDRS.
Los resultados clave están resumidos en la tabla 3 y la figura 2.
Tabla 3. Resultados a los 12 meses en el estudio D2301 (RESTORE).
|
Criterio de valoración |
Ranibizumab 0.5 mg (n = 115) |
Ranibizumab 0.5 mg + láser (n = 118) |
Láser (n = 110) |
|
Cambio promedio del AVMC desde el mes 1 al mes 12 respecto a la basal (Letras) (DE)b |
6.1 (6.43) |
5.9 (7.92) |
0.8 (8.56) |
|
Cambio medio del AVMC en 12 meses en comparación con basal (letras) (SD)b |
6.8 (8.25)b |
6.4 (11.77)c |
0.9 (11.44) |
|
Ganancia de ³ 10 letras en AVMC (% de pacientes) en el mes 12 |
37.4d |
43.2b |
15.5 |
|
Ganancia de ³ 15 letras en AVMC (% de pacientes) en el mes 12 |
22.6e |
22.9f |
8.2 |
b p < 0.0001, c p = 0.0004, d p = 0.0001, e p = 0.0032, f p = 0.0021.
Figura 2. Variación promedio de la AVMC a partir del tiempo basal en el estudio D2301 (RESTORE).
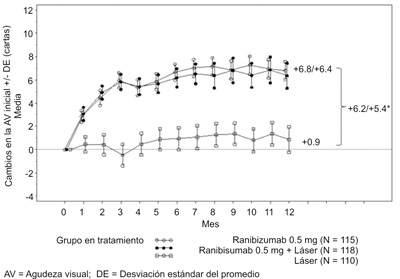
AV= Agudeza visual; DE= Desviación estándar del promedio
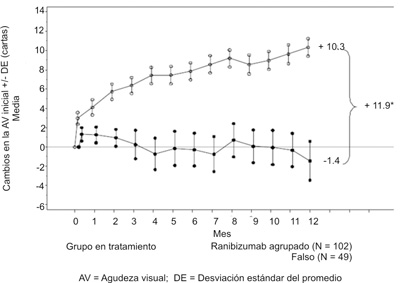
En el estudio D2201 (RESOLVE) participaron 151 pacientes con afectación del centro de la mácula causante de pérdida de visión, fueron tratados con ranibizumab (6 mg/mL, n = 51, 10 mg/mL, n = 51) o tratamiento de simulación (n = 49) mediante inyecciones intravítreas mensuales hasta que se cumplieron los criterios predefinidos para la suspensión del tratamiento. La dosis inicial de ranibizumab (0.3 mg o 0.5 mg) podía duplicarse en cualquier momento durante el estudio tras la primera inyección si el investigador consideraba que la respuesta al tratamiento era insuficiente. Para ambos grupos que recibieron tratamiento activo como en el grupo de control se permitió un tratamiento de rescate con fotocoagulación con láser después del tercer mes de tratamiento.
El estudio constó de dos partes: Una parte exploratoria (integrada por los primeros 42 pacientes que fueron analizados a los 6 meses), y otra parte confirmatoria (integrada por los restantes 109 pacientes analizados a los 12 meses).
Los resultados de importancia de la parte confirmatoria del estudio (2/3 del total de pacientes) están resumidos en la tabla 4 y en la figura 3.
Tabla 4. Resultados a los 12 meses en el estudio D2201 (RESOLVE) (población total del estudio).
|
Criterio de valoración |
Agrupación de ranibizumab (n = 102) |
Simulación (n = 49) |
|
Cambio medio en el AVMC desde el mes 1 hasta 12 meses en comparación con los valores iniciales (letras) (SD)b |
+7.8 (7.72) |
-0.1 (9.77) |
|
Cambio medio en el AVMC hasta los 12 meses en comparación con los valores iniciales (letras) (SD)b |
+10.3 (9.14) |
-1.4 (14.16) |
|
Ganancia de ³ 10 letras en AVMC (% pacientes) en el duodécimo mesb |
60.8 |
18.4 |
|
Ganancia de ³ 15 letras en AVMC (% pacientes) en el duodécimo mesg. |
32.4 |
10.2 |
b p < 0.0001, g p = 0.0043.
Figura 3. Variación promedio en la agudeza visual a partir del valor inicial en el estudio D2201 (RESOLVE), (población general).
Los pacientes tratados con ranibizumab experimentaron una reducción continua del ERC. A los 12 meses, la variación media del ERC desde el inicio fue de -194 micrómetros con el ranibizumab y de -48 micrómetros con el tratamiento simulado.
En general, los hallazgos de seguridad oculares y no oculares en los pacientes con EMD en ambos estudios D2201 y D2301 fueron comparables con el perfil de seguridad conocido y previamente observado para los pacientes que padecen DMRE húmeda.
Tratamiento de la pérdida de visión por edema macular secundario a OVR: La seguridad y la eficacia clínica de LUCENTIS® en pacientes con pérdida de visión por edema macular secundario a OVR se han evaluado en dos estudios aleatorizados, doblemente enmascarados y controlados: BRAVO y CRUISE que reclutaron pacientes con ORVR (n = 397) y OVC (n = 392), respectivamente. En ambos estudios, los pacientes recibieron tanto 0.3 mg o 0.5 mg de ranibizumab o tratamiento de simulación** por vía intravitreal. Después de 6 meses, los pacientes con tratamiento de simulación fueron administrados con 0.5 mg de ranibizumab. En el estudio BRAVO, el tratamiento por fotocoagulación de rescate estaba permitido a partir del tercer mes de tratamiento.
Los resultados de importancia de los estudios BRAVO y CRUISE están resumidos en las tablas 5 y 6 y en las figuras 4 y 5.
Tabla 5. Resultados a los 6 y 12 meses (BRAVO).
|
Tratamiento simulado/LUCENTIS® 0.5 mg (n = 132) |
LUCENTIS® 0.5 mg (n = 131) |
|
|
Variación promedio de la agudeza visual desde el inicio hasta el sexto mesb (letras) (criterio primario) |
+7.3 |
+18.3 |
|
Variación promedio de la agudeza visual desde el inicio hasta el doceavo mes (letras) |
+12.1 |
+18.3 |
|
Proporción de pacientes con mejoría de la AVMC ³ 15 letras con respecto a los valores iniciales hasta el sexto mesb |
28.8% |
61.1% |
|
Proporción de pacientes con mejoría de la AVMC ³ 15 letras con respecto a los valores iniciales hasta el doceavo mes |
43.9% |
60.3% |
|
Proporción de pacientes que recibieron terapia de rescate con láser durante 12 meses |
61.4% |
34.3% |
b p < 0.0001.
Figura 4. Variación media de la AVMC desde el inicio, en 6 meses y hasta el mes 12 (BRAVO).
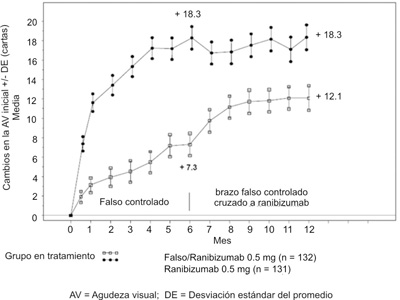
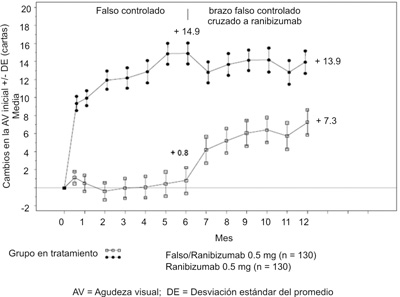
Tabla 6. Resultados a los 6 y 12 meses (CRUISE).
|
Tratamiento simulado/LUCENTIS® 0.5 mg (n = 130) |
LUCENTIS® 0.5 mg (n = 130) |
|
|
Variación media de la agudeza visual con respecto a los valores iniciales hasta el sexto mesb (letras) |
+0.8 |
+14.9 |
|
Variación media de la agudeza visual con respecto a los valores iniciales hasta el doceavo mes (letras) |
+7.3 |
+13.9 |
|
Proporción de pacientes con mejoría de la AVMC ³ 15 letras con respecto a los valores iniciales hasta el sexto mesb |
16.9% |
47.7% |
|
Proporción de pacientes con mejoría de la AVMC ³ 15 letras con respecto a los valores iniciales hasta el doceavo mes |
33.1% |
50.8% |
b p < 0.0001.
Figura 5. Variación media de la AVMC desde el inicio en 6 meses y hasta el mes 12 (CRUISE).
En ambos estudios, la mejoría de la visión se acompañó de una reducción continua del edema macular, medido por el ERC.
La mejoría de la agudeza visual observada con el ranibizumab a los 6 y a los 12 meses se acompañó de beneficios referidos por el paciente, es decir, mejorías en las subescalas de actividades relacionadas con la visión cercana y la visión lejana del Cuestionario de Funcionamiento Visual del Instituto Oftalmológico Nacional de los EE.UU.-25 (VFQ-25), que constituyeron otro criterio secundario de valoración de la eficacia definido de antemano. La diferencia entre el grupo tratado con 0.5 mg de ranibizumab y el grupo de control se evaluó a los 6 meses y presentó valores p de 0.02 a 0.0002.
CONTRAINDICACIONES:
• Hipersensibilidad al principio activo o a cualquiera de los excipientes de LUCENTIS®.
• Infecciones oculares o perioculares activas o sospecha en ellas.
• Inflamación intraocular en activa.
• Uso en menores de 18 años de edad.
• Uso durante el embarazo y la lactancia.
RESTRICCIONES DE USO DURANTE EL EMBARAZO Y LA LACTANCIA:
Mujeres en edad de procrear: Las mujeres en edad de procrear deben utilizar métodos anticonceptivos eficaces durante el tratamiento.
Embarazo: No se dispone de información suficiente acerca del uso de ranibizumab en las mujeres embarazadas.
Los estudios realizados en macacos no muestran efectos dañinos directos o indirectos sobre el embarazo o desarrollo embrional/fetal (ver Precauciones en relación con efectos de carcinogénesis, mutagénesis, teratogénesis y sobre la fertilidad). La exposición sistémica al ranibizumab posterior a la administración ocular es baja, pero debido al mecanismo de acción, se debe considerar al ranibizumab como un agente potencialmente teratogénico, embriotóxico y fetotóxico.
Por lo tanto, el ranibizumab no debe ser utilizado durante el embarazo a menos que el beneficio supere al riesgo potencial para el feto. Para las mujeres que deseen embarazarse y que hayan sido tratadas con ranibizumab, se recomienda esperar al menos 3 meses después de la última dosis antes de concebir.
Lactancia: No se sabe si el ranibizumab (LUCENTIS®) se excreta en la leche humana. Como medida de precaución, no se recomienda amamantar durante el tratamiento con LUCENTIS®.
Fertilidad: No existen datos disponibles sobre la fertilidad.
REACCIONES SECUNDARIAS Y ADVERSAS:
Resumen del perfil de seguridad:
Población tratada para DMRE: Un total de 1,315 pacientes constituyeron la población de seguridad en los tres estudios de fase III para DMRE con 24 meses de exposición a LUCENTIS® y 440 pacientes fueron tratados con la dosis recomendada de 0.5 mg.
Entre los acontecimientos adversos graves relacionados con el procedimiento de inyección figuran la endoftalmitis, el desprendimiento de retina regmatógeno, el desgarro retiniano y la catarata por traumatismo iatrogénico (ver Precauciones generales).
Otros acontecimientos oculares graves que se han observado en pacientes tratados con LUCENTIS® son la inflamación intraocular y la presión intraocular elevada (ver Precauciones generales).
Los acontecimientos adversos que se enumeran a continuación sucedieron con una frecuencia mayor (al menos de 2 puntos porcentuales) en los pacientes que recibieron tratamiento con 0.5 mg de ranibizumab (LUCENTIS®) que en los que recibieron el tratamiento de control (tratamiento simulado o terapia fotodinámica —TFD— con verteporfina) según los datos agrupados de los tres estudios controlados de fase III: FVF2598g (MARINA), FVF2587g (ANCHOR) y FVF3192g (PIER). Por consiguiente, se han considerado posibles reacciones adversas al medicamento (RAM). Los datos de seguridad expuestos a continuación incluyen también todos los acontecimientos adversos registrados en los 440 pacientes que recibieron la dosis de 0.5 mg y de los que se sospechó que estaban al menos potencialmente relacionados con el procedimiento de inyección o con el medicamento en la DMRE húmeda.
Población tratada para EMD: La seguridad de LUCENTIS® se estudió en un estudio controlado con tratamiento simulado de un año de duración (RESOLVE) y en un estudio controlado con laserterapia de un año de duración (RESTORE) en los que participaron, respectivamente, 102 y 235 pacientes con pérdida de visión por EMD, a quienes se administró ranibizumab (ver Estudios clínicos). La infección de las vías urinarias, incluida en la categoría de acontecimientos frecuentes, cumplía los criterios de reacciones adversas para ser incluida en la tabla 1; por lo demás, la frecuencia y la gravedad de las reacciones oculares y no oculares descritas en los ensayos RESOLVE y RESTORE fueron similares a las de las reacciones observadas en los estudios clínicos realizados en la DMRE húmeda.
Población tratada para OVR: La seguridad de LUCENTIS® se estudió en dos ensayos de 12 meses de duración (BRAVO y CRUISE) realizados, respectivamente, en 264 y 261 pacientes tratados con ranibizumab que presentaban pérdida de visión por edema macular secundario a oclusión venosa central (OVC) o de rama (ORVR) (ver Estudios clínicos). En los estudios BRAVO y CRUISE los acontecimientos adversos oculares y no oculares notificados presentaron una frecuencia y una gravedad similares a las observadas en los estudios sobre la DMRE húmeda.
Resumen de las reacciones adversas agrupadas de los estudios clínicos: Las reacciones adversas de los estudios clínicos se enumeran por la clasificación órgano-sistema de MedDRA. En cada clase órgano-sistema, las reacciones adversas están acomodadas por frecuencia, siendo las frecuentes las primeras. En cada grupo de frecuencia, las reacciones adversas se presentan en orden decreciente de gravedad. Adicionalmente, cada categoría de frecuencia para cada reacción adversa esta basada en la siguiente convención, (CIOMS III): muy frecuente (³ 1/10), frecuente (³ 1/100 a < 1/10), infrecuente (³ 1/1,000 a < 1/100), raro (³ 1/10,000 a < 1/1,000), muy raro (< 1/10,000).
Tabla 1. Reacciones adversas de los estudios clínicos.
|
Infecciones e infestaciones |
|
|
Muy frecuentes |
Nasofaringitis. |
|
Frecuentes |
Gripe, infección del conducto urinario* |
|
Trastornos hematopoyéticos y linfáticos |
|
|
Frecuentes |
Anemia |
|
Trastornos psiquiátricos |
|
|
Frecuentes |
Ansiedad |
|
Trastornos del sistema nervioso |
|
|
Muy frecuentes |
Cefalea |
|
Frecuentes |
Accidente cerebrovascular |
|
Trastornos visuales |
|
|
Muy frecuentes |
Inflamación intraocular, vitreítis, desprendimiento del vítreo, hemorragia retiniana, alteración de la visión, dolor ocular, partículas flotantes en vítreo, hemorragia conjuntival, irritación ocular, sensación de cuerpo extraño en los ojos, aumento del lagrimeo, blefaritis, ojo seco, hiperemia ocular, prurito ocular |
|
Frecuentes |
Degeneración de la retina, trastorno retinal, desprendimiento de retina, rasgadura de la retina, desprendimiento del epitelio pigmentario de la retina, rasgadura del epitelio pigmentario de la retina, agudeza visual disminuida, hemorragia vítrea, trastorno vítreo, uveítis, iritis, iridociclitis, cataratas, catarata subcapsular, opacidad de la cápsula posterior, queratitis punctata, abrasión en la córnea (Flare) de la cámara anterior, visión borrosa, hemorragia en el lugar de inyección, hemorragia ocular, conjuntivitis, conjuntivitis alérgica, secreción ocular, fotopsia, fotofobia, molestia ocular, edema del párpado, dolor del párpado, hiperemia conjuntival |
|
Infrecuentes |
Ceguera, endoftalmitis, hipopión, hipema, queratopatía, adherencias del iris, depósitos corneales, edema corneal, estrías corneales, dolor en el lugar de la inyección, irritación en el sitio de inyección, sensación anormal en el ojo, irritación del párpado |
|
Trastornos respiratorios, torácicos y del mediastino |
|
|
Frecuentes |
Tos |
|
Trastornos gastrointestinales |
|
|
Frecuentes |
Náusea |
|
Trastornos de la piel y del tejido subcutáneo |
|
|
Frecuentes |
Reacciones alérgicas (rash, urticaria, prurito y eritema) |
|
Trastornos músculo esqueléticos y del tejido conectivo |
|
|
Muy frecuentes |
Artralgia |
|
Exploraciones complementarias |
|
|
Muy frecuentes |
Incremento de la presión intraocular. |
* Observadas solamente en la población tratada para la EMD.
PRECAUCIONES EN RELACIÓN CON EFECTOS DE CARCINOGÉNESIS, MUTAGÉNESIS, TERATOGÉNESIS Y SOBRE LA FERTILIDAD: La administración intravítrea bilateral de ranibizumab en dosis de entre 0.25 y 2.0 mg/ojo a macacos (Macaca fascicularis) una vez cada dos semanas durante 26 semanas produjo efectos dependientes de la dosis.
A nivel intraocular, se registraron aumentos del flare y la celularidad de la cámara anterior dependientes de la dosis, que alcanzaban el máximo dos días después de la inyección. En general, la intensidad de la reacción inflamatoria disminuyó con las inyecciones posteriores o durante la recuperación. En el segmento posterior se observaron infiltración celular y cuerpos flotantes en el vítreo, que también tendían a ser dependientes de la dosis y generalmente persistían al final del periodo de tratamiento. En el estudio de 26 semanas, la intensidad de la inflamación vítrea aumentó con el número de inyecciones. Sin embargo, se observaron signos de reversibilidad tras la recuperación. La naturaleza y cronología de la inflamación en el segmento posterior es signo de una reacción inmunitaria mediada por anticuerpos que puede carecer de significación clínica. En algunos animales se observó la formación de cataratas tras un periodo relativamente largo de intensa inflamación, lo que parece indicar que las alteraciones del cristalino eran secundarias a inflamación grave. Tras las inyecciones intravítreas se observó un incremento pasajero de la presión intraocular, el cual es independiente de la dosis.
Las alteraciones oculares microscópicas están relacionadas con la inflamación y no son indicativas de procesos degenerativos. Se percibieron alteraciones inflamatorias granulomatosas en la papila de algunos ojos. Estas alteraciones en el segmento posterior disminuyeron (y en algunos casos desaparecieron) durante el periodo de recuperación. Tras la administración intravítrea no se detectaron signos de toxicidad sistémica. Se detectaron anticuerpos contra el ranibizumab en el suero y el vítreo de un subgrupo de animales tratados.
No se dispone de datos sobre carcinogenia o mutagenia.
En monos preñados, el tratamiento in vitro con ranibizumab no provocó toxicidad progresiva o teratogenicidad, y no tuvo efecto sobre el peso o la estructura de la placenta, aunque, basándose en el efecto farmacológico de ranibizumab, debe considerarse como un agente potencialmente teratogénico, fetotóxico y embriotóxico.
Sin embargo, debido a las restricciones dictadas para la vía de administración intravítrea, las dosis utilizadas en este estudio no alcanzan el rango de la toxicidad materna, pero sólo en un múltiplo con respecto a la exposición sistémica en humanos. La ausencia de efectos provocados por ranibizumab sobre el desarrollo embriofetal es plausible si es relacionado principalmente con la incapacidad del fragmento Fab de cruzar la placenta.
Sin embargo, un caso fue descrito con altos niveles séricos de ranibizumab en la madre y la presencia de ranibizumab en el suero fetal, lo que sugiere que el anticuerpo anti-ranibizumab actuó (Fc, región que contiene) como una proteína transportadora de ranibizumab, disminuyendo así la depuración en los niveles séricos de la madre y permitiendo su transferencia placentaria. A medida que se realizaron las investigaciones sobre el desarrollo embriofetal en animales sanos preñados y con enfermedades (como por ejemplo, diabetes) pueden modificar la permeabilidad de la placenta hacia el fragmento Fab, (ver Restricciones de uso durante el embarazo y lactancia).
INTERACCIONES MEDICAMENTOSAS Y DE OTRO GÉNERO: No se han efectuado estudios de interacción propiamente dichos.
Para el uso conjunto de la terapia fotodinámica con verteporfina (TFD) y LUCENTIS® en DMRE ver Estudios clínicos.
Para el uso conjunto de la fotocoagulación con láser y LUCENTIS® en EMD y la OVR ver Estudios clínicos y Dosis y vía de administración.
ALTERACIONES DE LAS PRUEBAS DE LABORATORIO: No se han reportado alteraciones de pruebas de laboratorio.
PRECAUCIONES GENERALES: Las inyecciones intravítreas, como las de LUCENTIS® se han asociado con endoftalmitis, inflamación intraocular, desprendimiento de la retina regmatógeno, desgarros retinianos y catarata traumática iatrogénica (ver Reacciones secundarias y adversas). Siempre que se administre LUCENTIS® se deben emplear técnicas asépticas adecuadas. Además, debe vigilarse a los pacientes durante la semana posterior a la inyección para poder administrar tratamiento temprano en caso de infección. Se les debe indicar que notifiquen sin demora cualquier síntoma indicativo de endoftalmitis o cualquiera de los acontecimientos mencionados.
Se han observado aumentos transitorios de la presión intraocular en los 60 minutos siguientes a la inyección de LUCENTIS® (ver Reacciones secundarias y adversas). También se han reportado aumentos prolongados de la presión intraocular. Por consiguiente, se deben vigilar y tratar apropiadamente, tanto la presión intraocular como la perfusión de la papila del nervio óptico.
Existe un riesgo potencial de trastornos tromboembólicos arteriales tras el uso de inhibidores intravítreos del FCVE (Factor de Crecimiento del Endotelio Vascular). En estudios de fase III realizados para degeneración macular asociada a la edad, en general, la frecuencia de acontecimientos tromboembólicos arteriales fue similar entre el ranibizumab y el control. Se observó una tasa numéricamente mayor de incidentes cerebrovasculares en los pacientes tratados con ranibizumab 0.5 mg, en comparación con 0.3 mg de ranibizumab o los del grupo control; sin embargo, las diferencias no fueron estadísticamente significativas. La diferencia radica en que el número de incidentes cerebrovasculares puede ser mayor en pacientes con factores de riesgo conocidos, incluyendo los antecedentes de incidentes cerebrovasculares previos o por un ataque isquémico transitorio. Es importante el control glucémico del paciente y tener especial atención en pacientes con retinopatía proliferativa. Por lo tanto, los pacientes deben ser evaluados cuidadosamente por sus médicos para evaluar si el tratamiento con LUCENTIS® es adecuado y genera un beneficio mayor que el riesgo potencial.
Como todas las proteínas terapéuticas, LUCENTIS® tiene capacidad inmunógena.
No se ha estudiado la seguridad ni la eficacia de la administración del tratamiento con LUCENTIS® en ambos ojos a la vez.
LUCENTIS® no ha sido estudiado en pacientes con infecciones sistémicas activas o con trastornos oculares concurrentes, como el desprendimiento de la retina o el agujero macular.
Existe experiencia limitada en el tratamiento de pacientes con episodios previos de OVR y de pacientes con episodios de ORVR isquémico y de OVC. No se recomienda el tratamiento de pacientes con OVR que presenten sintomatología clínica de pérdida irreversible de la función visual isquémica.
Efectos sobre la capacidad para conducir y utilizar máquinas: El procedimiento de administración de LUCENTIS® puede inducir trastornos visuales pasajeros, que pueden afectar a la capacidad para conducir o utilizar maquinaria (ver Reacciones secundarias y adversas). Los pacientes que experimentan signos de tales trastornos no deben conducir ni utilizar máquinas hasta que dichos trastornos hayan desaparecido.
DOSIS Y VÍA DE ADMINISTRACIÓN:
Vía de administración: Intravítrea.
Dosis: Vial de uso individual únicamente para administración intravítrea. El utilizar más de una inyección de un solo vial puede generar contaminación y una subsecuente infección.
LUCENTIS® debe ser administrado por un oftalmólogo calificado y con experiencia en inyecciones intravítreas.
La dosis recomendada de LUCENTIS® es 0.5 mg (0.05 mL). El intervalo entre dos dosis no debe ser menor a un mes.
Población general objetivo:
Tratamiento de la DMRE: El tratamiento se administra mensualmente y debe de mantenerse hasta que se alcance la máxima agudeza visual, confirmada por tres evaluaciones consecutivas de la estabilidad de la agudeza visual realizadas durante el tratamiento con LUCENTIS®.
El tratamiento deberá reiniciarse con inyecciones mensuales cuando la vigilancia indique una pérdida en la agudeza visual debido a la DMRE y deberá continuar hasta que la estabilidad de la agudeza visual se alcance, evaluándola por tres determinaciones consecutivas.
Tratamiento de la deficiencia visual por Edema Macular Diabético (EMD): El tratamiento se administra mensualmente y de forma continua hasta que se alcance la agudeza visual máxima, confirmada por la estabilidad de la agudeza visual evaluada en tres determinaciones consecutivas mensuales realizadas durante el tratamiento con LUCENTIS®.
El tratamiento se reanudará con inyecciones mensuales cuando la evaluación indique una pérdida de la agudeza visual debido a la EMD y se debe continuar hasta que se alcance la estabilidad en la agudeza visual al monitorearla por tres evaluaciones mensuales consecutivas.
LUCENTIS® y fotocoagulación con láser en EMD: LUCENTIS® ha sido utilizado concomitantemente con la fotocoagulación con láser en los estudios clínicos (ver Estudios clínicos). Cuando se administra el mismo día, LUCENTIS® debe ser administrado al menos 30 minutos después de la fotocoagulación con láser. LUCENTIS® puede administrarse en pacientes que ya hayan recibido terapia de fotocoagulación con láser previamente.
Tratamiento de la pérdida de visión por edema macular secundario a OVR: El tratamiento se administra una vez por mes hasta que se logre la máxima agudeza visual, confirmada por su estabilidad durante tres evaluaciones mensuales consecutivas realizadas durante el tratamiento con LUCENTIS®.
El tratamiento se reanudará con inyecciones mensuales cuando la evaluación indique una disminución de la agudeza visual por edema macular secundario a OVR, y se sigue administrando hasta que la agudeza visual vuelva a estabilizarse en tres evaluaciones mensuales consecutivas.
LUCENTIS® y fotocoagulación con láser en la oclusión de rama venosa retiniana (ORVR): LUCENTIS® ha sido utilizado concomitantemente con la fotocoagulación con láser en estudios clínicos (ver Estudios clínicos). Si LUCENTIS® se administra el mismo día, debe administrarse al menos 30 minutos después de la fotocoagulación con láser. LUCENTIS® puede administrarse en pacientes que ya hayan recibido terapia de fotocoagulación con láser.
Modo de administración: Como con todos los medicamentos por vía parenteral, antes de administrar LUCENTIS® se debe comprobar visualmente que no contiene partículas ni ha sufrido cambios de color.
La inyección debe realizarse en condiciones de asepsia, lo que comprende la antisepsia quirúrgica de las manos, el uso de guantes estériles, un campo estéril y un blefarostato estéril (o equivalente), y la disponibilidad de material para realizar una paracentesis estéril (en caso necesario). Antes de administrar la inyección intravítrea deben considerarse detenidamente los antecedentes personales del paciente en lo relativo a reacciones de hipersensibilidad (ver Contraindicaciones). Se debe desinfectar la piel de la región periocular y los párpados, así como la superficie ocular. Antes de la inyección deben aplicarse una anestesia adecuada y un microbicida tópico de amplio espectro.
Se le debe enseñar al paciente a instilarse él mismo un colirio antimicrobiano cuatro veces al día durante los tres días anteriores y posteriores a cada inyección.
Antes de extraer el contenido del vial es preciso desinfectar la parte externa del tapón de goma. Utilizando una técnica aséptica, monte la aguja de filtro de 5 micrómetros en la jeringuilla de 1 mL. Se debe extraer todo el contenido del vial de LUCENTIS® manteniendo éste en posición vertical. Una vez extraído, la aguja de filtro debe desecharse y no utilizarse para la inyección intravítrea. En su lugar se coloca una aguja estéril para inyección intravítrea. Se ha de expulsar el contenido hasta que la punta del émbolo coincida con la línea que marca los 0.05 mL en la jeringuilla.
La aguja de inyección debe introducirse en la cámara vítrea penetrando entre 3.5 y 4.0 mm por detrás del limbo esclerocorneal, evitando el meridiano horizontal y en dirección al centro del globo ocular. Posteriormente se libera el volumen inyectable de 0.05 mL; las inyecciones siguientes deberán aplicarse cada vez en un meridiano escleral distinto.
Poblaciones especiales:
Insuficiencia hepática: No se ha estudiado la administración de LUCENTIS® en pacientes con insuficiencia hepática, aun así, dado que la exposición sistémica es insignificante, no se considera necesario adoptar medidas especiales en esta población.
Insuficiencia renal: No es necesario ajustar la dosis en los pacientes con insuficiencia renal (ver Farmacocinética).
Pacientes pediátricos: Ante la falta de datos de inocuidad y eficacia, no se recomienda el uso de LUCENTIS® en los niños y adolescentes.
Pacientes geriátricos: No es necesario ajustar la dosis en los pacientes de edad avanzada.
MANIFESTACIONES Y MANEJO DE LA SOBREDOSIFICACIÓN O INGESTA ACCIDENTAL: Los casos de sobredosis accidental se han registrado en los estudios clínicos para la DMRE y en los datos posteriores a la comercialización. Las reacciones adversas más frecuentemente asociadas con los casos reportados fueron aumento de la presión intraocular y dolor en los ojos. En caso de sobredosis se debe vigilar la presión intraocular, y tratarla si el médico responsable lo considera necesario.
PRESENTACIÓN:
Caja con un frasco vial de vidrio, una aguja de filtro para retirar el contenido del vial, una aguja de inyección y una jeringuilla para inyección intravítrea e instructivo anexo.
RECOMENDACIONES SOBRE ALMACENAMIENTO: Consérvese en refrigeración entre 2° y 8°C. No congelar.
LEYENDAS DE PROTECCIÓN:
No se deje al alcance de los niños. Su venta requiere receta médica. Dosis: La que el médico señale. Vía de administración: Intravítrea. El empleo de este medicamento durante el embarazo, queda bajo la responsabilidad del médico. Sólo debe ser administrado por un médico oftalmólogo experimentado. Literatura exclusiva para médicos.
Para mayor información comuníquese al Centro de Atención a Clientes de Novartis Farmacéutica S.A. de C.V., Calzada de Tlalpan No. 1779 Col. San Diego Churubusco, Coyoacán, C.P. 04120, teléfono 5420-8685, en el Interior de la República 01 800-718-5459.
Hecho en Suiza por:
Novartis Pharma Stein AG
Schaffhauserstrasse
4332 Stein, Suiza
Acondicionado y/o distribuido por:
NOVARTIS FARMACÉUTICA, S. A. de C. V.
Calz. de Tlalpan No. 1779
Col. San Diego Churubusco
C.P. 04120, Deleg. Coyoacán
D.F., México
Reg. Núm. 052M2007, SSA IV
®Marca registrada


