
LIXIANA
EDOXABÁN
Tabletas
1 Caja, 14 Tabletas, 15 mg
1 Caja, 28 Tabletas, 30 mg
1 Caja, 28 Tabletas, 60 mg
FORMA FARMACÉUTICA Y FORMULACIÓN:
Cada TABLETA de LIXIANA® contiene:
Tosilato de edoxabán monohidratado 20.2 mg, 40.4 mg, 80.8 mg equivalente a 15 mg, 30 mg, 60 mg de Edoxabán
Excipiente cbp 1 tableta
INDICACIONES TERAPÉUTICAS:
Edoxabán está indicado para:
• Reducir el riesgo de evento vascular cerebral (EVC) y embolismo sistémico en pacientes con fibrilación auricular no valvular (FANV).
• Para el tratamiento de tromboembolias venosas (TVE) incluida la trombosis venosa profunda (TVP) y la embolia pulmonar (EP) y la prevención de TVE recurrente (TVP y/o EP).
Pacientes geriátricos (≥ 65 años de edad):
Los estudios clínicos sobre la prevención de accidentes cerebrovasculares en pacientes con fibrilación auricular (FA), tratamiento de TEV y prevención de TVP y EP recurrentes incluyeron a pacientes ≥ 65 años de edad.
Pacientes pediátricos < 18 años de edad:
Todavía no se estableció la seguridad y la eficacia de LIXIANA en niños con menos de 18 años de edad. Por tanto, no se recomienda el uso de LIXIANA en dichos pacientes.
FARMACOCINÉTICA Y FARMACODINAMIA:
FARMACOCINÉTICA:
Tabla 1. Resumen de los Parámetros Farmacocinéticos en Pacientes sanos (Dosis Única)
|
Cmax |
t1/2 (h) |
AUC0-∞ |
Depuración |
Volumen de Distribución |
|
|
Promedio de dosis única |
309 ± 97 ng/mL |
10-14 |
63,1 ± 12,5% (media aritmética ± DE) |
21,8±3,03 L/h |
107±19,9 L |
Absorción:
Edoxabán se absorbe con concentraciones plasmáticas máximas alcanzadas en 1-2 horas. La biodisponibilidad absoluta es del 62%. Los alimentos aumentan la exposición máxima en diversos grados, pero tienen un efecto mínimo sobre la exposición total. LIXIANA se administró con o sin alimentos en los estudios ENGAGE AF-TIMI 48 y HOKUSAI-VTE. Edoxabán es insuficientemente soluble a un pH de 6,0 o más. Se absorbe predominantemente en el tracto gastrointestinal superior. Por tanto, los fármacos o condiciones de enfermedad que aumentan el pH del estómago o aumentan el vaciado gástrico y la motilidad intestinal tienen la posibilidad de reducir la disolución y la absorción de edoxabán. Sin embargo, la administración concomitante de inhibidores de la bomba de protones (esomeprazol) no afectó la exposición a edoxabán.
Distribución:
La disposición es bifásica. El volumen medio de distribución es de 107 ±19,9 L (DE). La unión a proteínas plasmáticas in vitro es de aproximadamente el 55%. No hay acumulación clínicamente relevante de edoxabán (índice de acumulación 1,14) con una dosis diaria. Las concentraciones en estado de equilibrio se logran en 3 días.
Metabolismo:
El edoxabán inalterado es la forma predominante en plasma. Hay un metabolismo mínimo (<10%) por medio de hidrólisis (mediada por la carboxilesterasa 1), conjugación u oxidación por CYP3A4. El metabolito predominante (M-4), formado por hidrólisis, es activo y alcanza <10% de la exposición del compuesto original en pacientes sanos. La exposición a los demás metabolitos es <5%.
Excreción:
En pacientes sanos, el edoxabán se elimina tanto a través del metabolismo como a través del fármaco inalterado en la orina y las heces. El aclaramiento renal (11 L/hora) del fármaco sin cambios contribuye aproximadamente al 50% del aclaramiento total (22 L/hora), y el 50% restante del aclaramiento no renal se produce a través del metabolismo y la secreción biliar. El t½ para la administración oral es de 10-14 horas.
Linealidad/No Linealidad:
Edoxabán muestra una farmacocinética aproximadamente proporcional a la dosis para dosis de 15 mg a 60 mg en pacientes sanos.
Relación(es) farmacocinética/farmacodinámica:
El TP, el INR, el TTPa y el Anti-factor Xa se correlacionan linealmente con las concentraciones de edoxabán.
Poblaciones y Condiciones Especiales:
Pacientes Mayores:
Después de tener en cuenta la función renal y el peso corporal, la edad no tuvo ningún efecto clínicamente significativo adicional sobre la farmacocinética de edoxabán en un análisis farmacocinético poblacional de pacientes ≥ 75 años de edad en el estudio ENGAGE AF-TIMI 48.
Sexo:
Después de tener en cuenta el peso corporal, el sexo no tuvo ningún efecto clínicamente significativo adicional sobre la farmacocinética de LIXIANA en un análisis farmacocinético poblacional del estudio ENGAGE AF-TIMI 48.
Raza:
En un análisis farmacocinético poblacional del estudio ENGAGE AF-TIMI 48, la exposición máxima y total en pacientes asiáticos y no asiáticos fue similar.
Insuficiencia Hepática:
Los pacientes con insuficiencia hepática leve o moderada (clasificados como Child Pugh A o Child Pugh B) mostraron una farmacocinética y una farmacodinámica comparables a su grupo control de salud compatible. No se estudió LIXIANA en pacientes con insuficiencia hepática grave.
Insuficiencia renal:
El 50% del edoxabán inalterado se elimina por el riñón. Los AUCs plasmáticos en pacientes con insuficiencia renal leve (50-80 mL/min), moderada (30-50 mL/min) y grave (<30 mL/min, pero que no se someten a diálisis) aumentaron en 32%, 74% y 72%, respectivamente, en comparación con los pacientes con función renal normal. El modelo de población PK indica que la exposición aproximadamente se duplica en pacientes con insuficiencia renal grave CrCL 15-29 mL/min en comparación con pacientes con función renal normal.
Las respuestas de eficacia y seguridad previstas en pacientes con diferentes funciones renales basadas en modelos y simulaciones se muestran en la figura a continuación. La Figura 1 describe la relación modelada entre la exposición al fármaco y los resultados. Las dos curvas representan la probabilidad promedio prevista de un accidente cerebrovascular/SEE o un evento de sangrado importante dado un paciente promedio del ENGAGE AF, es decir, utilizando la edad promedio (72 años) y ponderada con la probabilidad de factores de riesgo de accidente cerebrovascular/SEE (accidente cerebrovascular previo/AIT versus ningún accidente cerebrovascular previo/AIT) o hemorragia importante (uso concomitante de aspirina/agente antiplaquetario (ASA) o uso no concomitante de ASA). El modelo de respuesta a la exposición prevé diferencias mínimas en la eficacia dentro del rango de concentraciones de LIXIANA observadas en los grupos de función renal, pero prevé un aumento significativo en el sangrado importante en este mismo rango. No se establecieron las implicaciones terapéuticas del uso de dichos datos en el seguimiento de pacientes con edoxabán en estudios clínicos.
Figura 1. Respuesta de Eficacia y Seguridad Prevista para Pacientes con FA mediana
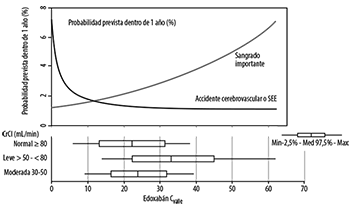
Nota: Las concentraciones de valle de Edoxabán fueron previstas utilizando el modelo PK poblacional; las concentraciones inmediatamente antes de los resultados no estaban disponibles.
Las barras horizontales representan el modelo predicho Cvalle en la función renal normal (dosis de edoxabán 60 mg), insuficiencia renal leve (dosis de edoxabán 60 mg) e insuficiencia renal moderada (dosis de edoxabán 30 mg).
Hemodiálisis:
Una sesión de 4 horas de hemodiálisis redujo las exposiciones totales a LIXIANA en menos del 7%.
Polimorfismo Genético:
Las variantes del gen ABCB1, que codifica la P-gp, no tuvieron efecto sobre la farmacocinética de LIXIANA en pacientes sanos.
Bajo Peso Corporal:
En un análisis farmacocinético poblacional del estudio ENGAGE AF-TIMI 48, la Cmax y el AUC en pacientes con peso corporal mediano bajo (55 kg) aumentaron en 40% y 13%, respectivamente, en comparación con los pacientes con peso corporal mediano alto (84 kg). En los estudios clínicos de fase 3 (indicaciones SPAF y TEV), los pacientes con peso corporal ≤ 60 kg tuvieron una reducción de la dosis de LIXIANA del 50% y tuvieron resultados consistentes de eficacia y seguridad con los resultados generales.
Tabla 2. Resumen de la Farmacocinética y de la Farmacodinámica de LIXIANA en los subgrupos de interés en el Estudio ENGAGE AF TIMI 48
|
Edoxabán 60 mg (dosis reducida a 30 mg) |
Edoxabán Cvalle (ng/mL) |
Edoxabán–Actividad anti-FXa Mina (UI/mL) |
Edoxabán-Actividad anti-FXa Maxb (UI/mL) |
|
Mediana [2,5-97,5%] |
|||
|
Edoxabán 60 mg dosis completa |
27,3 [14,6–45,5] |
0,65 (0,11, 3,56) |
3,96 (0,22, 8,0) |
|
Edoxabán–dosis reducida a 30 mg (a causa de factores únicos o múltiples) |
21,0 [10,2–30,7] |
0,53 (0,05, 2,16) |
2,88 (0,24, 6,20) |
|
Función renal por CrCL en el periodo inicial |
|||
|
30-50 mL/min* |
23,7 [16,5–31,5] |
0,56 (0,05, 2,12) |
2,80 (0,24, 6,40) |
|
> 50-80 mL/min |
32,8 [22,4–45,2] |
0,74 (0,05, 3,32) |
4,34 (0,23, 8,00) |
|
> 80 mL/min |
22,1 [13,0–31,2] |
0,51 (0,05, 3,92) |
3,44 (0,19, 7,60) |
|
Peso ≤ 60 kg solo* |
19,6 [9,43–30,9] |
0,43 (0,05, 2,52) |
3,20 (0,31, 6,40) |
|
Uso concomitante de inhibidores de la P-gp solo* |
17,2 [9,24–32,4] |
0,63 (0,05, 3,32) |
3,28 (0,19, 8,00) |
|
CrCl ≤ 50 y P-gp |
27,2 [15,7–36,7] |
1,22 (0,24, 2,52) |
3,52 (1,81, 6,52) |
|
Peso ≤ 60 kg y P-gp |
22,4 [13,5–36,6] |
0,66 (0,05, 2,52) |
3,52 (0,24, 5,88) |
|
Norteamérica |
26,2 [14,1–45,0] |
0,69 (0,11, 2,12) |
3,44 (0,33, 7,96) |
|
Edad ≥ 75 años |
28,2 [15,1–46,6] |
0,68 (0,05, 2,56) |
3,52 (0,28, 8,00) |
|
Pacientes frágiles** |
26,1 [15,2–49,3] |
0,63 (0,05, 2,16) |
3,18 (0,24, 8,00) |
* Dosis reducida a 30 mg.
** Pacientes frágiles definidos como ≥ 80 años de edad, peso ≤ 50 kg, CrCL ≤ 50 mL/min y/o historial de queda.
a En el ENGAGE-AF, se evaluó la actividad Anti-Xa Min de Edoxabán el día 29 predosis.
b En el ENGAGE AF, se evaluó la actividad Anti-Xa Max de Edoxabán el día 29 posdosis.
FARMACODINAMIA:
Mecanismo de Acción:
LIXIANA es un inhibidor altamente selectivo, directo y reversible del factor Xa, la serina proteasa ubicada en la vía común final de la cascada de coagulación. LIXIANA inhibe el factor Xa libre y la actividad de la protrombinasa. La inhibición del factor Xa en la cascada de coagulación reduce la generación de trombina y prolonga el tiempo de coagulación, además de reducir el riesgo de formación o de formación de trombos provocada.
Farmacodinama:
LIXIANA produce efectos farmacodinámicos de inicio rápido en 1-2 horas, lo que corresponde con la exposición máxima a LIXIANA (Cmax). Los efectos farmacodinámicos medidos por el estudio anti-factor Xa son predecibles y se correlacionan con la dosis y la concentración de LIXIANA. Como resultado de la inhibición de FXa, LIXIANA también prolonga el tiempo de coagulación en pruebas como el tiempo de protrombina (TP) y el tiempo de tromboplastina parcial activada (TTPa). Sin embargo, los cambios observados en dichas pruebas de coagulación en la dosis terapéutica esperada son pequeños y están sujetos a un alto grado de variabilidad. No son útiles para monitorear el efecto anticoagulante de LIXIANA.
Tabla 3. Predicción de la exposición al estado estable de edoxabán y actividad anti-FXa
|
ENGAGE-AF |
Edoxabán Cmin (ng/mL) |
Edoxabán Cmax (ng/mL) |
Edoxabán-Actividad Anti-FXa Mina (UI/mL) |
Edoxabán- Actividad Anti-FXa Maxb, (UI/mL) |
|
Mediana (percentiles 2,5°–97,5°) |
||||
|
Edoxabán 60 mg dosis completa |
27,3 (14,6–45,5) |
217 (129–302) |
0,65 (0,11–3,50) |
3,96 (0,23–8,0) |
|
Edoxabán dosis reducida a 30 mg |
21,0 (10,2–30,7) |
143 (91,1-198) |
0,53 (0,05–2,15) |
2,88 (0,24–6,15) |
|
HOKUSAI VTE |
Edoxabán Cmin (ng/mL) |
Edoxabán Cmax (ng/mL) |
Edoxabán-Actividad Anti-Xa Minc, (UI/mL) |
Edoxabán- Actividad Anti-Xa Maxd, (UI/mL) |
|
Mediana (percentiles 2,5°–97,5°) |
||||
|
Edoxabán 60 mg dosis completa |
15,2 (8,37-31,1) |
211 (135-296) |
0,28 (0,10-2,73) |
2,79 (0,21-5,67) |
|
Edoxabán dosis reducida a 30 mg |
11,7 (4,55-23,5) |
141 (91,9-190) |
0,26 (0,10-1,66) |
1,95 (0,19-4,98) |
a En el ENGAGE-AF se evaluó la actividad Anti-Xa Min de Edoxabán en el día 29 predosis.
b En el ENGAGE-AF se evaluó la actividad Anti-Xa Max de Edoxabán en el día 29 posdosis.
c Para el HOKUSAI-VTE se evaluó la actividad Anti-Xa Min de Edoxabán durante 3 meses, predosis.
d Para el HOKUSAI-VTE se evaluó la actividad Anti-Xa Max de Edoxabán durante 3 meses, posdosis.
Efectos sobre los marcadores de la coagulación al cambiar de rivaroxabán, dabigatrán y apixabán a LIXIANA:
En estudios de farmacología clínica, los pacientes sanos recibieron rivaroxabán 20 mg una vez al día, dabigatrán 150 mg dos veces al día, o apixabán 5 mg dos veces al día, seguidos de una dosis única de LIXIANA 60 mg el día 4. Después del cambio a LIXIANA en el día 4, los efectos sobre el TP, TPPa y anti-FXa (rivaroxabán o apixabán) fueron comparables a aquellos observados cuando se administró LIXIANA en monoterapia durante 4 días. Después de cambiar de dabigatrán a LIXIANA, los valores de TTPa fueron comparables a aquellos observados en dabigatrán. Basado en estos datos, se puede iniciar la primera dosis de LIXIANA en la siguiente dosis programada del anticoagulante anterior.
Las variantes de la subunidad 1 del complejo de reductasa epóxido de la vitamina K (VKORC1) y los genes CYP2C9 que se sabe que afectan la sensibilidad a la warfarina no tuvieron efecto sobre el sangrado en pacientes tratados con LIXIANA.
Efectos sobre los marcadores de coagulación al cambiar de warfarina a LIXIANA:
En un estudio doble ciego después de una dosis única de 60 mg de edoxabán administrada 24 horas después de la última dosis de warfarina, los valores medios de RNI aumentaron de 2,25 (momento de 24 horas) a niveles máximos de aproximadamente 3,7. Los valores medios de RNI luego disminuyeron y alcanzaron niveles cercanos al valor medio de predosis aproximadamente 12 horas tras la dosis (36 h).
Eficacia y Seguridad Clínica:
Prevención del accidente cerebrovascular y de la embolia sistémica en pacientes con fibrilación auricular (FA): Estudio ENGAGE- AF TIMI 48:
Diseño del Estudio y Datos Demográficos del Estudio:
El programa clínico de LIXIANA para la fibrilación auricular fue diseñado para demostrar la eficacia y la seguridad de dos regímenes de dosis de LIXIANA en comparación con la warfarina para la prevención de accidentes cerebrovasculares y de embolismo sistémico (SEE) en pacientes con fibrilación auricular no valvular y con riesgo moderado a alto de accidente cerebrovascular y SEE.
En el estudio pivotal ENGAGE AF-TIMI 48 (estudio de grupos paralelos, dirigido al evento, de fase 3, multicéntrico, aleatorizado, doble ciego, doble enmascarado), 21.105 pacientes (21.026 de los cuales recibieron el fármaco del estudio), con una puntuación CHADS2 media de 2,8 fueron asignados al azar para recibir 30 mg de LIXIANA (15 mg de dosis reducida) una vez al día, en el grupo de tratamiento de LIXIANA 60 mg (dosis reducida a 30 mg) una vez al día o warfarina. Los pacientes en ambos grupos de LIXIANA se redujeron la dosis a la mitad si uno o más de los siguientes factores clínicos, conocidos por aumentar la exposición al fármaco, estuvieran presentes en la asignación al azar o durante el estudio: insuficiencia renal moderada (CrCL30–50 mL/min), bajo peso corporal (≤ 60 kg) o uso concomitante de inhibidores específicos de la P-gp (verapamilo, quinidina, dronedarona). La razón más común para la reducción de la dosis fue CrCL ≤ 50 mL/min en la asignación al azar (19% de los pacientes).
Tabla 4. Resumen de los datos demográficos de los pacientes para los estudios clínicos en FA
|
Diseño del Estudio |
Dosificación, vía de administración y duración |
Pacientes del Estudio |
Edad promedio (Rango) años |
Sexo (M/F)% |
|
Aleatorizado, doble ciego, doble enmascarado, de grupos paralelos, controlado con activo |
LIXIANAa: 30 mg 1 vez al día, VO |
n = 7002 |
70,6 (27–95) |
61,2/38,8 |
|
LIXIANAa: 60 mg, 1 vez al día, VO |
n = 7012 |
70,6 (25–96) |
62,1/37,9 |
|
|
Warfarina: 1 vez al día, VO Dosis ajustada para mantener la RNI entre 2,0 y 3,0 |
n = 7012 |
70,5 (27–95) |
62,5/37,5 |
|
|
Duración promedio del tratamiento de 2,5 años. |
Total = 21,026 |
FA = fibrilación atrial.
a Reducción de la dosis (30 mg a 15 mg una vez al día; 60 mg a 30 mg una vez al día) para insuficiencia renal moderada, bajo peso corporal o medicamentos concomitantes específicos.
b Todos los pacientes tratados que recibieron el fármaco dentro de 3 días desde la última dosis administrada.
Los pacientes estaban bien equilibrados con respecto a los datos demográficos y las características del periodo basal. Los porcentajes de pacientes de edad ≥ 75 años y ≥ 80 años fueron aproximadamente el 40% y el 17%, respectivamente. Las enfermedades concomitantes de los pacientes en este estudio incluyeron hipertensión (94%), insuficiencia cardiaca congestiva (58%) y accidente cerebrovascular previo o ataque isquémico transitorio (28%). En el periodo basal, aproximadamente el 30% de los pacientes administraban aspirina y aproximadamente el 2% de los pacientes administraban una tienopiridina.
Los pacientes fueron excluidos si tenían una depuración de creatinina <30 mL/min, enfermedad hepática significativa, cáncer, sangrado activo, síndrome coronario agudo o intervención coronaria percutánea (ICP) (dentro de los 30 días anteriores). Los pacientes con válvulas cardiacas protésicas, o aquellos con cardiopatía reumática hemodinámicamente significativa, especialmente estenosis mitral, también fueron excluidos del estudio y, por tanto, no se evaluaron. Se debe señalar que aproximadamente el 20% de los pacientes tenían otra cardiopatía valvular que incluía estenosis aórtica, regurgitación aórtica y/o regurgitación mitral. Los pacientes con historial de reparación de la válvula mitral no se excluyeron del estudio.
El criterio de valoración de eficacia primario fue el compuesto de accidente cerebrovascular y eventos embólicos sistémicos (SEE) que ocurrieron durante el tratamiento o dentro de los 3 días posteriores a la última dosis administrada (durante el tratamiento ITTm: véase la Tabla 5 para la definición). Los criterios de valoración de eficacia secundarios incluyeron: Compuesto de accidente cerebrovascular, SEE y mortalidad cardiovascular (CV); evento cardiovascular adverso importante (MACE), que es el compuesto de IM no fatal, accidente cerebrovascular no fatal, SEE no fatal y muerte a causa de CV o sangrado; compuesto de accidente cerebrovascular, SEE y mortalidad por todas las causas.
La mediana de la exposición al fármaco del estudio para los grupos de tratamiento con LIXIANA 60 mg y 30 mg fue de 2,5 años. La mediana de seguimiento del estudio para los grupos de tratamiento con LIXIANA 60 mg y 30 mg fue de 2,8 años.
Eficacia en FA:
En el estudio ENGAGE AF-TIMI 48, los regímenes de grupo de LIXIANA 30 mg y 60 mg no fueron inferiores a la warfarina para el criterio de valoración de eficacia primario con el límite superior de IC del 97,5%, inferior al margen de no inferioridad preespecificado de 1,38. Sin embargo, el régimen de 30 mg fue numéricamente menos efectivo que la warfarina para el criterio de valoración primario, y también fue notablemente inferior en la reducción de la tasa de accidente cerebrovascular isquémico (Tabla 5).
En el grupo de warfarina, el TRT mediano (tiempo en el rango terapéutico, RNI 2,0 a 3,0) fue del 68,4%.
Tabla 5. Resultados de Eficacia del Estudio ENGAGE AF-TIMI 48 (conjunto de análisis ITTm durante el tratamiento)
|
Criterio de Valoración Primario |
LIXIANA 30 mg (Dosis reducida a 15 mg) (N = 7002) |
LIXIANA 60 mg (Dosis reducida a 30 mg) (N = 7,012) |
Warfarina (N = 7,012) |
|
Primer accidente cerebrovascular o SEEa |
|||
|
n (%/año)b |
253 (1,61) |
182 (1,18) |
232 (1,5) |
|
TR (97,5% IC) |
1,07 (0,874, 1,314) |
0,79 (0,632, 0,985) |
|
|
Valor pc |
0,0055 |
<0,0001 |
|
|
Primer accidente cerebrovascular isquémico |
|||
|
n (%/año)b |
226 (1,43) |
135 (0,87) |
144 (0,93) |
|
HR (IC del 95%) |
1,54 (1,253, 1,903) |
0,94 (0,746, 1,193) |
|
|
Primer accidente cerebrovascular hemorrágico |
|||
|
n (%/año)b |
18 (0,11) |
40 (0,26) |
76 (0,49) |
|
HR (IC del 95%) |
0,23 (0,139, 0,389) |
0,53 (0,362, 0,778) |
|
|
Primer SEE |
|||
|
n (%/año)a |
11 (0,07) |
8 (0,05) |
13 (0,08) |
|
HR (IC del 95%) |
0,83 (0,370, 1,850) |
0,62 (0,257, 1,497) |
|
Abreviaturas: HR= Tasa de Riesgo versus warfarina, IC= Intervalo de Confianza, n = número de eventos, ITTm = intención de tratar modificada, N = número de pacientes en la población de ITTm, SEE= Embolismo Sistémico.
Nota: La población de ITTm incluyó sólo a pacientes que recibieron al menos una dosis del fármaco; y el periodo durante el tratamiento fue el periodo en el que el paciente administró el fármaco del estudio, a menos que el paciente haya discontinuado prematuramente el fármaco, caso en el que el periodo durante el tratamiento incluyó los 3 días después de la discontinuación.
a Un paciente puede ser representado en múltiples líneas.
b La tasa de evento (%/año) se calcula como el n.º de eventos/exposición paciente-año.
c El valor p bilateral se basa en el margen de no inferioridad de 1,38.
Los pacientes que recibieron LIXIANA 30 mg (pacientes con dosis reducida en el grupo de 60 mg) tuvieron una tasa de eventos del 1,79% al año para el criterio de valoración primario, en comparación con una tasa de eventos del 2,21% al año para los pacientes con la dosis correspondiente reducida en el grupo de warfarina. En comparación con los pacientes tratados con warfarina, la HR en LIXIANA 30 mg (pacientes con dosis reducida en el grupo de 60 mg) fue de 0,81 (IC del 95%: 0,58, 1,13).
Figura 2. Estimación de la Curva de Kaplan-Meier de las Tasas de Eventos Acumulativos para el Criterio de Valoración Primario (primera aparición de accidente cerebrovascular o SEE) (conjunto de análisis ITTm–durante el periodo del tratamiento del estudio) en el estudio ENGAGE AF-TIMI 48
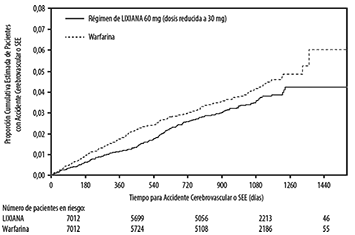
La HR para los criterios de valoración compuestos para la comparación del grupo de LIXIANA 60 mg (dosis reducida a 30 mg) y warfarina para el accidente cerebrovascular, la SEE y la mortalidad cardiovascular (CV) fue de 0,87 (IC del 95%: 0,79, 0,96), MACE fue de 0,89 (IC del 95%: 0,81, 0,97) y accidente cerebrovascular, el SEE y la mortalidad por todas las causas fueron de 0,90 (IC del 95%: 0,82 a 0,98).
Resultados en Subgrupos de Interés:
Los resultados de eficacia para los subgrupos principales pre especificados (con reducción de dosis según sea necesario), incluidos la edad, el peso corporal, el accidente cerebrovascular anterior o el AIT, la diabetes y los inhibidores de la P-gp fueron generalmente consistentes con los resultados de eficacia primarios para los estudios de población general en el estudio.
Sin embargo, hubo una interacción estadísticamente significativa entre el efecto de edoxabán versus warfarina en el criterio de valoración de eficacia primario basado en la función renal (HR fue de 1,41 a favor de la warfarina para el subgrupo con CrCL ≥ 80 mL/min) y regiones geográficas (la HR fue de 1,47 a favor de la warfarina para Europa Occidental) (Figura 3).
Figura 3. Estudio ENGAGE AF-TIMI 48: Criterio de Valoración de Eficacia Primario por Subgrupos (tratamiento de ITTm)
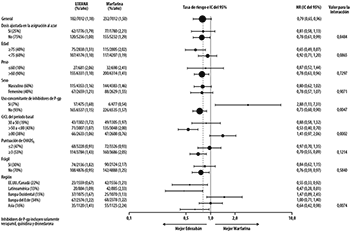
Nota: En los siguientes grupos de pacientes, la dosis de edoxabán se redujo a 30 mg: Peso ≤60 kg, CrCL 30 a 50 mL/min y uso concomitante de inhibidores de la P-gp.
Los pacientes frágiles incluyeron a los pacientes que tenían ≥80 años, peso ≤50 kg, CrCL ≤50 mL/min y/o historial de caída.
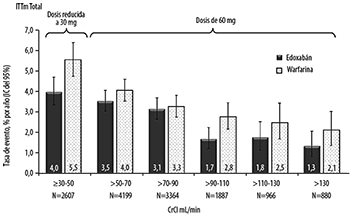
Se llevó a cabo un análisis exploratorio adicional para los criterios de valoración primarios de eficacia y seguridad mediante intervalos de CrCl de 20 mL/min. Las diferencias porcentuales observadas en la eficacia en los grupos con mayor depuración de creatinina entre edoxabán y warfarina fueron numéricamente pequeñas y, en particular, con intervalos de confianza superpuestos. Las tasas del evento de accidente cerebrovascular/SEE en el grupo de edoxabán se mantuvieron en pacientes con CrCL entre 70 y 130 mL/min. Hubo un efecto desfavorable, pero no significativo de edoxabán en comparación con la warfarina en pacientes con CrCl por encima de 130 mL/min para los cuales hubo menos eventos (Figura 4). Para hemorragias importantes, la tendencia a favor de edoxabán 60 mg (30 mg de dosis reducida) en comparación con la warfarina se mantuvo en todo el continuo de la función renal (Figura 5).
Figura 4. Tasa de Evento de Accidente Cerebrovascular/SEE por el Conjunto de Análisis de ITTm de CrCl del Periodo Basal, Periodo de Estudio Total-Estudio ENGAGE AF-TIMI 48
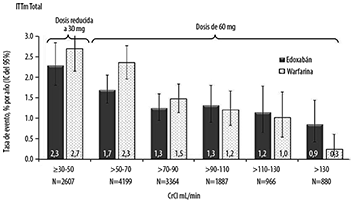
Figura 5. Sangrados Importantes por Categoría de CrCl del Periodo Basal en el Estudio ENGAGE AF-TIMI 48
Transición a Otros Anticoagulantes:
En el estudio ENGAGE AF-TIMI 48, los esquemas de transición descritos en la Tabla 16 fueron efectivos al realizar la transición a VKA, inhibidores del factor Xa y de IIa al final del estudio. El esquema de transición incluyó la mitad de la dosis de LIXIANA durante ≤14 días concomitantemente con VKA. La tasa de accidente cerebrovascular y SEE durante los 30 días posteriores a la última dosis del fármaco del estudio ciego fue similar para aquellos que hicieron la transición fuera de LIXIANA y para aquellos que hicieron la transición fuera de warfarina. En el grupo de LIXIANA 60 mg, 7 de 4529 (0,2%) pacientes tuvieron un accidente cerebrovascular o SEE en comparación con 7 de 4506 (0,2%) pacientes en el brazo de warfarina.
Seguridad en FA:
El criterio de valoración primario de seguridad fue el sangrado importante. El criterio de valoración secundario de seguridad fue sangrado importante o sangrado no importante clínicamente relevante (CRNM).
Tabla 9 resume los eventos de sangrado adjudicados para el análisis de seguridad establecido en el periodo de tratamiento. Los pacientes del grupo de LIXIANA 60 mg (30 mg de dosis reducida) presentaron eventos significativamente menores de sangrado para todas las categorías de sangrado (Sangrado importante, CRNM y cualquier sangrado confirmado) en comparación con la warfarina.
La tasa de hemorragia importante fue significativamente menor en el grupo de LIXIANA 60 mg (dosis reducida a 30 mg) en comparación con el grupo de warfarina (2,75% y 3,43% al año, respectivamente) [HR (IC del 95%): 0,80 (0,71, 0,91)]; p = 0,0009]. Se observaron beneficios similares a favor del grupo de LIXIANA 60 mg (reducción de dosis a 30 mg) en comparación con el grupo de warfarina para el subconjunto de pacientes que presentan ICH (0,39% y 0,85%), respectivamente [HR (IC del 95%): 0,47 (0,34, 0,63); p <0,0001].
La tasa en hemorragias fatales también fue significativamente menor en el grupo de LIXIANA 60 mg (dosis reducida a 30 mg) en comparación con el grupo de warfarina (0,21% y 0,38%) [HR (IC del 95%): 0,55 (0,36, 0,84); p = 0,0059 para superioridad.
Los pacientes que recibieron LIXIANA 30 mg (pacientes con dosis reducida en el grupo de 60 mg) tuvieron una tasa de evento del 3,05% al año para sangrados importantes, en comparación con la tasa de eventos del 4,85% al año para los pacientes correspondientes con dosis reducida en el grupo de warfarina. En comparación con los pacientes tratados con warfarina, la HR en el grupo de LIXIANA 30 mg (pacientes con dosis reducida en el grupo de 60 mg) fue de 0,63 (IC del 95%: 0,50, 0,81).
Los análisis de subgrupos mostraron que el grupo de LIXIANA 60 mg (dosis reducida a 30 mg) tuvo una tasa de evento menor y una tasa de riesgo de menos de 1 para sangrado importante en comparación con el grupo de warfarina para todos los subgrupos, a excepción de los pacientes con historial de AIT solo. En el subgrupo de pacientes con un alto riesgo de sangrado, como edad ≥ 75 años, CrCL 30 a ≤ 50 y >50 a <80 mL/min y puntuación CHADS2 ≥3, el grupo de LIXIANA 60 mg (dosis reducida a 30 mg) tuvo una tasa de riesgo inferior a 1 para sangrado importante en comparación con la warfarina.
Tratamiento del TEV y prevención de TVP y EP recurrentes:
El estudio HOKUSAI VTE:
Diseño del Estudio y Datos Demográficos del Estudio:
El programa clínico de LIXIANA para el tromboembolismo venoso (TEV) se diseñó para demostrar la eficacia y la seguridad de LIXIANA en el tratamiento de la trombosis venosa profunda (TVP) y la embolia pulmonar (EP), y la prevención de la TVP recurrente y de la EP.
En el estudio pivotal HOKUSAI-VTE, 8,292 pacientes se asignaron al azar para recibir tratamiento inicial con heparina (enoxaparina o heparina no fraccionada durante 5-10 días), seguido de LIXIANA 60 mg una vez al día o el comparador. En el brazo del comparador, los pacientes recibieron tratamiento inicial con heparina concomitantemente con warfarina, titulada en una razón normalizada internacional (RNI) de 2,0 a 3,0, seguida de warfarina sola. La duración del tratamiento fue de 3 meses a 12 meses, determinada por el investigador en función de las características clínicas del paciente. Los pacientes eran excluidos si requirieran trombectomía, inserción de un filtro caval, uso de un agente fibrinolítico, presentaran una depuración de creatinina < 30 mL/min, enfermedad hepática significativa o sangrado activo. El criterio de valoración de eficacia primario fue la recurrencia de la TEV sintomática, definida como el compuesto de TVP sintomática recurrente, EP sintomática no fatal y EP fatal en pacientes durante el periodo de estudio de 12 meses. Los resultados de eficacia secundaria incluyeron el resultado clínico compuesto de TEV recurrente y la mortalidad por todas las causas.
Tabla 6. Resumen de los datos demográficos de los pacientes para los estudios clínicos de TEV
|
Diseño del estudio |
Dosificación, vía de administración y duración |
Pacientes del Estudio (n = número) |
Edad promedio (Rango) años |
Sexo (M/F) % |
|
Aleatorizado, doble ciego, placebo correspondiente, de grupos paralelos, controlado con activo |
LIXIANA 60 mg 1 vez al día VOa Duración promedio del tratamiento = 267 días |
N = 4118 |
55,7 (18–106) |
57,3/42,7 |
|
Warfarina 1 vez al día VOb Duración promedio del tratamiento = 266 días |
N = 4122 |
55,9 (18–95) |
57,2/42,8 |
|
|
Total = 8240 |
TEV = tromboembolismo venoso.
a La dosis de LIXIANA reducida a la mitad para pacientes con insuficiencia renal moderada [CrCL ≥ 30 y ≤ 50 mL/min], bajo peso corporal [≤60 kg], o con inhibidor fuerte de la P-gp concomitante [ej., verapamilo, quinidina].
b Dosis de warfarina ajustada para mantener la RNI entre 2,0 y 3,0, incluso.
Los pacientes en el grupo de tratamiento con LIXIANA 60 mg redujeron su dosis a la mitad si uno o más de los siguientes estuvieran presentes: insuficiencia renal moderada (CrCL30-50 mL/min); peso corporal ≤ 60 kg; uso concomitante de inhibidores específicos de la P-gp (verapamilo y quinidina o la administración concomitante a corto plazo de azitromicina, claritromicina, eritromicina, itraconazol oral o ketoconazol oral).
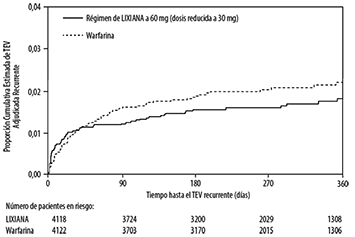
Eficacia en el TEV:
En el estudio HOKUSAI-VTE (Tabla 7), se demostró que LIXIANA es no inferior a la warfarina para el resultado de eficacia primaria, TEV recurrente, que ocurrió en 130 de 4118 pacientes (3,2%) en el grupo de LIXIANA versus 146 de 4122 pacientes (3,5%) en el grupo de warfarina [HR (IC del 95%): 0,89 (0,70, 1,13); p <0,0001 para la no inferioridad en un margen pre especificado de 1,5]. En el grupo de warfarina, el TRT (tiempo en el rango terapéutico, RNI 2,0 a 3,0) mediano fue del 63,5%. Para los pacientes que presentaron PE (con o sin TVP), 47 (2,8%) de los pacientes de LIXIANA y 65 (3,9%) de los pacientes de warfarina tuvieron un TEV recurrente [HR (IC del 95%): 0,73 (0,50, 1,06)]. Para los pacientes que presentaron TVP, 83 (3,4%) de los pacientes de LIXIANA y 81 (3,3%) de los pacientes de warfarina tuvieron un TEV recurrente [HR (IC del 95%): 1,02 (0,75, 1,38)].
Para los pacientes que recibieron la dosis de 30 mg (predominantemente pacientes con peso corporal ≤ 60 kg o insuficiencia renal moderada), 22 (3,0%) de LIXIANA y 30 (4,2%) de los pacientes con warfarina tuvieron un TEV recurrente.
El criterio de valoración compuesto de TEV recurrente y la mortalidad por todas las causas se produjeron en 228 de los pacientes (5,5%) en el grupo de LIXIANA y en 228 pacientes (5,5%) en el grupo de warfarina [HR: (IC del 95%): 1,00 (0,83, 1,20)].
En el estudio HOKUSAI-VTE, la duración de la exposición al fármaco para LIXIANA 60 mg fue ≤ 6 meses para 1561 (37,9%) pacientes, > 6 meses para 2557 (62,1%) pacientes y 12 meses para 1661 (40,3%) pacientes.
Tabla 7. Resultados de Eficacia del Estudio HOKUSAI-VTE (Periodo Total de ITTm del Estudio)
|
LIXIANA 60 mg (Dosis reducida a 30 mg) (N = 4118) |
Warfarina (N = 4122) |
LIXIANA vs. Warfarina HR (IC del 95%) |
|
|
Todos los pacientes con TEV sintomática recurrente,a n (%) |
130 (3,2) |
146 (3,5) |
0,89 (0,70, 1,13) valor p<0,0001 (no inferioridad) |
|
EP con o sin TVP |
73 (1,8) |
83 (2,0) |
|
|
Óbito/EP fatal no se puede descartar el EP |
24 (0,6) |
24 (0,6) |
|
|
EP no fatal |
49 (1,2) |
59 (1,4) |
|
|
TVP solo |
57 (1,4) |
63 (1,5) |
Abreviaturas: ITTm = intención de tratar modificada; HR= Tasa de Riesgo vs. warfarina; IC= Intervalo de confianza; N= número de pacientes en la población con ITTm; n= número de eventos.
a Criterio de Valoración de Eficacia Primaria: TVE sintomática recurrente (es decir, el compuesto del criterio de valoración de TVP, EP no fatal y EP fatal).
Nota: El análisis de eficacia primaria se llevó a cabo en el Conjunto de Análisis de ITTm, Periodo de Estudio Total-(todos los eventos que ocurren durante el Periodo de Estudio Total se incluyen independientemente del estado de la administración del fármaco del estudio).
Figura 6. Estimación de la Curva de Kaplan-Meier Respecto a las Tasas Cumulativas de Eventos para el Criterio de Valoración de Eficacia Primario (tratamiento con ITTm en curso) para el Estudio HOKUSAI
Resultados en subgrupos de interés:
Los resultados de eficacia para los subgrupos principales pre-especificados (con una reducción de la dosis según sea necesario), incluidos la edad, el peso corporal y los inhibidores de la P-gp fueron generalmente consistentes con los resultados de eficacia primarios para los estudios de población total en el estudio (Figura 7).
Figura 7. Estudio HOKUSAI-VTE: Criterio de Valoración de Eficacia Primario por Subgrupos (ITTm-total)

Nota: ‘Pacientes frágiles’ incluyó pacientes que tenían ≥ 75 años de edad y/o peso corporal ≤ 50 kg y/o CrCL ≥ 30 a ≤ 50 mL/min, cada uno según se determinó en la asignación al azar.
Seguridad en TEV:
El criterio de valoración principal de seguridad fue el sangrado clínicamente relevante (importante o clínicamente relevante no importante-CRNM) que ocurrió durante o dentro de los tres días de la interrupción o suspensión del tratamiento del estudio. Un criterio de valoración adicional incluyó Eventos Cardiovasculares Adversos Importantes: MACE (infarto de miocardio no fatal, accidente cerebrovascular no fatal, eventos embólicos sistémicos no fatales y muerte cardiovascular).
La Tabla 10 resume los eventos de sangrado adjudicados para el análisis de seguridad establecido en el periodo de tratamiento. LIXIANA demostró ser superior a la warfarina para el criterio de valoración primario de seguridad del sangrado clínicamente relevante, un compuesto de sangrado importante o CRNM, que ocurrió en 349 de 4118 pacientes (8,5%) en el grupo de LIXIANA y en 423 de 4122 pacientes (10,3%) en el grupo de warfarina [HR (IC del 95%): 0,81 (0,71 a 0,94); p = 0,004 para superioridad].
El criterio de valoración compuesto de MACE fue del 1,2% en el grupo de LIXIANA y del 1,0% en el grupo de warfarina.
Los pacientes que recibieron LIXIANA 30 mg (pacientes con dosis reducida en el grupo de 60 mg) tuvieron una tasa de eventos del 7,9% para el sangrado clínicamente relevante, en comparación con la tasa de eventos del 12,8% para los pacientes con dosis reducidas correspondientes en el grupo de la warfarina. En comparación con los pacientes tratados con warfarina, la tasa de riesgo (HR) de LIXIANA 30 mg (pacientes con dosis reducida en el grupo de 60 mg) fue de 0,62 (IC del 95%: 0,44, 0,86).
Los análisis de subgrupos de pacientes frágiles, mayores y pacientes con historial de cáncer demostraron un resultado hemorrágico favorable con la terapia con LIXIANA. Sin embargo, en el grupo de LIXIANA se observaron tasas numéricamente más altas de eventos de sangrado del tracto GI y vaginal.
FARMACOLOGÍA DETALLADA:
Prolongación de QT/QTc:
En un estudio aleatorizado, doble ciego, de dosis única, controlado con placebo y con activo, cruzado de cuatro periodos, las dosis de 90 mg y 180 mg de LIXIANA no afectaron el intervalo QTc, la duración de QRS, el intervalo PR o la frecuencia cardiaca en pacientes sanos (N = 62).
CONTRAINDICACIONES:
El uso de LIXIANA está contraindicado en las siguientes condiciones:
• Sangrado activo clínicamente significativo, incluyendo el sangrado gastrointestinal.
• Lesiones o afecciones con mayor riesgo de sangrado clínicamente significativo, por ejemplo, infarto cerebral reciente (hemorrágico o isquémico), enfermedad ulcerosa péptica activa con sangrado reciente, pacientes con deterioro espontáneo o adquirido de la hemostasia.
• Enfermedad hepática asociada con coagulopatía y riesgo de sangrado clínicamente relevante.
• Tratamiento concomitante con cualquier otro anticoagulante, incluidos:
- heparina no fraccionada (HNF), a excepción de las dosis utilizadas para mantener un catéter arterial o venoso central permeable,
- heparinas de bajo peso molecular (LMWH), como la enoxaparina y la dalteparina,
- derivados de la heparina, como fondaparinux, y
- los anticoagulantes orales, como warfarina, dabigatrán, apixabán, rivaroxabán, excepto en circunstancias de cambio de terapia para o desde LIXIANA.
• Embarazo.
• Mujeres Lactantes.
• Hipersensibilidad a edoxabán o a cualquiera de los ingredientes de la formulación.
RESTRICCIONES DE USO DURANTE EL EMBARAZO Y LA LACTANCIA:
Uso durante el embarazo:
No hay datos disponibles sobre el uso de LIXIANA en mujeres embarazadas. Basado en datos de animales, el uso de LIXIANA está contraindicado durante todo el embarazo.
Si LIXIANA se va a utilizar en mujeres en edad fértil, se debe evitar el embarazo.
La seguridad y la eficacia de LIXIANA durante el trabajo de parto y el parto no se han estudiado en los estudios clínicos. El riesgo de hemorragia relacionada con el embarazo y/o parto urgente aumenta con el uso de un anticoagulante que no es fácilmente reversible.
Los estudios de toxicidad reproductiva y para desarrollo en animales mostraron toxicidades maternas y embriofetales en ratas y conejos en dosis más altas. El rendimiento reproductivo no se vio afectado tanto en ratas como en conejos.
Uso durante la lactancia:
No hay datos disponibles sobre el uso de LIXIANA en madres lactantes. En un estudio no clínico, LIXIANA se excreta en la leche materna de ratas. LIXIANA sólo debe administrarse después de discontinuar la lactancia.
No se sabe si LIXIANA o sus metabolitos se excretan en la leche humana. Debido a que muchos medicamentos se excretan en la leche humana, se debe tener cautela y se debe decidir discontinuar la lactancia o el tratamiento con LIXIANA, teniendo en cuenta la importancia del medicamento para la madre.
REACCIONES SECUNDARIAS Y ADVERSAS:
Visión General de la Reacción Adversa al Fármaco:
FA:
En el estudio pivotal doble ciego aleatorizado ENGAGE AF-TIMI 48, un total de 21,026 pacientes con fibrilación auricular (FA) documentada recibió al menos una dosis de LIXIANA 60 mg (N=7012), LIXIANA 30 mg (N=7002) o warfarina (N=7012). La duración de la exposición a LIXIANA fue ≥360 días para 11,479 pacientes y ≥720 días para 10,075 pacientes. La exposición al fármaco en el estudio mediana para el grupo de tratamiento con LIXIANA y warfarina fue de 2,5 años.
En el estudio ENGAGE AF-TIMI 48, 2256 (32,2%) de los pacientes tratados con LIXIANA 60 mg (30 mg de dosis reducida) presentaron reacciones adversas. Los eventos adversos no relacionados con el criterio de valoración llevaron a la discontinuación del fármaco del estudio en el 11,2% y el 11,0% de los pacientes tratados con LIXIANA 60 mg y los grupos de tratamiento con warfarina, respectivamente.
Tratamiento de TEV y Prevención de TVP y EP Recurrente:
En el estudio pivotal doble ciego aleatorizado HOKUSAI-VTE, los pacientes con TVP aguda sintomática que afecta a las venas poplítea, femoral o ilíaca, o EP que requiere tratamiento anticoagulante fueron tratados con LIXIANA (N=4118) o warfarina (N=4122) después de tratamiento inicial a base de heparina de ≥5 días. Estos 8240 pacientes incluían la población de seguridad. El tiempo medio del tratamiento fue de 8,8 meses en ambos grupos. La duración de la exposición al fármaco para LIXIANA fue ≤ 6 meses para 1561 (37,9%) pacientes, > 6 meses para 2557 (62,1%) de los pacientes y 12 meses para 1661 (40,3%) de los pacientes.
En el estudio HOKUSAI-VTE, en total, 1249 (30,3%) de los pacientes tratados con LIXIANA 60 mg (30 mg de dosis reducida) presentaron reacciones adversas. La frecuencia de eventos adversos no relacionados con el criterio de valoración que llevaron a la discontinuación permanente del fármaco del estudio fue del 5,7% en el grupo LIXIANA y del 5,4% en el grupo de warfarina.
Estudio Clínico de Reacciones Adversas al Fármaco:
Debido a que los estudios clínicos se realizan en condiciones muy específicas, las tasas de reacciones adversas observadas en los estudios clínicos pueden no reflejar las tasas observadas en la práctica y no deben compararse con las tasas en los estudios clínicos de otro fármaco. La información de reacciones adversas a los medicamentos de los estudios clínicos es útil para identificar eventos adversos relacionados con los medicamentos y para tasas aproximadas.
Eventos de Sangrado:
Las reacciones adversas más notables informadas con LIXIANA estaban relacionadas con el sangrado. El sangrado de cualquier tipo de gravedad ocurrió en una tasa del 14,2% al año entre los pacientes con FA tratados con LIXIANA en el estudio ENGAGE-AF TIMI 48 y del 21,7% en el estudio HOKUSAI-VTE. El sangrado puede ocurrir en cualquier sitio y puede ser grave, amenazante a la vida e incluso fatal. Se han reportado complicaciones conocidas secundarias a hemorragias graves, como daño renal aguda debido hipoperfusión.
En ambos estudios, las reacciones adversas más comunes relacionadas con el sangrado con LIXIANA 60 mg (30 mg de dosis reducida) incluyeron hemorragia cutánea de tejido blando (≤ 5,9%) y epistaxis (≤ 4,7%), mientras la hemorragia vaginal (9,0%) fue la reacción adversa relacionada con el sangrado más común en el estudio HOKUSAI-VTE solo.
Debido a que las poblaciones de pacientes tratados con LIXIANA para diferentes indicaciones no son intercambiables, a continuación se proporciona una descripción resumida del sangrado importante y total mediante la indicación y el estudio pivotal.
Estudio ENGAGE AF-TIMI 48:
Tabla 9. Eventos de sangrado adjudicados en pacientes con FA, estudio ENGAGE AF-TIMI 48
|
LIXIANA 60 mg (dosis reducida de 30 mg) (N=7012) |
Warfarina (N=7012) |
LIXIANA 60 mg (dosis reducida de 30 mg) vs. Warfarina |
||
|
Categoría de Sangrado–Primer Evento |
n (%/años) [a] |
n (%/años) [a] |
HR (IC del 95%) |
valor-p |
|
Importante[b] |
418 (2,75) |
524 (3,43) |
0,80 (0,707, 0,914) |
0,0009 |
|
ICH[c] |
61 (0,39) |
132 (0,85) |
0,47 (0,344, 0,631) |
<0,0001 |
|
Gastrointestinal |
232 (1,51) |
190 (1,23) |
1,23 (1,019, 1,496) |
0,0311 |
|
Fatal |
32 (0,21) |
59 (0,38) |
0,55 (0,355, 0,840) |
0,0059 |
|
ICH[c] |
24 (0,15) |
42 (0,27) |
0,58 (0,349, 0,951) |
0,0312 |
|
No-ICH |
8 (0,05) |
17 (0,11) |
0,47 (0,204, 1,095) |
0,0804 |
|
CRNM[d] |
1214 (8,67) |
1396 (10,15) |
0,86 (0,795, 0,927) |
0,0001 |
|
Cualquier Sangrado Confirmado[c] |
1865 (14,15) |
2114 (16,40) |
0,87 (0,816, 0,924) |
<0,0001 |
Abreviaturas: IC = Intervalo de Confianza; ICH = Hemorragia Intracraneal; HR = Tasa de Riesgo versus Warfarina; CRNM= Clínicamente Relevante No Importante.
Nota: Los eventos de sangrado adjudicados incluyen eventos durante el tratamiento o dentro de los 3 días posteriores a la interrupción del tratamiento del estudio.
Un paciente puede ser incluido en múltiples subcategorías si el mismo tuvo un evento para dichas categorías. El primer evento de cada categoría se incluye en el análisis.
[a]: La tasa de evento (%/año) se calcula como n.º de eventos/exposición paciente-año.
[b]: Un evento de Sangrado Importante (el criterio de valoración de seguridad primario del estudio) se definió como un sangrado clínico evidente que cumpliera con uno de los siguientes criterios: sangrado fatal; hemorragia sintomática en un sitio crítico como retroperitoneal, intracraneal, intraocular, intraespinal, intraarticular, pericárdico o intramuscular con síndrome compartimental; un evento de hemorragia clínicamente evidente que causó una caída en la hemoglobina de al menos 2,0 g/dL (o una caída en el hematócrito de al menos el 6,0% en la ausencia de datos de hemoglobina), cuando se ajustó para transfusiones (1 unidad de transfusión = 1,0 g/dL de caída en la hemoglobina).
[c]: La ICH incluye al accidente cerebrovascular hemorrágico primario, hemorragia subaracnoidea, hemorragia epi/subdural y accidente cerebrovascular isquémico con conversión hemorrágica importante.
[d]: CRNM (sangrado no importante clínicamente relevante) se definió como un evento de sangrado evidente que requirió atención médica, incluidos aquellos que pueden haber llevado a medidas diagnósticas o terapéuticas.
[e]: Cualquier sangrado confirmado incluye a aquellos que el adjudicador define como clínicamente evidente.
El sitio de sangrados importantes fue principalmente en el tracto gastrointestinal (GI), seguido de los sitios intracraneal e intraocular. Hubo más hemorragias GI Importantes en el grupo de LIXIANA 60 mg (reducción de dosis a 30 mg) que en el grupo de warfarina (1,5% y 1,2% por año, respectivamente).
Una mayor proporción de pacientes tratados con LIXIANA informó eventos relacionados con la anemia; 8,2% (578/7012) de los pacientes con 60 mg (reducción de dosis a 30 mg) en comparación con el 5,6% (396/7012) de los pacientes tratados con warfarina. Similarmente, se informaron más eventos de anemia y relacionados con anemia como serios o graves para el grupo de LIXIANA 60 mg (reducción de dosis a 30 mg) (1,4%) en comparación con el grupo de warfarina (0,7%). La mayoría de los eventos de sangrado que ocurrieron en los pacientes tratados con LIXIANA 60 mg (reducción de dosis a 30 mg) con anemia/eventos relacionados con la anemia seria o grave fueron eventos en el tracto GI. Se han identificado varios factores de riesgo de aumentar el riesgo de hemorragia, lo que puede ocasionar anemia post hemorrágica y están asociados con ajustes de dosis.
El porcentaje de pacientes que discontinuaron el fármaco del estudio a causa de eventos de sangrado informado por el Investigador fue del 3,9% y el 4,1%, respectivamente, para LIXIANA 60 mg (reducción de dosis a 30 mg) y los grupos de tratamiento con warfarina.
Estudio HOKUSAI-VTE:
Tabla 10. Eventos de hemorragia adjudicada en pacientes con VTE, Estudio HOKUSAI-VTE
|
Hemorragia adjudicada |
LIXIANA 60 mg (reducción de dosis a 30 mg) N=4118 |
Warfarina N=4122 |
LIXIANA vs. Warfarina |
|
|
HR (IC del 95%) |
valor-p |
|||
|
Sangrado Importante/CRNM, n (%) |
349 (8,5) |
423 (10,3) |
0,81 (0,705, 0,936) [a] |
0,0040 [a] |
|
ICH, n (%) |
5 (0,1) |
18 (0,4) |
- |
- |
|
Gastrointestinal, n (%) |
98 (2,4) |
94 (2,3) |
- |
- |
|
Sangrado Importante, n (%) [b] |
56 (1,4) |
66 (1,6) |
0,84 (0,592, 1,205) [a] |
0,3521 [a] |
|
ICH, n (%) |
5 (0,1) |
18 (0,4) |
- |
- |
|
ICH fatal, n (%) |
0 (0) |
6 (0,1) |
- |
- |
|
Gastrointestinal, n (%) |
27 (0,7) |
18 (0,4) |
- |
- |
|
Todos los sangrados, n (%) |
895 (21,7) |
1056 (25,6) |
0,82 (0,750, 0,896) |
<0,0001 |
Abreviaturas: IC = Intervalo de Confianza, CRNM = Clínicamente Relevante No Importante, HR = Tasa de Riesgo vs. Warfarina, ICH: hemorragia intracraneal; N = número de pacientes en el conjunto de análisis, n = número de pacientes que cumplieron los criterios del evento.
Nota: Los eventos de sangrado adjudicados incluyen eventos durante el tratamiento o dentro de los 3 días posteriores a la interrupción del tratamiento del estudio.
[a] La HR y el IC bilateral se basan en el modelo de regresión de riesgos proporcionales de Cox, que incluye el tratamiento y los siguientes factores de estratificación de asignación al azar como covariables: diagnóstico de presentación (EP con o sin TVP, TVP únicamente), factores de riesgo del periodo inicial (factores temporales, todos los demás) y la necesidad de 30 mg de LIXIANA/dosis de placebo de LIXIANA en asignación al azar (sí, no), valor p α = 0,01 [bilateral].
[b] Un Evento de Sangrado Importante se definió como una hemorragia clínicamente evidente que cumplía uno de los siguientes criterios: que estuviera asociada con una caída en el nivel de hemoglobina de 2,0 g/dL o más, o que requiriera la transfusión de dos o más unidades de glóbulos rojos empaquetados o sangre completa; que ocurriera en un sitio u órgano crítico: intracraneal, intraespinal, intraocular, pericárdico, intraarticular, intramuscular con síndrome compartimental, retroperitoneal; que contribuyera para la muerte.
El porcentaje de pacientes que discontinuaron el fármaco del estudio a causa de eventos de sangrado informados por el Investigador fue del 1,4 % en ambos grupos.
Reacción adversa a medicamentos más común:
Las reacciones adversas debidas al tratamiento de no sangrado más frecuentes informadas en el Estudio ENGAGE AF-TIMI 48 para el grupo de LIXIANA 60 mg (reducción de dosis a 30 mg) frente warfarina fueron erupción cutánea (4,2% vs. 4,1%) y pruebas de función hepática anormales (4,8% vs. 4,6%), respectivamente. Los resultados se presentan a continuación en la Tabla 11.
Tabla 11. Reacciones adversas al fármaco comunes observadas en ≥ 1% de los pacientes tratados con LIXIANA en el estudio ENGAGE AF-TIMI 48
|
LIXIANA 60 mg (Reducción de dosis a 30 mg) N = 7012 n (%)a |
Warfarina N = 7012 n (%)a |
|
|
Trastornos Respiratorios, Torácicos y Mediastínicos |
||
|
Epistaxis |
392 (2,6) |
359 (2,4) |
|
Trastornos Gastrointestinales |
||
|
Hemorragia GI Inferior |
411 (2,7) |
264 (1,7) |
|
Hemorragia GI Superior |
187 (1,2) |
144 (0,9) |
|
Trastornos de la Piel y del Tejido Subcutáneo |
||
|
Hemorragia cutánea de tejido blando |
577 (3,8) |
947 (6,6) |
|
Trastornos Renales y Urinarios |
||
|
Hematuria macroscópica/uretral |
293 (1,9) |
255 (1,7) |
|
Trastornos del Sistema Sanguíneo y Linfático |
||
|
Anemia |
368 (5,2) |
242 (3,5) |
|
Trastornos de la Piel y del Tejido Subcutáneo |
||
|
Erupción cutánea |
295 (4,2) |
289 (4,1) |
|
Investigaciones |
||
|
Prueba de función hepática anormal |
337 (4,8) |
326 (4,6) |
a Resumen de los eventos de sangrado adjudicados por sitio (%/año).
Las reacciones adversas al fármaco debidas al tratamiento más comunes en el Estudio HOKUSAI-VTE se presentan a continuación en la Tabla 12.
Tabla 12. Reacciones Adversas Comunes Observadas en ≥ 1% de los Pacientes Tratados con LIXIANA en el Estudio HOKUSAI-VTE
|
LIXIANA 60 mg (Reducción de dosis a 30 mg) N = 4118 n (%)a |
Warfarina N = 4122 n (%)a |
|
|
Trastornos Respiratorios, Torácicos y Mediastínicos |
||
|
Epistaxis |
195 (4,7) |
237 (5,7) |
|
Trastornos Gastrointestinales |
||
|
Hemorragia GI Inferior |
141 (3,4) |
126 (3,1) |
|
Hemorragia Oral/Faríngea |
138 (3,4) |
162 (3,9) |
|
General |
||
|
Hemorragia en el sitio de punción |
56 (1,4) |
99 (2,4) |
|
Trastornos de la Piel y del Tejido Subcutáneo |
||
|
Hemorragia cutánea del tejido blando |
245 (5,9) |
414 (10,0) |
|
Erupción cutánea |
147 (3,6) |
151 (3,7) |
|
Trastornos Renales y Urinarios |
||
|
Hematuria macroscópica/uretral |
91 (2,2) |
117 (2,8) |
|
Sistema Reproductivo y Trastornos de la Mama |
||
|
Hemorragia Vaginal |
158 (9,0) |
126 (7,1) |
|
Trastornos del Sistema Sanguíneo y Linfático |
||
|
Anemia |
72 (1,7) |
55 (1,3) |
|
Investigaciones |
||
|
Prueba de función hepática anormal |
322 (7,8) |
322 (7,8) |
a Resumen de los eventos de sangrado adjudicados por sitio (%). Para la categoría específica de sexo (sangrado vaginal), la tasa del evento se basa en los números de pacientes específicos del sexo.
Reacciones Adversas al Fármaco de los Estudios Clínicos Menos Comunes (<1%) (No se informa de otra manera):
ENGAGE AF-TIMI 48:
Trastornos Oculares: Hemorragia intraocular.
Trastornos Cardiacos: Hemorragia pericárdica.
Trastornos Respiratorios, Torácicos y Mediastínicos: Hemoptisis, Enfermedad Pulmonar Intersticial.
Trastornos Gastrointestinales: Hemorragia Oral/Faríngea, Hemorragia Retroperitoneal.
Trastornos Musculoesqueléticos y del Tejido Conectivo: Intramuscular (sin síndrome compartimental), Hemorragia intraarticular.
Sistema Reproductivo y Trastornos Mamarios: Hemorragia vaginal.
Vascular: Otra hemorragia (incluido el subconjuntivo, del oído, pleural).
General: Hemorragia en el sitio de punción.
Lesiones, Intoxicaciones y Complicaciones de Procedimiento: Hemorragia en el sitio quirúrgico.
Hokusai VTE:
Trastornos Oculares: Conjuntiva/Hemorragia escleral, Hemorragia intraocular.
Trastornos Cardiacos: Hemorragia pericárdica.
Trastornos Respiratorios, Torácicos y Mediastínicos: Hemoptisis.
Trastornos Musculoesqueléticos y del Tejido Conectivo: Intramuscular (sin síndrome compartimental), Hemorragia intraarticular.
Vascular: Otra hemorragia (incluyendo en el sitio quirúrgico, pleural).
Lesión, Intoxicación y Complicaciones de Procedimiento: Hemorragia Subdural, Hemorragia de procedimiento.
Reacciones adversas a medicamentos posteriores al comercialización: Las reacciones adversas a los medicamentos enumeradas a continuación provienen de todas las fuentes de informes.
Trastornos de la sangre y del sistema linfático: Trombocitopenia.
Trastornos gastrointestinales: Dolor abdominal.
Trastornos del sistema inmunitarios: Angioedema, edema alérgico, hipersensibilidad, urticaria.
Trastornos del sistema nervioso: Mareos, dolor de cabeza.
Trastornos de la piel y del tejido subcutáneo: Síndrome de Stevens-Johnson.
PRECAUCIONES EN RELACIÓN CON EFECTOS DE CARCINOGÉNESIS, MUTAGÉNESIS, TERATOGÉNESIS Y SOBRE LA FERTILIDAD:
Los datos no clínicos no revelan riesgos especiales para los seres humanos según los estudios convencionales de farmacología de seguridad, toxicidad a dosis repetidas, genotoxicidad, potencial carcinogénico o fototoxicidad.
Toxicidad a dosis Repetidas:
En los estudios de toxicidad oral a dosis repetidas en ratas, se observó una pequeña cantidad de lesiones hemorrágicas focales en el páncreas, el pulmón y el timo de ratas que recibieron edoxabán tosilato hidrato a ≥20 mg/kg/día. En estudios de toxicidad oral a dosis repetidas en monos cynomolgus, se observaron hallazgos hemorrágicos y anemia en algunos animales que recibieron el fármaco a ≥15 mg/kg/día, lo que llevó a un deterioro del estado del animal o al óbito de animales con dosis crónicas en algunos monos.
Se cree que dichos hallazgos están relacionados con el efecto anticoagulante del edoxabán tosilato hidrato (su principal acción farmacológica), que constituye la única toxicidad limitante de la dosis para este compuesto. Debido a que la actividad farmacológica del fármaco para el mono cynomolgus era comparable a aquella de los humanos, se estimaron los márgenes de seguridad para el riesgo hemorrágico mediante la comparación de las exposiciones entre los monos cynomolgus y los humanos. Los valores medios de AUC0-24h en NOAEL en el estudio de toxicidad oral de dosis repetidas de 52 semanas en monos cynomolgus fueron aproximadamente 2,1 veces más altas que las exposiciones en pacientes humanos que recibieron edoxabán en la dosis clínica máxima recomendada de 60 mg.
Carcinogénesis, Mutagénesis:
Edoxabán no fue carcinogénico cuando se administró diariamente a ratones y ratas mediante sonda oral durante ≤104 semanas. La dosis más alta probada (500 mg/kg/día) en ratones machos y hembras fue 3 y 6 veces, respectivamente, la exposición humana (AUC) a la dosis humana de 60 mg/día, y las dosis más altas probadas en ratas machos (600/400 mg/kg/día) y hembras (200 mg/kg/día) fueron 8 y 14 veces, respectivamente, la exposición humana a la dosis humana de 60 mg/día.
Genotoxicidad:
Basado en el peso de la evidencia, no se consideró que el edoxabán tosilato hidrato y su metabolito M-4 específico para el ser humano presenten cualquier riesgo genotóxico para los humanos.
Toxicología Reproductiva y Lactancia:
Edoxabán mostró hemorragia vaginal a dosis más altas en ratas y conejos, pero no tuvo efectos en el rendimiento reproductivo de las ratas progenitoras.
En ratas, no se observaron efectos sobre la fertilidad masculina o femenina.
En los estudios de reproducción animal, los conejos mostraron una mayor incidencia de variaciones de la vesícula biliar a una dosis de 200 mg/kg [aproximadamente 65 veces la dosis máxima recomendada en humanos (MRHD) de 60 mg/día en función del área de superficie corporal total en mg/m2]. Hubo aumento de las pérdidas en el embarazo después de la implantación en ratas a 300 mg/kg/día (aproximadamente 49 veces la MRHD) y en conejos a 200 mg/kg/día (aproximadamente 65 veces la MRHD), respectivamente.
Se encontró edoxabán en fetos de ratas preñadas y se excretó en la leche materna de ratas lactantes.
INTERACCIONES MEDICAMENTOSAS Y DE OTRO GÉNERO:
Visión General:
Los estudios in vitro indican que edoxabán es un sustrato del transportador de la glucoproteína-p (P-gp); por tanto, su concentración plasmática puede aumentar en presencia de inhibidores de la P-gp, como ciclosporina, dronedarona, eritromicina o ketoconazol (véase la Tabla 6). Edoxabán no inhibe las principales enzimas del citocromo P450 (CYP1A2, 2A6, 2B6, 2C8/9, 2C19, 2D6, 2E1 o 3A4) y no induce la CYP1A2, CYP3A4 o el transportador de la P-gp (MDR1). Los datos in vitro también indican que edoxabán no inhibe los siguientes transportadores en concentraciones clínicamente relevantes: P-gp, los transportadores de los aniones orgánicos OAT1 u OAT3; los transportadores de cationes orgánicos OCT1 u OCT2; o los polipéptidos transportadores de iones orgánicos OATP1B1 u OATP1B3.
Interacciones Medicamentosas:
Los fármacos enumerados en esta tabla se basan en los informes o estudios de casos de interacciones medicamentosas, o posibles interacciones debidas a la magnitud esperada y la gravedad de la interacción (es decir, aquellos identificados como contraindicados).
Tabla 13. Interacciones Medicamentosas Establecidas o Potenciales
|
Nombre Apropiado |
Ref |
Efecto |
Comentario clínico |
|
Inhibidores de la P-gp/Sustratos |
|||
|
Ciclosporina |
CT |
Administración concurrente de una dosis única de 500 mg de ciclosporina con una dosis única de LIXIANA 60 mg aumentó el AUC y la Cmax de LIXIANA en el 73% y el 74%, respectivamente. |
El uso concomitante de LIXIANA con ese fármaco requiere una reducción de la dosis a 30 mg una vez al día. |
|
Dronedarona |
CT |
Dronedarona 400 mg dos veces al día por 7 días con una dosis única concomitante de LIXIANA 60 mg en el Día 5 aumentó el AUC y la Cmax de LIXIANA en el 85% y el 46%, respectivamente. |
El uso concomitante de LIXIANA con este fármaco requiere la reducción de la dosis a 30 mg una vez al día. |
|
Eritromicina |
CT |
Eritromicina 500 mg cuatro veces al día por 8 días con una dosis única concomitante de LIXIANA 60 mg en el Día 7 aumentó el AUC y la Cmax de LIXIANA en el 85% y el 68%, respectivamente. |
El uso concomitante de LIXIANA con este fármaco requiere una reducción de la dosis a 30 mg una vez al día. |
|
Ketoconazol |
CT |
Ketoconazol 400 mg una vez al día por 7 días con una dosis única concomitante de LIXIANA 60 mg el Día 4, aumentó el AUC y la Cmax de LIXIANA en el 87% y el 89%, respectivamente. |
El uso concomitante de LIXIANA con este fármaco requiere una reducción de la dosis a 30 mg una vez al día. |
|
Quinidina |
CT |
Quinidina 300 mg una vez al día en los Días 1 y 4 y tres veces al día en los Días 2 y 3, con una dosis única concomitante de LIXIANA 60 mg en el Día 3, aumentó el AUC de LIXIANA durante 24 horas en el 77% y la Cmax en el 85%, respectivamente. LIXIANA no tuvo efecto sobre la Cmax y el AUC de quinidina. |
El uso concomitante de LIXIANA con este fármaco requiere una reducción de la dosis a 30 mg una vez al día. |
|
Verapamilo |
CT |
Verapamilo 240 mg una vez al día por 11 días con una dosis única concomitante de LIXIANA 60 mg en el Día 10 aumentó el AUC y la Cmax de LIXIANA en aproximadamente el 50%. LIXIANA disminuyó la Cmax y el AUC de Verapamilo administrado concomitantemente en el 14% y en el 16%, respectivamente. |
No se requiere ajuste de dosis. Use con cautela, teniendo en cuenta las características específicas individuales del paciente. |
|
Amiodarona |
CT |
Amiodarona 400 mg una vez al día por 4 días con una dosis única de LIXIANA 60 mg en el Día 4 aumentó el AUC y la Cmax de LIXIANA en aproximadamente el 40% y el 66%, respectivamente. Amiodarona no estaba en estado de equilibrio en este estudio. |
No se requiere ajuste de dosis. Use con cautela, teniendo en cuenta las características específicas individuales del paciente. |
|
Inductores de la CYP 3A4 y de la P-gp |
|||
|
Rifampicina |
CT |
Rifampicina 600 mg una vez al día por 7 días con una dosis única de LIXIANA 60 mg en el Día 7 disminuyó el AUC de LIXIANA en el 34% sin un efecto evidente sobre la Cmax. |
El uso combinado con inductores fuertes de la CYP3A4 y de la P-gp (ej. Fenitoína, carbamazepina y fenobarbital) normalmente debe evitarse, ya que esto puede afectar la eficacia de LIXIANA. |
|
Sustratos de la P-gp |
|||
|
Digoxina |
CT |
Dosis múltiples diarias de digoxina 0,25 mg con administración concomitante de LIXIANA 60 mg una vez al día durante los Días 8-14 aumentó la Cmax de LIXIANA en el 17%, sin efecto significativo sobre el AUC o la depuración renal en el estado de equilibrio. LIXIANA aumentó la Cmax de digoxina administrada concomitantemente en el 28%; sin embargo, el AUC no se vio afectado. |
No hay necesidad de modificar la dosis cuando se administra LIXIANA con digoxina. |
|
Inhibidores e Inductores de CYP3A4 |
CT |
Menos del 10% de una dosis de LIXIANA administrada oralmente se metaboliza vía CYP3A4 en pacientes con función renal normal. Por tanto, no se prevé interacción con inhibidores o inductores de CYP3A4. |
No hay necesidad de modificar la dosis para pacientes que administran inhibidores o inductores de CYP. |
|
Inhibidores de la Bomba de Protones (PPIs) |
|||
|
Esomeprazol |
CT |
Esomeprazol 40 mg una vez al día por 5 días con una dosis única concomitante de LIXIANA 60 mg en el Día 5 no tuvo efecto sobre el AUC de LIXIANA, pero la Cmax disminuyó en aproximadamente el 33%. |
No se necesita ningún cambio de la dosis cuando se administra LIXIANA con esomeprazol. |
|
Anticoagulantes |
CT, T |
Una dosis única subcutánea de 1 mg/kg de enoxaparina no tuvo efecto sobre la PK de una dosis oral única de LIXIANA 60 mg cuando se administró concomitantemente o con intervalo de 12 horas entre ambos. |
La administración concomitante de LIXIANA con otros anticoagulantes está contraindicada a causa del riesgo aumentado de sangrado. |
|
Inhibidores de Plaquetas |
|||
|
Ácido Acetilsalicílico (ASA) |
CT |
La administración concomitante de ASA (100 mg o 325 mg) y LIXIANA aumentó el tiempo de sangrado en relación con cada medicamento aisladamente. La administración concomitante de alta dosis de ASA (325 mg) aumentó el AUC y la Cmax en estado de equilibrio de LIXIANA en el 35% y el 32%, respectivamente. En los estudios clínicos, se autorizó el uso concomitante de ASA (dosis baja ≤ 100 mg/día) y llevó a un aumento cínicamente relevante de sangrado, aunque con un menor riesgo de sangrado con LIXIANA en comparación con la warfarina. |
LIXIANA puede administrarse concomitantemente con baja dosis de ASA (≤ 100 mg/día). Evaluar el riesgo de sangrado antes de la administración concomitante y usar con cautela, si se considera necesario. |
|
Tienopiridinas (ej. Clopidogrel) |
CT |
En el ENGAGE AF-TIMI 48 hubo una experiencia muy limitada con el uso de LIXIANA con terapia antiplaquetaria doble o agentes fibrinolíticos. |
El uso concomitante de fármacos afectando la hemostasia puede aumentar el riesgo de sangrado. Utilice con cautela, si se considera necesario. |
|
AINEs |
|||
|
Naproxeno |
CT |
La administración concomitante de naproxeno y LIXIANA aumentó el tiempo de sangrado relativo a cada medicamento aisladamente. Naproxeno no tuvo efecto en la Cmax y el AUC de LIXIANA. En los estudios clínicos, la administración concomitante de AINEs llevó a aumento de sangrado clínicamente significante. |
No se recomienda el uso crónico de AINEs con LIXIANA. El uso a corto plazo debe usarse con cautela, si se considera necesario. |
|
Otros fármacos |
|||
|
Atorvastatina |
CT |
Atorvastatina 80 mg una vez al día durante 8 días con una dosis única concomitante de LIXIANA 60 mg en el Día 7 disminuyó la Cmax o el AUC de edoxabán en el 15%. |
Utilice con cautela, teniendo en cuenta las características específicas individuales del paciente. |
|
Inhibidores de la proteasa del VIH, ej., darunavir/ritonavir/lopinavir/ritonavir |
T |
No se llevó a cabo ninguna interacción específica medicamentosa con los inhibidores de la proteasa de VIH en combinación con edoxabán. Los inhibidores de la proteasa del VIH pueden inhibir la P-gp (en lugar de CYP3A) y potencialmente aumentar la exposición a edoxabán en 1,5 a 2 veces. |
Utilice con cautela, si se considera necesawrio. |
|
ISRS"s/IRSN"s |
CT,T |
Al igual que con otros anticoagulantes, los pacientes bajo tratamiento con LIXIANA tienen un mayor riesgo de sangrado en caso de uso concomitante con ISRS o IRSN debido a su efecto sobre las plaquetas. |
Utilice con precaución, si se considera necesario. |
Leyenda: C = Estudio de Caso, CT = Estudio Clínico; T = Teórico.
Interacción Fármaco-Alimentos:
LIXIANA puede administrarse con o sin comida.
Interacción Fármaco-Hierba:
No se han establecido interacciones fármaco-hierba.
Interacción Fármaco-Laboratorio:
No se han establecido interacciones fármaco-laboratorio.
Interacciones fármaco-estilo de vida:
LIXIANA tiene una influencia nula o insignificante en la capacidad para conducir y utilizar máquinas.
ALTERACIONES EN LOS RESULTADOS DE PRUEBAS DE LABORATORIO:
Se observaron cambios en las pruebas de laboratorio hepáticas (ALT, AST, Bilirrubina Total) durante los ensayos clínicos. Sin embargo, los datos no indicaron ningún signo clínicamente preocupante para la lesión hepática inducida por fármacos.
PRECAUCIONES GENERALES:
LA DISCONTINUACIÓN PREMATURA DE CUALQUIER ANTICOAGULANTE ORAL, INCLUYENDO LIXIANA, AUMENTA EL RIESGO DE EVENTOS TROMBÓTICOS.
Para reducir este riesgo, considere la cobertura con otro anticoagulante, si se discontinúa LIXIANA por motivos diferentes de sangrado patológico o la finalización de un ciclo de terapia.
Sangrado:
LIXIANA aumenta el riesgo de sangrado y puede causar un sangrado grave y potencialmente fatal. Evaluar rápidamente cualquier signo o síntoma de pérdida de sangre. Discontinuar LIXIANA en pacientes con hemorragia activa clínicamente significativa. LIXIANA, al igual que otros anticoagulantes, debe usarse con precaución en pacientes con mayor riesgo de sangrado. A los pacientes con alto riesgo de sangrado no se les debe recetar LIXIANA.
Si ocurre una hemorragia grave, se debe discontinuar el tratamiento con LIXIANA e investigar inmediatamente el origen del sangrado.
Se recomienda una estrecha vigilancia clínica (en busca de signos de sangrado o anemia) durante todo el periodo de tratamiento, especialmente en presencia de múltiples factores de riesgo de sangrado (véase la Tabla 8 a continuación).
Tabla 8. Factores que aumentan el riesgo hemorrágico
|
Factores que aumentan los niveles plasmáticos de edoxabán* |
Insuficiencia renal grave (CrCl 15-29 mL/min) Insuficiencia renal moderada (CrCl 30-50 mL/min) |
|
Tratamiento sistémico concomitante con inhibidores de la P-gp |
|
|
Bajo peso corporal ≤ 60 kg |
|
|
Interacciones farmacodinámicas |
Uso crónico de AINEs |
|
Inhibidores de la agregación plaquetaria, incluso el ASA, clopidogrel, prasugrel, ticagrelor |
|
|
Inhibidores selectivos de la recaptación de serotonina (ISRS), inhibidores de la recaptación de serotonina norepinefrina (IRSN) |
|
|
Enfermedades/procedimientos con riesgos hemorrágicos especiales |
Trastornos de coagulación congénitos o adquiridos |
|
Trombocitopenia o defectos de la función plaquetaria |
|
|
Enfermedad gastrointestinal ulcerosa activa |
|
|
Sangrado gastrointestinal reciente |
|
|
Hemorragia intracraneal o isquemia recientes |
|
|
Cirugía cerebral, espinal u oftalmológica reciente |
|
|
Otros |
Edad > 75 años |
Administración concomitante con Anticoagulantes, Antiplaquetarios y Trombolíticos y ISRS/IRSN:
El uso concomitante de fármacos que afectan la hemostasia puede aumentar el riesgo de sangrado. Estos fármacos incluyen agentes antiplaquetarios, como la aspirina (ASA), los inhibidores de plaquetas P2Y12, otros agentes antitrombóticos, la terapia fibrinolítica, inhibidores selectivos de la recaptación de serotonina (ISRS), inhibidores de la recaptación de serotonina norepinefrina (IRSN) y los antiinflamatorios crónicos no esteroideos (AINEs). Por tanto, no se recomienda el tratamiento concomitante a largo plazo con LIXIANA y otros anticoagulantes a causa de un mayor riesgo de sangrado. La administración concomitante a corto plazo puede ser necesaria para pacientes en transición hacia o desde LIXIANA.
No se recomienda el uso concomitante de LIXIANA con heparina no fraccionada (HNF), excepto en las dosis utilizadas para mantener un catéter venoso central o arterial permeable.
En los estudios clínicos con LIXIANA, el uso concomitante de dosis bajas (≤ 100 mg/día) de ASA o las tienopiridinas (clopidogrel) y los AINEs dieron lugar a mayores tasas de sangrado clínicamente relevante. Otros inhibidores de la agregación plaquetaria, como prasugrel y ticagrelor, no se han estudiado con LIXIANA en ninguna población de pacientes, y no se recomiendan como terapia concomitante.
Cardiovascular:
Enfermedad valvular:
La seguridad y la eficacia de LIXIANA no se han estudiado en pacientes con válvulas cardiacas protésicas (mecánicas o biológicas) o en pacientes con enfermedad cardiaca reumática hemodinámicamente significativa, principalmente estenosis mitral. Por tanto, no se recomienda LIXIANA en dichos pacientes. Cabe señalar que el estudio ENGAGE AF-TIMI 48 que evaluó LIXIANA en SPAF, incluyó a pacientes con otras enfermedades cardiacas valvulares (ej., estenosis aórtica, regurgitación aórtica o mitral).
Cardioversión:
Los pacientes pueden mantenerse en el tratamiento con LIXIANA mientras se someten a cardioversión.
Insuficiencia Hepática/Biliar:
Insuficiencia Hepática:
Los pacientes con enfermedad hepática significativa (ej., hepatitis aguda, hepatitis crónica activa, cirrosis) fueron excluidos de los estudios clínicos de LIXIANA. Por tanto, LIXIANA está contraindicado en pacientes con enfermedad hepática asociada con coagulopatía y riesgo de hemorragia clínicamente relevante.
No se estudió LIXIANA en pacientes con insuficiencia hepática grave; por tanto no se recomienda. LIXIANA debe utilizarse con precaución en pacientes con insuficiencia hepática leve a moderada.
Monitoreo y Pruebas de Laboratorio:
Un agente de reversión anticoagulante específico para LIXIANA no está disponible comercialmente.
Los efectos farmacodinámicos medidos por el estudio anti-factor Xa (FXa) son predecibles y se correlacionan con la dosis y la concentración de LIXIANA. Como resultado de la inhibición de FXa, LIXIANA también prolonga el tiempo de coagulación en pruebas como el tiempo de protrombina (TP) y el tiempo de tromboplastina parcial activada (TTPa). Sin embargo, los cambios observados en dichas pruebas de coagulación en la dosis terapéutica esperada son pequeños y están sujetos a un alto grado de variabilidad y no son útiles para monitorear el efecto anticoagulante de LIXIANA.
Aunque la terapia con LIXIANA llevará a una RNI elevada, dependiendo del momento de la medición, la RNI no es una medida válida para evaluar la actividad anticoagulante de LIXIANA. La RNI está calibrada y validada solamente para los antagonistas de la vitamina K (VKA) y no debe utilizarse para ningún otro anticoagulante, incluida LIXIANA.
Aunque no se necesite monitorear el efecto anticoagulante de LIXIANA durante la práctica clínica rutinaria, en determinadas situaciones infrecuentes como sobredosis, sangrado agudo, cirugía urgente, en casos de sospecha de incumplimiento o en otras circunstancias inusuales, la evaluación del efecto anticoagulante de LIXIANA puede ser apropiada. Por consiguiente, un estudio calibrado cuantitativo de anti-FXa puede ser útil para informar decisiones clínicas en dichas circunstancias para el nivel máximo de estado de equilibrio previsto y durante la actividad anti-FXa en distintas indicaciones y para distintas dosis de LIXIANA).
Renal:
Insuficiencia Renal:
La concentración plasmática de LIXIANA aumentó con el grado de insuficiencia renal. Por tanto, la función renal: CrCL debe monitorearse al inicio del tratamiento en todos los pacientes y después, cuando esté clínicamente indicado. Hay datos limitados en pacientes con insuficiencia renal grave (CrCl<30 mL/min) o en diálisis, ya que estos pacientes se excluyeron de los estudios pivotales de Fase III. Así que no se recomienda LIXIANA en dichos pacientes. Los pacientes que desarrollan insuficiencia renal aguda mientras están en tratamiento con LIXIANA deben discontinuar el tratamiento.
Consideraciones Peri-Operativas/Procedimentales:
Al igual que con cualquier anticoagulante, los pacientes con LIXIANA que se someten a cirugía o procedimientos invasivos tienen un mayor riesgo de sangrado. En dichas circunstancias, puede ser necesaria la discontinuación temporal de LIXIANA.
Fase Pre-operativa:
Si se requiere un procedimiento invasivo o una intervención quirúrgica, se debe interrumpir LIXIANA al menos 24 horas antes de la intervención, si es posible, a causa del mayor riesgo de sangrado, y basándose en el juicio clínico del médico. Si no se puede retrasar el procedimiento, el mayor riesgo de sangrado debe evaluarse en función de la urgencia de la intervención. Aunque los datos sean limitados en pacientes con mayor riesgo de sangrado o en cirugía importante que puede requerir hemostasia completa, considere la interrupción del tratamiento con LIXIANA al menos 48 horas antes de la cirugía, dependiendo de las circunstancias clínicas. LIXIANA debe reiniciarse después de la cirugía o de los procedimientos de intervención tan pronto como se haya determinado la hemostasia adecuada.
Anestesia Peri-Operatoria Espinal/Epidural, Punción Lumbar:
Cuando se lleva a cabo anestesia neuroaxial (epidural/espinal) o punción espinal, los pacientes tratados con antitrombóticos para la prevención de complicaciones tromboembólicas corren el riesgo de desarrollar un hematoma epidural o espinal que puede provocar una lesión neurológica a largo plazo o una parálisis permanente.
El riesgo de dichos eventos aumenta aún más con el uso de catéteres permanentes o el uso concomitante de medicamentos que afectan la hemostasia. Por consiguiente, los catéteres epidurales o intratecales permanentes deben retirarse al menos 5 horas antes de la primera dosis de LIXIANA. El riesgo también puede aumentar por punción epidural o espinal traumática o repetida. Si se ocurre una punción traumática, se debe retrasar la administración de LIXIANA por 24 horas.
Los pacientes que se sometieron a una punción epidural y que reciben LIXIANA deben monitorearse con frecuencia para detectar signos y síntomas de deterioro neurológico (ej., entumecimiento o debilidad de las piernas, disfunción intestinal o vesical). Si se observan deficiencias neurológicas, se necesita un diagnóstico y tratamiento urgente.
El médico debe considerar el beneficio potencial frente al riesgo antes de la intervención neuroaxial en pacientes anticoagulados o que deben estar anticoagulados para la tromboprofilaxis y usar LIXIANA sólo cuando los beneficios superen claramente los posibles riesgos. Un catéter epidural no debe retirarse antes de las 24 horas posteriores a la última administración de LIXIANA.
Periodo Post-procedimiento:
LIXIANA debe reiniciarse después de un procedimiento invasivo o intervención quirúrgica tan pronto como se haya establecido una hemostasia adecuada y la situación clínica lo permita, para evitar un aumento innecesario del riesgo de trombosis.
Cirugía Ortopédica:
No se recomienda LIXIANA para la prevención del TEV en pacientes sometidos a cirugía total electiva de rodilla o cadera, ya que no se estableció la seguridad y la eficacia de LIXIANA en dichas situaciones clínicas.
Pulmonar:
No se recomienda LIXIANA como una alternativa a la heparina no fraccionada en pacientes con embolia pulmonar que son hemodinámicamente inestables o pueden recibir trombólisis o embolectomía pulmonar, ya que no se estableció la seguridad y la eficacia de LIXIANA en estas situaciones clínicas.
Pacientes con síndrome antifosfolípido:
Los anticoagulantes orales directos (DOACs), incluido el edoxabán, no se recomiendan para pacientes con antecedentes de trombosis diagnosticados con síndrome antifosfolípido. En particular, para los pacientes que son triple positivos (para el anticoagulante lúpico, los anticuerpos anticardiolipina y los anticuerpos anti-beta 2-glucoproteína I), el tratamiento con DOAC podría asociarse con mayores tasas de eventos trombóticos recurrentes en comparación con la terapia con antagonistas de la vitamina K.
Poblaciones Especiales:
Pacientes Pediátricos (<18 años de edad):
Todavía no se estableció la seguridad y la eficacia de LIXIANA en niños menores de 18 años. Por tanto, no se recomienda el uso de LIXIANA en dichos pacientes.
Pacientes Geriátricos (>65 años de edad):
Normalmente, no se requiere ningún cambio en la dosis en función de la edad.
Pacientes con cáncer activo:
No se estableció la eficacia y seguridad de edoxabán en el tratamiento y/o en la prevención del TEV en pacientes con cáncer activo. Por tanto, no se recomienda el uso de LIXIANA en dichos pacientes.
DOSIS Y VÍA DE ADMINISTRACIÓN:
Consideraciones sobre dosificación:
Al igual que cualquier fármaco anticoagulante oral no antagonista de la vitamina K (NOAC), antes de iniciar LIXIANA, se debe garantizar que el paciente comprende y está preparado para aceptar la adherencia a la terapia con NOAC, según las indicaciones.
LIXIANA debe administrarse regularmente, según lo prescrito, para garantizar una eficacia óptima. Se deben evitar todas las discontinuaciones temporales, a menos que estén clínicamente indicadas.
Determine la depuración estimada de la creatinina (eCrCl) en todos los pacientes antes de iniciar LIXIANA y monitoree la función renal durante el tratamiento con LIXIANA, según sea clínicamente apropiado. La determinación de la función renal por eCrCL debe ocurrir al menos una vez al año, y especialmente en circunstancias en las que se puede esperar que la función renal se vea comprometida, es decir, infarto agudo de miocardio (IAM), insuficiencia cardiaca aguda (ICA) descompensada, mayor uso de diuréticos, deshidratación, hipovolemia, etc. El deterioro clínicamente relevante de la función renal puede requerir un ajuste de la dosis o la discontinuación de LIXIANA.
El método utilizado para estimar la función renal (CrCL en mL/min) durante el desarrollo clínico de LIXIANA fue el método de Cockcroft-Gault. La fórmula es la siguiente:
|
en el sexo masculino |
(140-edad) (años) x peso (kg) x 1,23 |
o, |
(140-edad) (años) x peso (kg) |
|
creatinina en suero (μmol/L) |
72 x creatinina en suero (mg/100 mL) |
|
en el sexo femenino |
(140-edad) (años) x peso (kg) x 1,04 |
o, |
(140-edad) (años) x peso (kg) x 0,85) |
|
Creatinina en suero (μmol/L) |
72 x creatinina en suero (mg/100 mL) |
Dosis Recomendada y Ajuste de Dosis:
FA:
La dosis normal recomendada de LIXIANA es 60 mg diariamente.
El tratamiento de TEV y la Prevención de TVP y EP Recurrentes:
La dosis recomendada de LIXIANA es 60 mg una vez al día después del uso inicial de un anticoagulante parenteral por 5-10 días.
La duración del tratamiento debe ser individualizada después de una evaluación cuidadosa del beneficio del tratamiento en comparación con el riesgo de sangrado. La corta duración del tratamiento (al menos 3 meses) debe basarse en factores de riesgo transitorios (por ejemplo, cirugía, traumatismo, inmovilización), mientras la duración prolongada debe basarse en factores de riesgo permanentes o TVP idiopática o EP.
Reducciones de la dosis para FA y TEV:
La dosis recomendada de LIXIANA es 30 mg una vez al día en pacientes con uno o más de los factores clínicos a continuación:
• Insuficiencia renal moderada (depuración de creatinina (CrCl) 30-50 mL/min.
• Bajo peso corporal ≤ 60 kg (132 Ibs).
• Uso concomitante de inhibidores de la P-gp, a excepción de amiodarona y verapamilo.
Tabla 14. Resumen de la Dosificación en FA y TEV (TVP y EP)
|
Resumen de la Guía para Dosificación |
|
|
FA: Dosis recomendada |
60 mg una vez al día |
|
TEV: Dosis recomendada |
60 mg una vez al día (después de uso inicial de heparina) |
|
Insuficiencia Renal: Moderada (CrCl 30–50 mL/min) Bajo Peso Corporal: ≤ 60 kg, o Inhibidores de la P-gp, a excepción de amiodarona y verapamilo |
30 mg una vez al día |
Insuficiencia Renal:
Los datos son limitados en pacientes con insuficiencia renal grave (CrCl<30 mL/min) o en diálisis, ya que dichos pacientes fueron excluidos de los estudios pivotales de Fase III. Por tanto, no se recomienda LIXIANA en estos pacientes.
Tabla 15. Resumen de la posología en la insuficiencia renal para FA y VTE
|
Estatus Renal |
Depuración de Creatinina (CrCl) mL/min |
Dosis de LIXIANA Una vez al Día |
|
Leve |
50-80 |
60 mg |
|
Moderado |
30-50 |
30 mg |
|
Enfermedad Renal Grave o en Diálisis |
<30 |
No se recomienda |
Insuficiencia Hepática:
En pacientes con insuficiencia hepática leve a moderada, la dosis recomendada de LIXIANA es de 60 mg una vez al día.
No se estudió LIXIANA en pacientes con insuficiencia hepática grave. Por tanto, no se recomienda su uso en dichos pacientes.
LIXIANA está contraindicado en pacientes con enfermedad hepática asociada con anomalías de la coagulación intrínsecas.
Población Mayor:
A menudo no se requiere reducción de la dosis. El envejecimiento puede estar asociado con el deterioro de la función renal.
Cambiando a y de LIXIANA:
La terapia anticoagulante continua es importante en pacientes con FA y en el tratamiento de TEV. Puede haber situaciones que justifiquen un cambio en la terapia con anticoagulantes.
Tabla 16. Recomendación para el Cambio A y de LIXIANA
|
Cambiando de: |
A LIXIANA |
|
Antagonista de la Vitamina K (VKA) |
Discontinuar el VKA e iniciar LIXIANA cuando la razón normalizada internacional (INR) es ≤ 2,5. |
|
Anticoagulantes orales diferentes de VKA |
Discontinuar el anticoagulante oral no VKA e iniciar LIXIANA en el momento de la dosis siguiente de no VKA. |
|
Anticoagulante subcutáneo |
Discontinuar el anticoagulante subcutáneo e iniciar LIXIANA en el momento de la siguiente dosis programada de anticoagulante subcutáneo. |
|
Heparina no Fraccionada |
Discontinuar la infusión e iniciar LIXIANA 4 horas después. |
|
Cambiando de LIXIANA |
A: |
|
Opción oral de VKA: Administrar una dosis de LIXIANA de 30 mg (15 mg para pacientes con una dosis reducida para uno o más de los siguientes: insuficiencia renal moderada a grave (CrCL 15–50 mL/min), bajo peso corporal, o usar con inhibidores de la P-gp (a excepción de amiodarona y verapamilo), junto con una dosis adecuada de VKA. Se debe medir la INR al menos semanalmente e inmediatamente antes de la dosis diaria de LIXIANA para minimizar la influencia de LIXIANA en las mediciones de RNI. Una vez que se logra una RNI estable de ≥ 2,0, se debe discontinuar LIXIANA. |
|
|
Opción parenteral VKA: Discontinuar LIXIANA y administrar un anticoagulante parenteral y VKA en el momento de la próxima dosis programada de LIXIANA. Una vez que se logra una RNI estable de ≥ 2,0, se debe discontinuar el anticoagulante parenteral y mantener VKA. |
|
|
Anticoagulantes orales distintos de VKA: Discontinuar LIXIANA y comenzar el anticoagulante no VKA en el momento de la siguiente dosis programada de LIXIANA. |
|
|
Anticoagulantes parenterales: Discontinuar LIXIANA y comenzar el anticoagulante parenteral en el momento de la siguiente dosis programada de LIXIANA. |
Dosis olvidada:
Si se olvida una dosis de LIXIANA, se debe administrar la dosis lo antes posible el mismo día.
No se debe duplicar la dosis de LIXIANA para compensar una dosis olvidada. Retome la programación normal de dosificación al día siguiente.
MANIFESTACIONES Y MANEJO DE LA SOBREDOSIFICACIÓN O INGESTA ACCIDENTAL:
La sobredosis de LIXIANA puede provocar hemorragia. La experiencia con casos de sobredosis es muy limitada. En casos de sobredosis, según la situación clínica, se debe interrumpir LIXIANA o retrasar la dosis siguiente, teniendo en cuenta la vida media (t½) de LIXIANA (10-14 horas).
En casos de sangrado, inicie las medidas apropiadas, como los glóbulos rojos empaquetados y/o la hemostasia.
No hay un agente de reversión específico para LIXIANA. Aunque no se haya evaluado en pacientes, el concentrado de complejo protrombínico de 3 o 4 factores (CCP), los concentrados de complejo protrombínico activado (CCPas) o el factor VIIa recombinante podrían considerarse para la reversión del efecto anticoagulante de LIXIANA.
Concentrado de complejo protrombínico de 4 factores (CCP): En pacientes sanos, la administración de CCP de 4 factores a 50 UI/kg revirtió el efecto anticoagulante de LIXIANA en los 30 minutos posteriores a la finalización de la infusión.
CCP de 3 factores: En voluntarios sanos, un CCP de 3 factores restauró la generación de trombina, pero no normalizó el TP.
En modelos animales, los agentes de CCP, CCPa y Factor VIIa recombinante revirtieron los biomarcadores de coagulación y el sangrado.
No se espera que lo siguiente revierta los efectos anticoagulantes de LIXIANA: sulfato de protamina, vitamina K y ácido tranexámico.
La hemodiálisis no contribuye significativamente para la depuración de LIXIANA.
PRESENTACIONES:
Caja con 14 tabletas con 15 mg.
Caja con 28 tabletas con 30 mg.
Caja con 28 tabletas con 60 mg.
RECOMENDACIONES SOBRE ALMACENAMIENTO:
Consérvese a no más de 30°C.
LEYENDAS DE PROTECCIÓN:
Literatura exclusiva para médicos. Mantener fuera del alcance de los niños. Su venta requiere receta médica. No usar en el embarazo y la lactancia.
Reporte las sospechas de reacción adversa a los correos: farmacovigilancia@cofepris.gob.mx y
farmacovigilancia@menarini.com.mx
Hecho en Brasil por:
Daiichi Sankyo Brasil Farmacêutica Ltda.
Alameda Xingú, No 766,
Barueri, Sãn Paulo, Brasil
Representante legal e Importador:
A. MENARINI MÉXICO S.A. de C.V.
Avenida Periférico Sur 4118, Interior 301,
Col. Jardines del Pedregal, C.P. 01900,
Alcaldía Álvaro Obregón, Ciudad de México, México.
Reg. Núm. 017M2020, SSA IV
®Marca Registrada


