
KYPROLIS
CARFILZOMIB
Polvo liofilizado
1 Caja, 1 Frasco ámpula con polvo liofilizado, 60 Miligramos
FORMA FARMACÉUTICA Y FORMULACIÓN:
KYPROLIS® es un polvo liofilizado blanco a blanquecino estéril para solución para inyección/mediante infusión.
El frasco ámpula con polvo liofilizado contiene:
Carfilzomib 60 mg
Excipiente cbp
INDICACIONES TERAPÉUTICAS:
Mieloma Múltiple en Recaída o Refractario: KYPROLIS® está indicado para el tratamiento de pacientes adultos con mieloma múltiple en recaída o refractario que han recibido de una a tres líneas de terapia en combinación con:
• Lenalidomida y dexametasona; o
• Dexametasona; o
• Daratumumab y dexametasona
KYPROLIS® está indicado como agente único para el tratamiento de pacientes adultos con mieloma múltiple en recaída o refractario, y que hayan recibido una o más líneas de terapia.
FARMACOCINÉTICA Y FARMACODINAMIA:
Farmacocinética: Carfilzomib a dosis entre 20 mg/m2 y 70 mg/m2 administrados en pacientes con mieloma múltiple mediante infusión de 30 minutos, dio lugar a aumentos en las concentraciones plasmáticas máximas (Cmáx) y el área bajo la curva con tiempo hasta el infinito (ABC0 a INF) dependientes de la dosis. También se observó un aumento dependiente de la dosis en Cmáx y ABC0 a INF entre dosis de carfilzomib de 20 mg/m2 y 56 mg/m2 mediante infusión de 2 a 10 minutos en pacientes con mieloma múltiple en recaída o refractaria. Una infusión de 30 minutos dió como resultado una ABC0 a INF similar, pero con una Cmáx de 2 a 3 veces más baja que la observada mediante infusión de 2 a 10 minutos con la misma dosis. No hubo evidencia de acumulación de carfilzomib después de la administración repetida de 70 mg/m2 de carfilzomib mediante infusión semanal de 30 minutos o 15 mg/m2 y 20 mg/m2 mediante infusión de 2 a 10 minutos dos veces por semana.
La Tabla 1 muestra la media estimada del promedio del área bajo la curva diaria en el primer ciclo (ABCC1, avg), el promedio del área bajo la curva diaria en estado estable (ABCSS) y la Cmáx en la dosis más alta en el primer ciclo (Cmáx, C1) para los diferentes regímenes de dosificación.
Tabla 1. Parámetros de exposición a carfilzomib para diferentes regímenes de dosificación
|
Parámetros estimados (%CV) |
20/27 mg/m2 dos veces a la semana con infusión de 2 a 10-minutos |
20/56 mg/m2 dos veces a la semana con infusión de 30-minutos |
20/70 mg/m2 una vez a la semana con infusión de 30-minutos |
|
ABCC1, avg (ng• hr/mL) |
95 (40) |
170 (35) |
114 (36) |
|
ABCSS (ng• hr/mL) |
111 (34) |
228 (28) |
150 (35) |
|
Cmáx, C1 (ng/mL) |
1282 (17) |
1166 (29) |
1595 (36) |
CV= Coeficiente de variación.
Distribución: El volumen de distribución promedio en estado estable de una dosis de carfilzomib de 20 mg/m2 fue de 28 L. Carfilzomib se une en un 97% a las proteínas plasmáticas humanas en el rango de concentraciones de 0.4 micromolar a 4 micromolar in vitro.
Eliminación: Carfilzomib tiene una vida media de ≤ 1 hora en el Día 1 del Ciclo 1 después de las dosis intravenosas ≥15 mg/m2. La vida media fue similar cuando se administró mediante una infusión de 30 minutos o una infusión de 2 a 10 minutos. El aclaramiento sistémico osciló entre 151 L/hora y 263 L/hora.
Metabolismo: El carfilzomib se metaboliza rápidamente por la escisión de la peptidasa y la hidrólisis del epóxido fueron las principales vías del metabolismo. Los mecanismos mediados por el citocromo P450 (CYP) contribuyeron con un papel menor en el metabolismo general de carfilzomib. Excreción: Aproximadamente el 25% de la dosis administrada de carfilzomib se excreta en la orina como metabolitos en 24 horas. La excreción urinaria y fecal del compuesto original fue insignificante (0.3% de la dosis total).
Poblaciones específicas: Edad (35 a 89 años), sexo, raza u origen étnico (80% blancos, 11% negros, 6% asiáticos, 3% hispanos) e insuficiencia renal leve a grave (depuración de creatinina de 15 mL/min a 89 mL/min) no tuvo efectos clínicamente significativos en la farmacocinética de carfilzomib.
Pacientes con insuficiencia hepática: En comparación con los pacientes con función hepática normal, los pacientes con insuficiencia hepática leve (bilirrubina total 1 a 1.5 x ULN y cualquier AST o bilirrubina total ≤ ULN y AST> ULN) e insuficiencia hepática moderada (bilirrubina total > 1.5 a 3 x ULN y cualquier AST) tenían un ABC de carfilzomib aproximadamente 50% más alto. La farmacocinética de carfilzomib no se ha evaluado en pacientes con insuficiencia hepática grave (bilirrubina total > 3 × ULN y cualquier AST).
Pacientes con insuficiencia renal: En relación con los pacientes con función renal normal, los pacientes con ESRD (pacientes con enfermedad renal en etapa terminal, ESRD por sus siglas en inglés) en hemodiálisis mostraron un ABC de carfilzomib 33% mayor. Dado que la depuración de las concentraciones de KYPROLIS® en hemodiálisis no se ha estudiado, el medicamento debe administrarse después del procedimiento de hemodiálisis.
Estudios de interacción con medicamentos:
Estudios clínicos:
Efecto de carfilzomib sobre la sensibilidad del sustrato CYP3A: La farmacocinética de midazolam (un sustrato sensible del CYP3A) no se vió afectada por la administración concomitante de carfilzomib.
Estudios in vitro:
Efecto de carfilzomib en las enzimas del citocromo P450 (CYP): Carfilzomib mostró una inhibición directa y dependiente del tiempo de CYP3A pero no indujo CYP1A2 y CYP3A4 in vitro.
Efecto de los transportadores sobre carfilzomib: Carfilzomib es un sustrato de P-glicoproteína (P-gp) in vitro.
Efecto de carfilzomib sobre los transportadores: Carfilzomib inhibe la P-gp in vitro. Sin embargo, dado que KYPROLIS® se administra por vía intravenosa y se metaboliza ampliamente, es poco probable que la farmacocinética de KYPROLIS® se vea afectada por los inhibidores o inductores de la P-gp.
Mecanismo de Acción: Carfilzomib es una epoxicetona tetrapeptídica inhibidora del proteosoma que se une irreversiblemente a la región N-terminal de los residuos de treonina en los sitios activos del proteosoma 20S, el cual es la partícula proteolítica central del proteosoma 26S. Carfilzomib mostró actividades antiproliferativas y proapoptóticas in vitro en células de tumores sólidos y hematológicos. En animales, carfilzomib inhibió la actividad proteosomal en sangre y tejidos y retrasó el crecimiento tumoral en modelos de mieloma múltiple, de tumores hematológicos y de tumores sólidos.
Farmacodinamia: La administración de carfilzomib por vía intravenosa resultó en la supresión de la actividad tipo quimotripsina (CT-L por sus siglas en inglés) del proteosoma medida en la sangre 1 hora después de la primera dosis. Dosis de carfilzomib ≥ 15 mg/m2 con o sin lenalidomida y dexametasona indujeron una inhibición ≥80% de la actividad CT-L del proteosoma. Asimismo, la administración de carfilzomib a 20 mg/m2 intravenosamente como agente único, resultó en la inhibición media de las subunidades del proteosoma polipéptido de baja masa molecular 2 (LMP2) y complejo de tipo multicatalítico endopeptidasa 1 (MECL1) en un rango de 26% al 32% y de 41% al 49%, respectivamente. La inhibición del proteosoma se mantuvo durante ≥ 48 horas posteriores a la primera dosis de carfilzomib por cada semana de dosificación.
Estudios clínicos:
En Combinación con Lenalidomida y Dexametasona para Mieloma Múltiple en Recaída o Refractario ASPIRE
ASPIRE fue un estudio aleatorizado, abierto, multicéntrico, el cual evaluó la combinación de KYPROLIS® con lenalidomida y dexametasona (KRd) versus lenalidomida y dexametasona solos (Rd) en pacientes con mieloma múltiple en recaída o refractario que habían recibido 1 a 3 líneas de terapia. (Una línea de terapia es un curso planeado de tratamiento [incluyendo inducción secuencial, trasplante, consolidación y/o mantenimiento] sin una interrupción por falta de eficacia, tal como recaída o enfermedad progresiva). Los pacientes que presentaron lo siguiente, fueron excluidos del estudio: refractarios a bortezomib en el régimen más reciente, refractarios a lenalidomida y dexametasona en el régimen más reciente, que no respondieron a ningún régimen previo, depuración de creatinina < 50 mL/min, ALT/AST > 3.5 x ULN y bilirrubina > 2 x ULN, insuficiencia cardiaca congestiva de Clases III a IV conforme a la New York Heart Association, o infarto al miocardio en el lapso de los últimos 4 meses. En el brazo KRd, se evaluó KYPROLIS® en una dosis de arranque de 20 mg/m2, la cual fue aumentada a 27 mg/m2 en el Ciclo 1, Día 8 en adelante. Se administró KYPROLIS® mediante una infusión de 10 minutos en los Días 1, 2, 8, 9, 15 y 16 de cada ciclo de 28 días del Ciclo 1 al 12. Sólo se dosificó KYPROLIS® en los Días 1, 2, 15 y 16 de cada ciclo de 28 días a partir del Ciclo 13 y hasta el 18.
Se administró Dexametasona 40 mg oral o intravenosamente en los Días 1, 8, 15 y 22 de cada ciclo. Se dieron oralmente 25 mg de lenalidomida en los Días 1 a 21 de cada ciclo de 28 días. El brazo de tratamiento Rd tuvo el mismo régimen para lenalidomida y dexametasona que el brazo de tratamiento KRd. Se administró KYPROLIS® por un máximo de 18 ciclos a menos que se hubiera interrumpido de forma temprana por progresión de la enfermedad o toxicidad inaceptable. La administración de lenalidomida y dexametasona pudo continuar hasta progresión o toxicidad inaceptable. Para ambos brazos se requirió el uso concurrente de tromboprofilaxis y un inhibidor de bomba de protones, mientras que se requirió profilaxis antiviral para el brazo KRd.
Los 792 pacientes en ASPIRE fueron aleatorizados 1:1 al brazo KRd o Rd. Las características demográficas y basales estuvieron bien balanceadas entre los dos brazos (ver Tabla 2). Sólo 53% de los pacientes tuvieron pruebas para mutaciones genéticas; se identificó una mutación genética de alto riesgo en 12% de pacientes en el brazo KRd y en 13% en el brazo Rd.
Tabla 2: Características demográficas y basales en ASPIRE
|
Características |
KRd (N = 396) |
Rd (N = 396) |
|
Edad, Mediana, Años (mínimo, máximo) |
64 (38, 87) |
65 (31, 91) |
|
Edad ≥ 75 años, n (%) |
43 (11) |
53 (13) |
|
Hombres, n (%) |
215 (54) |
232 (59) |
|
Razas, n (%) |
||
|
Blanca |
377 (95) |
377 (95) |
|
Negra |
12 (3) |
11 (3) |
|
Otra o no reportado |
7 (2) |
8 (2) |
|
Número de regímenes previos, n(%) |
||
|
1 |
184 (46) |
157 (40) |
|
2 |
120 (30) |
139 (35) |
|
3a |
92 (23) |
100 (25) |
|
Trasplante previo |
217 (55) |
229 (58) |
|
Estado de desempeño ECOG |
||
|
0 |
165 (42) |
175 (44) |
|
1 |
191 (48) |
186 (47) |
|
2 |
40 (10) |
35 (9) |
|
Etapa ISS en línea basal del estudio, n (%) |
||
|
I |
167 (42) |
154 (39) |
|
II |
148 (37) |
153 (39) |
|
III |
73 (18) |
82 (21) |
|
Desconocido |
8 (2) |
7 (2) |
|
Duración de creatinina mL/min, mediana (mínimo, máximo) |
79 (39, 212) |
79 (30, 208) |
|
30 a < 50, n (%) |
19 (5) |
32 (8) |
|
50 a < 80, n (%) |
185 (47) |
170 (43) |
|
Refractario a la última terapia, n (%) |
110 (28) |
119 (30) |
|
Refractario en cualquier momento a, n (%) |
||
|
Bortezomib |
60 (15) |
58 (15) |
|
Lenalidomida |
29 (7) |
28 (7) |
|
Bortezomib + agente inmunomodulador |
24 (6) |
27 (7) |
ECOG = Grupo Oncológico Cooperativo del Este; IgG = Inmunoglobulina G; ISS = Sistema Internacional de Estadificación; KRd = KYPROLIS®, Lenalidomida, y Dexametasona; Rd = Lenalidomida y Dexametasona.
a Incluyendo 2 pacientes con 4 regímenes previos.
Los pacientes en el brazo KRd demostraron mejora en la supervivencia libre de progresión (PFS, por sus siglas en Inglés) comparado con aquellos en el brazo Rd (HR = 0.69, con valor P bilateral = 0.0001) según lo determinado usando los criterios de respuesta del Grupo Internacional de Trabajo sobre Mieloma (IMWG)/Grupo Europeo de Trasplante de Sangre y Médula (EBMT) por parte de un Comité de Revisión Independiente (IRC). La mediana de PFS fue 26.3 meses en el brazo KRd versus 17.6 meses en el brazo Rd (ver Tabla 3 y Figura 1).
Se realizó un análisis preestablecido de la supervivencia global (OS por sus siglas en inglés) después de 246 muertes en el brazo de KRd y 267 muertes en el brazo de Rd. La mediana de seguimiento fue de aproximadamente 67 meses. Se observó una ventaja estadísticamente significativa en la OS en pacientes del brazo de KRd en comparación con los pacientes del brazo de Rd (ver Tabla 3 y Figura 2).
Tabla 3: Resultados de eficacia en ASPIREa
|
Terapia de combinación |
||
|
KRd (N = 396) |
Rd (N = 396) |
|
|
PFSb |
||
|
Medianac, Meses (IC 95%) |
26.3 (23.3, 30.5) |
17.6 (15.0, 20.6) |
|
HR (IC 95%)d |
0.69 (0.57, 0.83) |
|
|
Valor P (bilateral)e |
0.0001 |
|
|
OS |
||
|
Medianac, Meses (IC 95%) |
48.3 (42.4, 52.8) |
40.4 (33.6, 44.4) |
|
HR (IC 95%)d |
0.79 (0.67, 0.95) |
|
|
Valor P (bilateral)e |
0.0091 |
|
|
Respuesta globalb |
||
|
N con respuesta |
345 |
264 |
|
ORR (%) (IC 95%)f |
87 (83, 90) |
67 (62, 71) |
|
Valor P (bilateral)g |
<0.0001 |
|
|
Categoría de respuesta, n (%) |
||
|
sCR |
56 (14) |
17 (4) |
|
CR |
70 (18) |
20 (5) |
|
VGPR |
151 (38) |
123 (31) |
|
PR |
68 (17) |
104 (26) |
IC = intervalo de confianza; CR = respuesta completa; KRd = KYPROLIS®, Lenalidomida y Dexametasona; ORR = Tasa de respuesta global; PFS = supervivencia libre de progresión; Rd = Lenalidomida y Dexametasona; sCR = CR estricta; VGPR = muy buena respuesta parcial.
a Los pacientes elegibles tuvieron 1 a 3 líneas de terapia previas.
b Acorde a lo determinado por el Comité de Revisión Independiente.
c Basado en estimados de Kaplan-Meier.
d Basado en modelo estratificado de Cox.
e El valor P fue derivado usando prueba estratificada log-rank.
f Intervalo de confianza exacto.
g El valor P fue derivado usando la prueba Cochran Mantel Haenszel.
La mediana de duración de respuesta (DOR, por sus siglas en inglés) fue de 28.6 meses (IC 95%: 24.9, 31.3) para los 345 pacientes que alcanzaron una respuesta en el brazo KRd y 21.2 meses (IC 95%: 16.7, 25.8) para los 264 pacientes que alcanzaron una respuesta en el brazo Rd. La mediana de tiempo para respuesta fue 1 mes (rango de 1 a 14 meses) en el brazo KRd y 1 mes (rango 1 a 16 meses) en el brazo Rd.
Figura 1. Curva Kaplan-Meier de supervivencia libre de progresión en ASPIRE
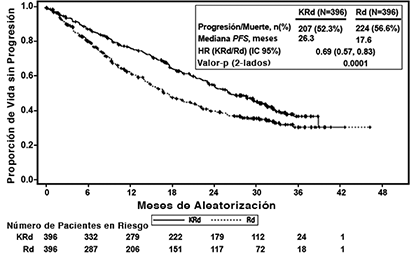
IC = intervalo de confianza; EBMT = Grupo Europeo de Trasplante de Sangre y Médula Ósea; HR = índice de riesgo; IMWG = Grupo Internacional de Trabajo sobre Mieloma; KRd = KYPROLIS®, lenalidomida, y dexametasona; mo = meses; PFS = supervivencia libre de progresión; Rd = brazo de lenalidomida y dexametasona.
Nota: La respuesta y los resultados de PD fueron determinados usando los criterios de respuesta objetiva estándar IMWG/EBMT.
Figura 2. Curva Kaplan-Meier de supervivencia global en ASPIRE
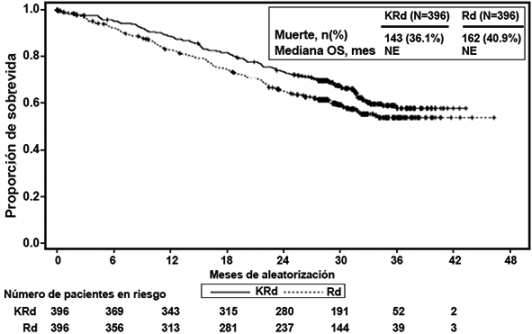
IC = intervalo de confianza; HR = índice de riesgo; KRd = KYPROLIS®, Lenalidomida, y Dexametasona; me = mes; OS = supervivencia global; Rd = brazo de lenalidomida y dexametasona.
En combinación con dexametasona para mieloma múltiple en recaída o refractario: La eficacia de KYPROLIS® en combinación con dexametasona fue evaluada en dos estudios abiertos, aleatorizados (ENDEAVOR y A.R.R.O.W.).
ENDEAVOR
ENDEAVOR fue un estudio aleatorizado, abierto, multicéntrico, de KYPROLIS® y dexametasona (Kd) versus bortezomib y dexametasona (Vd) en pacientes con mieloma múltiple en recaída o refractario que han recibido 1 a 3 líneas de terapia. Un total de 929 pacientes fueron enrolados y aleatorizados (464 en el brazo Kd; 465 en el brazo Vd). La aleatorización fue estratificada por terapia previa de inhibidor de proteosoma (sí versus no), líneas de terapia previas (1 versus 2 ó 3), etapa vigente del Sistema Internacional de Estadificación (1 versus 2 ó 3) y vía planeada de administración de bortezomib. Los pacientes fueron excluidos si tuvieron menos de una respuesta parcial PR a todos los regímenes previos; depuración de creatinina < 15 mL/min; transaminasas hepáticas ≥ 3 x ULN; o fracción de eyección ventricular izquierda < 40% u otra condición cardiaca significativa.
Este estudio evaluó KYPROLIS® en una dosis de arranque de 20 mg/m2, la cual se incrementó a 56 mg/m2 en el Ciclo 1, del Día 8 en adelante. KYPROLIS® fue administrado dos veces semanalmente mediante una infusión de 30 minutos en los Días 1, 2, 8, 9, 15 y 16 de cada ciclo de 28 días. Se administró dexametasona 20 mg oral e intravenoso en los Días, 1, 2, 8, 9, 15, 16, 22 y 23 de cada ciclo. En el brazo Vd, se dosificó bortezomib a 1.3 mg/m2 intravenoso y subcutáneo en los Días 1, 4, 8 y 11 de un ciclo de 21 días, y se administró dexametasona 20 mg oral o intravenoso en los Días 1, 2, 4, 5, 8, 9, 11 y 12 de cada ciclo. El uso concurrente de tromboprofilaxis fue opcional, y se requirió profilaxis con un agente antiviral y un inhibidor de bomba de protones. De los 465 pacientes en el brazo Vd, 381 recibieron bortezomib subcutáneo. El tratamiento continuó hasta progresión de la enfermedad o toxicidad inaceptable.
Las características demográficas y basales están resumidas en la Tabla 3.
Tabla 4: Características demográficas y basales en ENDEAVOR
|
Características |
Kd (N = 464) |
Vd (N = 465) |
|
Edad, años |
||
|
Mediana (min, max) |
65 (35, 89) |
65 (30, 88) |
|
< 65, n (%) |
223 (48) |
210 (45) |
|
65 a 74, n (%) |
164 (35) |
189 (41) |
|
≥ 75, n (%) |
77 (17) |
66 (14) |
|
Género, n (%) |
||
|
Femenino |
224 (48) |
236 (51) |
|
Masculino |
240 (52) |
229 (49) |
|
Raza, n (%) |
||
|
Blanca |
353 (76) |
361 (78) |
|
Negra |
7 (2) |
9 (2) |
|
Asiática |
56 (12) |
57 (12) |
|
Otra o no reportado |
48 (10) |
38 (8) |
|
Estado de desempeño ECOG, n (%) |
||
|
0 |
221 (48) |
232 (50) |
|
1 |
210 (45) |
203 (44) |
|
2 |
33 (7) |
30 (6) |
|
Depuración de creatinina (mL/min) |
||
|
Mediana (mínimo, máximo) |
73 (14, 185) |
72 (12, 208) |
|
< 30, n (%) |
28 (6) |
28 (6) |
|
30 a < 50, n (%) |
57 (12) |
71 (15) |
|
50 a < 80, n (%) |
186 (40) |
177 (38) |
|
≥ 80, n (%) |
193 (42) |
189 (41) |
|
FISH, n (%) |
||
|
Riesgo alto |
97 (21) |
113 (24) |
|
Riesgo estándar |
284 (61) |
291 (63) |
|
Riesgo desconocido |
83 (18) |
61 (13) |
|
Etapa ISS en línea basal del estudio, n (%) |
||
|
ISS I |
219 (47) |
212 (46) |
|
ISS II |
138 (30) |
153 (33) |
|
ISS III |
107 (23) |
100 (22) |
|
Número de regímenes previos |
||
|
1 |
232 (50) |
231 (50) |
|
2 |
158 (34) |
144 (31) |
|
3 |
74 (16) |
88 (19) |
|
4 |
0 (0) |
2 (0.4) |
|
Terapias previas, n (%) |
464 (100) |
465 (100) |
|
Bortezomib |
250 (54) |
252 (54) |
|
Trasplante por mieloma Múltiple |
266 (57) |
272 (59) |
|
Talidomida |
212 (46) |
249 (54) |
|
Lenalidomida |
177 (38) |
178 (38) |
|
Bortezomib + agente inmunomodulador |
159 (34) |
168 (36) |
|
Refractario a la última terapia previa, n (%)a |
184 (40) |
189 (41) |
ECOG = Grupo Oncológico Cooperativo del Este; FISH = Hibridación fluorescente in situ; ISS = Sistema Internacional de Estadificación; Kd = KYPROLIS® y Dexametasona; Vd = Bortezomib y Dexametasona.
a Refractario = enfermedad que no alcanza una respuesta mínima o mejor, que progresa durante la terapia o que progresa en un lapso de 60 días después de completar la terapia.
La eficacia de KYPROLIS® fue evaluada mediante PFS acorde a lo determinado por un IRC usando los criterios de respuesta IMWG. El estudio mostró una mediana de PFS de 18.7 meses en el brazo Kd versus 9.4 meses en el brazo Vd (ver Tabla 5 y Figura 3).
Figura 3: Gráfica Kaplan-Meier de supervivencia libre de progresión en ENDEAVOR
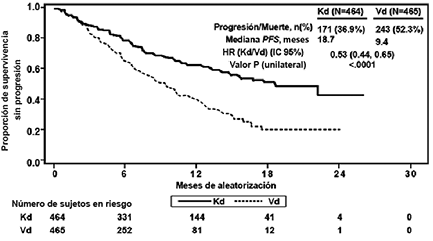
IC = intervalo de confianza; HR = índice de riesgo; Kd = KYPROLIS® y Dexametasona; me= mes; PFS = supervivencia libre de progresión; Vd = Bortezomib y Dexametasona.
Otros criterios de valoración incluyeron la OS y la tasa de respuesta global (ORR, por sus siglas en inglés).
Se realizó un análisis de la OS pre-planificado después de 189 muertes en el brazo de Kd y 209 muertes en el brazo de Vd. La mediana de seguimiento fue de aproximadamente 37 meses. Se observó una OS significativamente más prolongada en los pacientes del brazo de Kd en comparación con los pacientes en el brazo de Vd (HR = 0.79; IC del 95%: 0.65, 0.96; valor p = 0.01). Los resultados se proporcionan en la Tabla 5 y la Figura 4).
Tabla 5: Resumen de resultados clave en ENDEAVOR
(Población con intención de tratar)a
|
Kd (N = 464) |
Vd (N = 465) |
|
|
PFSb |
||
|
Número de eventos (%) |
171 (37) |
243 (52) |
|
Medianac, Meses (IC 95%) |
18.7 (15.6, NE) |
9.4 (8.4, 10.4) |
|
HR (Kd/Vd) (IC 95%)d |
0.53 (0.44, 0.65) |
|
|
Valor P (unilateral)e |
< 0.0001 |
|
|
Supervivencia global |
||
|
Número de muertes (%) |
189 (41) |
209 (45) |
|
Medianac, meses (IC 95%) |
47.6 (42.5, NE) |
40.0 (32.6, 42.3) |
|
HR (Kd/Vd) (IC 95%)d |
0.79 (0.65, 0.96) |
|
|
Valor P (unilateral)e |
0.01 |
|
|
Respuesta generalb |
||
|
N con respuesta |
357 |
291 |
|
ORR (%) (IC 95%)f |
77 (73, 81) |
63 (58, 67) |
|
Valor P (unilateral)g |
< 0.0001 |
|
|
Categoría de respuesta, n (%) |
||
|
sCR |
8 (2) |
9 (2) |
|
CR |
50 (11) |
20 (4) |
|
VGPR |
194 (42) |
104 (22) |
|
PRh |
105 (23) |
158 (34) |
IC = intervalo de confianza; CR = respuesta completa; HR = índice de riesgo; Kd = KYPROLIS® y dexametasona; ORR = Tasa de respuesta global; PFS = supervivencia libre de progresión; PR = respuesta parcial; sCR = CR estricta; Vd = bortezomib y dexametasona; VGPR = muy buena respuesta parcial; NE = no estimable.
a Los pacientes elegibles tuvieron 1 a 3 líneas de terapia previas.
b PFS y ORR fueron determinados por parte de un Comité de Revisión Independiente.
c En base a estimados Kaplan-Meier.
d En base a un modelo estratificado de Cox.
e El valor P fue derivado usando una prueba estratificada log-rank.
f Intervalo de confianza exacto.
g El valor P fue derivado usando la prueba de Cochran Mantel Haenszel.
h Incluye un paciente en cada brazo con una PR confirmada la cual puede no haber sido la mejor respuesta.
Figura 4: Gráfica de Kaplan-Meier de la supervivencia global en ENDEAVOR

IC = intervalo de confianza; HR = índice de riesgo; Kd = KYPROLIS® y Dexametasona; me = mes; OS = supervivencia global; Vd = Bortezomib y Dexametasona.
La mediana de DOR en sujetos que alcanzan PR o mejor, fue 21.3 meses (IC 95%: 21.3, no estimable) en el brazo Kd y 10.4 meses (IC 95%: 9.3, 13.8) en el brazo Vd. La mediana de tiempo para respuesta fue 1 mes (rango <1 a 8 meses) en ambos brazos.
A.R.R.O.W.:
A.R.R.O.W. fue un estudio aleatorizado, abierto, multicéntrico, de superioridad de KYPROLIS® más dexametasona (Kd) una vez por semana (20/70 mg/m2) versus Kd dos veces por semana (20/27 mg/m2) en pacientes con mieloma múltiple en recaída y refractario que habían recibido 2 a 3 líneas previas de terapia. Los pacientes se excluyeron si tenían menos de PR (remisión parcial, por sus siglas en inglés) en al menos una línea previa; depuración de creatinina < 30 mL/min; transaminasas hepáticas ≥ 3 × ULN; o fracción de eyección del ventrículo izquierdo < 40% u otras afecciones cardiacas significativas. Un total de 478 pacientes fueron reclutados y aleatorizados (240 en el brazo de 20/70 mg/m2; 238 en el brazo de 20/27 mg/m2). La aleatorizazión se estratificó en estadios por el actual Sistema Internacional de clasificación de estadios (estadio 1 versus estadios 2 ó 3), refractaria al tratamiento con bortezomib (sí versus no) y edad (< 65 versus ≥ 65 años).
El brazo 1 de este estudio evaluó KYPROLIS® a una dosis inicial de 20 mg/m2, que se incrementó a 70 mg/m2 en el Ciclo 1, desde el día 8 en adelante. En el brazo 1 KYPROLIS® se administró una vez a la semana mediante una infusión de 30 minutos los días 1, 8 y 15 de cada ciclo de 28 días. El brazo 2 de este estudio evaluó KYPROLIS a una dosis inicial de 20 mg/m2, que se incrementó a 27 mg/m2 en el Ciclo 1, desde el día 8 en adelante. En el brazo 2 KYPROLIS® se administró dos veces por semana mediante una infusión de 10 minutos en los días 1, 2, 8, 9, 15 y 16 de cada ciclo de 28 días. En ambos regímenes, se administraron 40 mg de dexametasona por vía oral o intravenosa en los días 1, 8, 15 para todos los ciclos y en el día 22 para los ciclos 1 a 9 solamente. El uso simultáneo de tromboprofilaxis fue opcional, se recomendó la profilaxis con un agente antiviral y se requirió la profilaxis con un inhibidor de la bomba de protones. El tratamiento continuó hasta la progresión de la enfermedad o una toxicidad inaceptable.
Las características demográficas y basales se resumen en la Tabla 6.
Tabla 6: Demografía y características basales en A.R.R.O.W.
|
Características |
Kd 20/70 mg/m2 una vez por semana (N = 240) |
Kd 20/27 mg/m2 dos veces por semana (N = 238) |
|
Edad, años |
||
|
Mediana (min, max) |
66 (39, 85) |
66 (35, 83) |
|
< 65, n (%) |
104 (43.3) |
104 (43.7) |
|
65 a 74, n (%) |
90 (37.5) |
102 (42.9) |
|
≥ 75, n (%) |
46 (19.2) |
32 (13.4) |
|
Género, n (%) |
||
|
Femenino |
108 (45.0) |
110 (46.2) |
|
Masculino |
132 (55.0) |
128 (53.8) |
|
Raza, n (%) |
||
|
Blanca |
200 (83.3) |
202 (84.9) |
|
Negra |
3 (1.3) |
2 (0.8) |
|
Asiática |
30 (12.5) |
15 (6.3) |
|
Otras o no reportada |
7 (3.0) |
19 (8.0) |
|
Estado de desempeño ECOG, n (%) |
||
|
0 |
118 (49.2) |
118 (49.6) |
|
1 |
121 (50.4) |
120 (50.4) |
|
2 |
1 (0.4) |
0 (0.0) |
|
Depuración de creatinina (mL/min) |
||
|
Mediana (min, max) |
70.80 (27.6, 211.8) |
73.20 (28.8, 181.2) |
|
< 30, n (%) |
2 (0.8) |
1 (0.4) |
|
30 a < 50, n (%) |
48 (20.0) |
34 (14.3) |
|
50 a < 80, n (%) |
91 (37.9) |
111 (46.6) |
|
≥ 80, n (%) |
99 (41.3) |
91 (38.2) |
|
FISH, n (%) |
||
|
Riesgo alto |
34 (14.2) |
47 (19.7) |
|
Riesgo estándar |
47 (19.6) |
53 (22.3) |
|
Riesgo desconocido |
159 (66.3) |
138 (58.0) |
|
Etapa ISS al basal, n (%) |
||
|
ISS I |
94 (39.2) |
99 (41.6) |
|
ISS II |
80 (33.3) |
81 (34.0) |
|
ISS III |
63 (26.3) |
54 (22.7) |
|
Número de regímenes previos |
||
|
2 |
116 (48.3) |
125 (52.5) |
|
3 |
124 (51.7) |
112 (47.1) |
|
> 3 |
0 (0.0) |
1 (0.4) |
|
Terapias previas, n (%) |
||
|
Bortezomib |
236 (98.3) |
237 (99.6) |
|
Trasplante |
146 (60.8) |
157 (66.0) |
|
Talidomida |
119 (49.6) |
119 (50.0) |
|
Lenalidomida |
207 (86.3) |
194 (81.5) |
ECOG = Grupo Oncológico Cooperativo del Este; FISH = Hibridación in situ fluorescente; ISS = Sistema Internacional de Estadificación; Kd = KYPROLIS® y Dexametasona.
La eficacia de KYPROLIS® se evaluó por la PFS utilizando los criterios de respuesta del IMWG. Los resultados de eficacia se proporcionan en la Tabla 7 y la Figura 5.
Figura 5: Gráfico de Kaplan Meier de supervivencia libre de progresión en A.R.R.O.W.

IC = intervalo de confianza; HR = índice de riesgo; Kd = KYPROLIS® y Dexametasona; PFS = supervivencia libre de progresión.
Tabla 7: Resumen de los resultados clave en A.R.R.O.W. (Población ITT)
|
Kd 20/70 mg/m2 Una vez por semana (N = 240) |
Kd 20/27 mg/m2 Dos veces por semana (N = 238) |
|
|
PFS |
||
|
Número de eventos, n (%) |
126 (52.5) |
148 (62.2) |
|
Mediana, meses (IC 95%) |
11.2 (8.6, 13.0) |
7.6 (5.8, 9.2) |
|
HR (IC 95%) |
0.69 (0.54, 0.88) |
|
|
Valor-p (unilateral) |
0.0014 |
|
|
Respuesta globala |
||
|
N con Respuesta |
151 |
97 |
|
ORR (%) (IC 95%) |
62.9 (56.5, 69.0) |
40.8 (34.5, 47.3) |
|
Valor-p (unilateral) |
<0.0001 |
|
|
Categoría de respuesta, n (%) |
||
|
sCR |
4 (1.7) |
0 (0.0) |
|
CR |
13 (5.4) |
4 (1.7) |
|
VGPR |
65 (27.1) |
28 (11.8) |
|
PR |
69 (28.8) |
65 (27.3) |
IC = intervalo de confianza; CR = respuesta completa; HR = índice de riesgo; Kd = KYPROLIS® y Dexametasona; ORR = Tasa de respuesta global; PFS = supervivencia libre de progresión; PR = respuesta parcial; sCR = respuesta completar estricta; VGPR = muy buena respuesta parcial.
a La respuesta global se define como lograr la mejor respuesta global de PR, VGPR, CR o sCR.
La mediana de DOR en sujetos que alcanzaron PR o mejor fue de 15 meses (IC 95%: 12.2, no estimable) en el brazo de Kd 20/70 mg/m2 y 13.8 meses (IC 95%: 9.5, no estimable) en el brazo de Kd 20/27 mg/m2. El tiempo medio de respuesta fue de 1.1 meses en el brazo de Kd 20/70 mg/m2 y de 1.9 meses en el brazo de Kd 20/27 mg/m2.
En combinación con daratumumab intravenoso y dexametasona para mieloma múltiple en recaída o refractario: La seguridad de KYPROLIS® en combinación con daratumumab y dexametasona (DKd) fue evaluada en dos estudios abiertos, aleatorizados (CANDOR y Equuleus).
CANDOR:
CANDOR fue un estudio aleatorizado, abierto, multicéntrico que evaluó la combinación de KYPROLIS® 20/56 mg/m2 dos veces a la semana con daratumumab intravenoso y dexametasona (DKd) frente a KYPROLIS® 20/56 mg/m2 dos veces a la semana y dexametasona (Kd) en pacientes con mieloma múltiple en recaída o refractario que habían recibido de 1 a 3 líneas de terapia anteriores. Se excluyó del estudio a los pacientes que presentaban las siguientes patologías: asma persistente moderada o grave en los últimos 2 años, enfermedad pulmonar obstructiva crónica (EPOC) con VEF 1 < 50% de insuficiencia cardíaca congestiva normal y activa predictiva. La aleatorización se estratificó según el ISS (estadio 1 o 2 versus estadio 3) en la selección, antes de la exposición al inhibidor de proteasoma (sí versus no), cantidad de líneas de terapia anteriores (1 versus ≥ 2) o terapia de anticuerpos de antígeno de diferenciación 38 (CD38) anterior (sí versus no).
KYPROLIS® se administró por vía intravenosa durante 30 minutos en una dosis de 20 mg/m2 en el Ciclo 1 los Días 1 y 2; en una dosis de 56 mg/m2 en el Ciclo 1 los Días 8, 9, 15 y 16; y los Días 1, 2, 8, 9, 15 y 16 de cada ciclo de 28 días, y de ahí en adelante. Se administraron 20 mg de dexametasona por vía oral o intravenosa los Días 1, 2, 8, 9, 15 y 16 y, luego, 40 mg por vía oral o intravenosa el Día 22 de cada ciclo de 28 días. En el brazo de DKd, se administró daratumumab por vía intravenosa en una dosis de 8 mg/kg en el Ciclo 1 los Días 1 y 2. A partir de allí, se administró daratumumab por vía intravenosa en una dosis de 16 mg/kg los Días 8, 15 y 22 del Ciclo 1; los Días 1, 8, 15 y 22 del Ciclo 2; los Días 1 y 15 de los Ciclos 3 a 6; y el Día 1 para los ciclos restantes o hasta la progresión de la enfermedad. En el caso de los pacientes mayores de 75 años a los que se administraba una dosis reducida de dexametasona de 20 mg, la dosis completa de 20 mg se administró como un medicamento preinfusión de daratumumab los días en que se administraba daratumumab. En los otros casos, la dosis de dexametasona se dividió en los días en que se administraba KYPROLIS® en ambos brazos del estudio. Se continuó el tratamiento hasta la progresión de la enfermedad o una toxicidad inaceptable.
Se aleatorizó un total de 466 pacientes; 312 en el brazo DKd y 154 en el brazo Kd. Las características demográficas y basales se resumen en la Tabla 8.
Tabla 8: Características demográficas y basales en CANDOR
|
Características |
DKd (N = 312) |
Kd (N = 154) |
|
Edad en la aleatorización (años) |
||
|
Mediana (mín., máx.) |
64 (29; 84) |
65 (29; 84) |
|
Grupo de edad, n (%) |
||
|
18 a 64 años |
163 (52) |
77 (50) |
|
65 a 74 años |
121 (39) |
55 (36) |
|
75 años o más |
28 (9) |
22 (14) |
|
Sexo, n (%) |
||
|
Masculino |
177 (57) |
91 (59) |
|
Femenino |
135 (43) |
63 (41) |
|
Raza, n (%) |
||
|
Asiática |
46 (15) |
20 (13) |
|
Negra o afroamericana |
7 (2.2) |
2 (1.3) |
|
Blanca |
243 (78) |
123 (80) |
|
Otra |
16 (5) |
9 (6) |
|
Región geográfica, n (%) |
||
|
Norteamérica |
21 (7) |
12 (8) |
|
Europa |
207 (66) |
103 (67) |
|
Asia Pacífico |
84 (27) |
39 (25) |
|
Estado de desempeño ECOG, n (%) |
||
|
0 o 1 |
295 (95) |
147 (95) |
|
2 |
15 (4.8) |
7 (4.5) |
|
Faltante |
2 (0.6) |
0 (0.0) |
|
Grupo de riesgo según lo determinado por FISH, n (%) |
||
|
Riesgo alto |
48 (15) |
26 (17) |
|
Riesgo estándar |
104 (33) |
52 (34) |
|
Desconocido |
160 (51) |
76 (49) |
|
Estadio de ISS según I x RS en la selección, n (%) |
||
|
I o II |
252 (81) |
127 (82) |
|
III |
60 (19) |
27 (17) |
|
Número de regímenes previos, n (%) |
||
|
1 |
144 (46) |
70 (45) |
|
2 |
99 (32) |
46 (30) |
|
3 |
69 (22) |
37 (24) |
|
Terapias previas |
||
|
Lenalidomida |
123 (39) |
74 (48) |
|
Refractario a la lenalidomida |
99 (32) |
55 (36) |
|
Bortezomib |
287 (92) |
134 (87) |
|
Terapia de anticuerpos CD38 anterior, n (%) |
1 (0.3) |
O (O.O) |
|
Trasplante de células madres (ASCT) anterior, n (%) |
195 (62) |
75 (49) |
ECOG = Grupo Oncológico Cooperativo del Este; FISH = Hibridación fluorescente in situ; ISS = Sistema Internacional de Estadificación; DKd = KYPROLIS®, daratumumab y dexametasona.
* Los sujetos con una cantidad de regímenes anteriores mayor que 3 fueron O en el brazo DKd y 1 en el brazo Kd.
Se evaluó la eficacia mediante una evaluación IRC de PFS con los criterios de respuesta de IMWG. Los resultados de eficacia se proporcionan en la Tabla 9 y la Figura 6. La mediana de la duración de respuesta no se alcanzó para el brazo DKd y fue de 16.6 meses (13.9, NE) para el brazo Kd. La mediana de tiempo (mín., máx.) para respuesta fue de 1.0 (1, 14) mes para el brazo DKd y 1.0 (1, 10) mes para el brazo Kd.
Figura 6: Gráfica de Kaplan-Meier de supervivencia libre de progresión en CANDOR

DKd = KYPROLIS®, Daratumumab y Dexametasona; Kd = Kyprolis y Dexametasona.
Tabla 9: Resumen de resultados clave en CANDOR (Población con intención de tratar)
|
DKd (N = 312) |
Kd (N = 154) |
|
|
PFS |
||
|
Número de eventos (%) |
110 (35%) |
68 (44%) |
|
Mediana, meses (IC 95%) |
NE (NE, NE) |
15.8 (12.1, NE) |
|
HR (IC 95%) |
0.63 (0.46; 0.85) |
|
|
Valor P (unilateral)a |
0.0014 |
|
|
Respuesta global |
||
|
N con respuesta |
263 |
115 |
|
ORR (%) (IC 95%) |
84% (80, 88%) |
75% (67%, 81%) |
|
Valor P (unilateral)b |
0.0040 |
|
|
CR |
89 (28%) |
16 (10%) |
|
VGPR |
127 (41%) |
59 (38%) |
|
PR |
47 (15%) |
40 (26%) |
|
MRD [-] tasa de CR a 12 meses n(%)C (IC 95%) |
39 (12%) (9%, 17%) |
2 (1,3%) (0.2%; 4.6%) |
|
Valor P (unilateral)b |
< 0.0001 |
|
|
MRD [-] CRd |
43 (14%) |
5 (3.2%) |
IC = intervalo de confianza; CR = respuesta completa; HR = índice de riesgo; DKd = KYPROLIS®, Daratumumab y Dexametasona; Kd = KYPROLIS® y Dexametasona; ORR = tasa de respuesta global; PFS = supervivencia libre de progresión; PR = respuesta parcial; MRD [-] CR = enfermedad mínima residual negativa-respuesta completa; NE = no estimable; VGPR = muy buena respuesta parcial.
ª El valor P fue derivado usando una prueba estratificada log-rank.
b El valor P fue derivado usando la prueba estratificada de Chi-cuadrado de Cocheran-Mantel-Haenszel.
c MRD [-] CR (en un nivel de 10-5) se define como el alcance de una CR por MWG URC y el estado de MRD[-] según se evaluó mediante el ensayo de secuenciación de próxima generación (ClonoSEQ) en el punto de referencia de 12 meses (un periodo de 8 a 13 meses).
d MRD[-]CR (en un nivel de 10-5) se define como el alcance de una CR por IMWG-URC y el estado de MRD[-] según se evaluó mediante el ensayo de secuenciación de próxima generación (ClonoSEQ) en cualquier fecha de evaluación durante el estudio.
EQUULEUS:
EQUULEUS fue un estudio abierto, de múltiples cohortes que evaluó la combinación de KYPROLIS® con daratumumab intravenoso y dexametasona en pacientes con mieloma múltiple en recaída o refractario que habían recibido de 1 a 3 líneas de terapia anteriores. Se excluyó del estudio a los pacientes que presentaban las siguientes patologías: asma persistente moderada o grave en los últimos 2 años, enfermedad pulmonar obstructiva crónica (EPOC) con VEF 1 < 50% de insuficiencia cardiaca congestiva normal o activa predictiva (definida como Clase III a IV por la New York Heart Association).
KYPROLIS® se administró por vía intravenosa durante 30 minutos una vez por semana en una dosis de 20 mg/m2 en el Ciclo 1, Día 1 y se aumentó a una dosis de 70 mg/m2 en el Ciclo 1, los Días 8 y 15; y los Días 1, 8, y 15 de cada ciclo de 28 días. Se administró daratumumab a diez pacientes en dosis de 16 mg/kg por vía intravenosa en el Ciclo 1, Día 1 y se administró daratumumab a los pacientes restantes en dosis de 8 mg/kg por vía intravenosa en el Ciclo 1, Días 1 y 2. A partir de allí, se administró daratumumab por vía intravenosa en dosis de 16 mg/kg los Días 8, 15 y 22 del Ciclo 1; los Días 1, 8, 15 y 22 del Ciclo 2; los Días 1 y 15 de los Ciclos 3 a 6; y el Día 1 para los ciclos restantes de cada ciclo de 28 días. En los Ciclos 1 y 2, se administró dexametasona en dosis de 20 mg por vía oral o intravenosa los Días 1, 2, 8, 9, 15, 16, 22 y 23; en los ciclos 3 a 6, se administró dexametasona de 20 mg por vía oral o intravenosa los Días 1, 2, 15 y 16 y en dosis de 40 mg el Día 8 y 22; y en los ciclos 7 y siguientes, se administró dexametasona de 20 mg por vía oral o intravenosa los Días 1 y 2 y en dosis de 40 mg los Días 8, 15 y 22. En los pacientes mayores de 75 años, se administraron 20 mg de dexametasona por vía oral o intravenosa semanalmente después de la primera semana. El tratamiento continuó hasta la progresión de la enfermedad o una toxicidad inaceptable.
85 pacientes se inscribieron en el estudio EQUULEUS. Las características demográficas y basales se resumen en la Tabla 10.
Tabla 10: Características demográficas y basales con un régimen de 20/70 mg/m2 de EQUULEUS (Terapia de combinación para mieloma múltiple en recaída o refractario)
|
Características |
Número de pacientes (%) |
|
Edad (años) |
|
|
Mediana (mín., máx.) |
66 (38; 85) |
|
Grupo de edad, n (%) |
|
|
< 65 años |
36 (42) |
|
65 a 75 años |
41 (48) |
|
≥ 75 años |
8 (9) |
|
Sexo, n (%) |
|
|
Masculino |
46 (54) |
|
Femenino |
39 (46) |
|
Raza, n (%) |
|
|
Asiática |
3 (3.5) |
|
Negra o afroamericana |
3 (3.5) |
|
Blanca |
68 (80) |
|
Escala ECOG, n (%) |
|
|
0 |
32 (38) |
|
1 |
46 (54) |
|
2 |
7 (8) |
|
FISH, n (%) |
|
|
n (%) |
67 |
|
Riesgo estándar |
54 (81) |
|
Riesgo alto |
13 (19) |
|
Número de regímenes previos |
|
|
1 |
20 (23) |
|
2 |
40 (47) |
|
3 |
23 (27) |
|
> 3 |
2 (2,4) |
|
Terapias previas |
|
|
Bortezomib |
85 (100) |
|
Lenalidomida |
81 (95) |
|
Trasplante de células madres (ASCT) anterior |
62 (73) |
|
Refractario a la lenalidomida |
50 (59) |
|
Refractario a PI e IMiD |
25 (29) |
ECOG = Grupo Oncológico Cooperativo del Este; FISH = hibridación fluorescente in situ. Los resultados de eficacia se basaron en la tasa de respuesta global mediante criterios IMWG; PI = inhibidor de proteosoma; IMiD = agente inmunomodulador.
Los resultados de eficacia se basaron en la tasa de respuesta global mediante criterios IMWG. Los resultados de eficacia se proporcionan en la Tabla 11 . La mediana de tiempo de respuesta fue de 0.95 meses (rango: 0.9; 14.3). La mediana de duración de respuesta fue de 28 meses (IC 95%: 20.5; no estimable).
Tabla 11: Resumen de resultados clave en EQUULEUS (Población con intención de tratar)
|
Pacientes del Estudio n % |
|
|
Respuesta global |
|
|
N con respuesta |
69 |
|
ORR (%) (IC 95%) |
81% (71, 89) |
|
Categoría de respuesta, n (%) |
|
|
sCR |
18 (21%) |
|
CR |
12 (14%) |
|
VGPR |
28 (33%) |
|
PR |
11 (13%) |
IC = intervalo de confianza; sCR = respuesta completa estricta; CR = respuesta completa, ORR = tasa de respuesta global; PR = respuesta parcial; VGPR = muy buena respuesta parcial.
Monoterapia para mieloma múltiple en recaída o refractario:
Estudio PX-171-007:
El estudio PX-171-007 fue un estudio multicéntrico, abierto, de escalamiento de dosis, de brazo único que evaluó la seguridad de la monoterapia con carfilzomib mediante una infusión de 30 minutos en pacientes con mieloma múltiple en recaída o refractario después de 2 ó más líneas de terapia. Los pacientes fueron excluidos si tenían una depuración de creatinina < 20 mL/min; ALT ≥ 3 x límite superior del normal (ULN); bilirrubina ≥ 1.5 x ULN; insuficiencia cardiaca congestiva de clases III o IV de la New York Heart Association, u otra condición cardiaca significativa. Un total de 24 sujetos con mieloma múltiple fueron enrolados en el nivel de dosis máxima tolerada de 20/56 mg/m2. Se administró carfilzomib dos veces semanalmente por 3 semanas consecutivas (Días 1, 2, 8, 9, 15 y 16) de un ciclo de 28 días. Del Ciclo 13 en adelante, las dosis de carfilzomib de los días 8 y 9 pudieron ser omitidas. Los pacientes recibieron carfilzomib en una dosis de arranque de 20 mg/m2 en los Días 1 y 2 del Ciclo 1, la cual se incrementó a 56 mg/m2 para todas las dosis subsecuentes. Se requirió dexametasona 8 mg oral o intravenoso antes de cada dosis de carfilzomib en el Ciclo 1 y fue opcional en ciclos subsecuentes. Se continuó el tratamiento hasta progresión de la enfermedad o toxicidad inaceptable.
La eficacia fue evaluada mediante ORR y DOR. La ORR por evaluación del investigador fue 50% (IC 95%: 29, 71) acorde a los criterios IMWG (ver Tabla 12). La mediana de DOR en sujetos que alcanzaron una PR o mejor fue 8.0 meses (Rango: 1.4, 32.5).
Tabla 12: Categorías de respuesta en el estudio PX-171-007 (Régimen de monoterapia de 20/56 mg/m2)
|
Características |
Pacientes del estudioa n (%) |
|
Número de pacientes (%) |
24 (100) |
|
Respuesta generalb |
12 (50) |
|
IC 95%c |
(29, 71) |
|
Categoría de Respuesta |
|
|
sCR |
1 (4) |
|
CR |
0 (0) |
|
VGPR |
4 (17) |
|
PR |
7 (29) |
IC = intervalo de confianza; CR = respuesta completa; PR = respuesta parcial; sCR = respuesta completa estricta; VGPR = muy buena respuesta parcial.
a Pacientes elegibles tuvieron 2 o más líneas de terapia previas.
b Según evaluación del investigador.
c Intervalo de confianza exacto.
Estudio PX-171-003 A1:
El Estudio PX-171-003 A1, fue un estudio de un solo brazo, multicéntrico de monoterapia con KYPROLIS® mediante infusión de hasta 10 minutos. Los pacientes elegibles fueron aquellos con mieloma múltiple en recaída y refractario que habían sido sometidos al menos a dos tratamientos previos (incluido el tratamiento con bortezomib y talidomida y/o lenalidomida) y tuvieron ≤25% de respuesta a la terapia más reciente o tuvieron progresión de la enfermedad durante o en un lapso de 60 días de la terapia más reciente. Los pacientes excluidos del ensayo fueron aquellos refractarios a las terapias anteriores o con bilirrubina total ≥ 2 x ULN; depuración de creatinina < 30 mL/min; insuficiencia cardiaca congestiva Clases III a IV conforme a la New York Heart Association (NYHA, por sus siglas en Inglés); isquemia cardiaca sintomática; infarto de miocardio dentro de los últimos 6 meses; neuropatía periférica Grado 3 o 4; o neuropatía periférica Grado 2 con dolor; infecciones activas que requerían tratamiento; o derrame pleural.
Se administró KYPROLIS® por vía intravenosa hasta 10 minutos en dos días consecutivos a la semana por tres semanas, seguido de un periodo de descanso de 12 días (ciclo de tratamiento de 28 días), hasta la progresión de la enfermedad, toxicidad inaceptable o por un máximo de 12 ciclos. Los pacientes recibieron 20 mg/m2 por dosis en el Ciclo 1, y 27 mg/m2 en los ciclos subsecuentes. Se administró dexametasona de 4 mg por vía oral o intravenosa antes de la administración de las dosis de KYPROLIS en el primer y segundo ciclo.
Un total de 266 pacientes fueron enrolados. Las características basales de los pacientes y de la enfermedad se resumen en la Tabla 13.
Tabla 13: Demografía y características basales en el estudio PX-171-003 A1 (Régimen de monoterapia de 20/27 mg/m2 )
|
Características |
Número de Pacientes (%) |
|
Características del Paciente |
|
|
Pacientes enrolados |
266 (100) |
|
Mediana de edad, años (rango) |
63 (37, 87) |
|
Grupo de edad, < 65/≥ 65 (años) |
146 (55)/120 (45) |
|
Sexo (masculino/femenino) |
155 (58)/111 (42) |
|
Raza (Blanca/Negra/Asiática/Otra) |
190 (71)/53 (20)/6 (2)/17 (6) |
|
Características de la enfermedad |
|
|
Número de regímenes previos (mediana) |
5a |
|
Trasplante Previo |
198 (74) |
|
Estado de refractariedad a terapia más recienteb |
|
|
Refractario: Progresión durante la terapia más reciente |
198 (74) |
|
Refractario: Progresión en el lapso de 60 días después de completar la terapia más reciente |
38 (14) |
|
Refractario: ≤ 25% de respuesta al tratamiento |
16 (6) |
|
En recaída: Progresión después de 60 días post-tratamiento |
14 (5) |
|
Años desde diagnóstico, mediana (rango) |
5.4 (0.5, 22.3) |
|
Involucramiento de célula plasmática (< 50%/≥ 50%/desconocido) |
143 (54)/106 (40)/17 (6) |
|
Etapa ISS en línea basal del estudio |
|
|
I |
76 (29) |
|
II |
102 (38) |
|
III |
81 (31) |
|
Desconocido |
7 (3) |
|
Citogenética o análisis FISH |
|
|
Normal/Favorable |
159 (60) |
|
Prognosis pobre |
75 (28) |
|
Desconocido |
32 (12) |
|
Depuración de creatinina < 30 mL/min |
6 (2) |
FISH = Hibridación fluorescente in situ; ISS = Sistema Internacional de Estadificación.
a Rango: 1, 20.
b Categorías para Estado de refractariedad se derivan mediante evaluación programática usando los datos de laboratorio disponibles.
La eficacia fue evaluada mediante ORR acorde a lo determinado por la evaluación del IRC usando los criterios del IMWG. Los resultados de eficacia se proporcionan en la Tabla 14). La mediana de la DOR fue de 7.8 meses (IC 95%: 5.6, 9.2).
Tabla 14: Categorías de respuesta en el estudio PX-171-003 A1 (Régimen de monoterapia de 20/27 mg/m2)
|
Características |
Pacientes en el estudioa n (%) |
|
Número de Pacientes (%) |
266 (100) |
|
Respuesta globalb |
61 (23) |
|
IC 95%c |
(18, 28) |
|
Categoría de respuesta |
|
|
CR |
1 (< 1) |
|
VGPR |
13 (5) |
|
PR |
47 (18) |
IC = intervalo de confianza; CR = respuesta completa; PR = respuesta parcial; VGPR = muy buena respuesta parcial.
a Los pacientes elegibles tuvieron 2 o más líneas de terapia previas y fueron refractarios al último régimen.
b Determinados por parte de un Comité de Revisión Independiente.
c Intervalo de confianza exacto.
Estudio PX-171-004 Parte 2:
El estudio PX-171-004 Parte 2 fue un estudio clínico de un solo brazo, multicéntrico de monoterapia con KYPROLIS® mediante infusión de hasta 10 minutos. Los pacientes elegibles fueron aquellos con mieloma múltiple en recaída o refractario que no tuvieron tratamientos previos con bortezomib, habían recibido de una a tres líneas de terapia previas y tuvieron ≤ 25% de respuesta o progresión durante la terapia o progresión en el lapso de 60 días después del término de la terapia. Los pacientes fueron excluidos del estudio si eran refractarios a la terapia estándar de primera línea o tenían bilirrubina total ≥ 2 × ULN; depuración de creatinina < 30 mL/min; falla cardiaca congestiva Clases III o IV conforme a la New York Heart Association; isquemia cardiaca sintomática; infarto al miocardio dentro de los últimos 6 meses; infecciones activas que requieren tratamiento; o derrame pleural.
Se administró KYPROLIS® intravenosamente hasta 10 minutos en dos días consecutivos cada semana por tres semanas, seguido por un periodo de descanso de 12 días (ciclo de tratamiento de 28 días), hasta la progresión de la enfermedad, toxicidad inaceptable, o por un máximo de 12 ciclos. Los pacientes recibieron 20 mg/m2 en cada dosis en el Ciclo 1, y 27 mg/m2 en ciclos subsecuentes. Se administró dexametasona 4 mg oral o intravenosa antes de las dosis de KYPROLIS® en el primero y segundo ciclo.
Se trataron un total de 70 pacientes con este régimen de 20/27 mg/m2. Las características basales del paciente y de la enfermedad se resumen en la Tabla 15.
Tabla 15: Demografía y características basales en el estudio PX-171-004 Parte 2 (Régimen de monoterapia de 20/27 mg/m2)
|
Características |
Número de Pacientes (%) |
|
Características de paciente |
|
|
Pacientes enrolados |
70 (100) |
|
Mediana de edad, años (rango) |
66 (45, 85) |
|
Grupo de edad, <65/≥65 (años) |
31 (44)/39 (56) |
|
Género (masculino/femenino) |
44 (63)/26 (37) |
|
Raza (Blanca/Negra/Asiática/Hispana/Otra) |
52 (74)/12 (17)/ |
|
Características de la enfermedad |
|
|
Número de regímenes previos (mediana) |
2a |
|
Trasplante previo |
47 (67) |
|
Estado de refractariedad a la terapia más recienteb |
|
|
Refractario: Progresión durante la terapia más reciente |
28 (40) |
|
Refractario: Progresión dentro de los 60 días después del término de la terapia más reciente |
7 (10) |
|
Refractario: ≤25% de respuesta al tratamiento |
10 (14) |
|
En recaída: Progresión después de 60 días post-tratamiento |
23 (33) |
|
Sin Signos de Progresión |
2 (3) |
|
Años desde el diagnóstico, mediana (rango) |
3.6 (0.7, 12.2) |
|
Participación de célula plasmática (< 50%/≥ 50%/desconocido) |
54 (77)/14 (20)/1 (1) |
|
Etapa ISS en Línea Basal del Estudio, n (%) |
|
|
I |
28 (40) |
|
II |
25 (36) |
|
III |
16 (23) |
|
Desconocido |
1 (1) |
|
Citogenética o Análisis FISH |
|
|
Normal/Favorable |
57 (81) |
|
Prognosis Pobre |
10 (14) |
|
Desconocida |
3 (4) |
|
Depuración de Creatinina <30 mL/min |
1 (1) |
FISH=Hibridación fluorescente in situ; ISS=Sistema Internacional de Estadificación.
a Rango: 1, 4.
b Categorías para estado de refractariedad se derivan mediante evaluación programática usando los datos de laboratorio disponibles.
Se evaluó la eficacia mediante ORR acorde a lo determinado por evaluación del IRC usando los criterios del IMWG. Los resultados de eficacia se proporcionan en la Tabla 16. No se alcanzó la mediana de la DOR.
Tabla 16: Categorías de respuesta en el estudio PX-171-004 parte 2 (Régimen de monoterapia de 20/27 mg/m2)
|
Características |
Pacientes del estudioa n (%) |
|
Número de pacientes (%) |
70 (100) |
|
Respuesta globalb |
35 (50) |
|
IC 95%c |
(38-62) |
|
Categoría de respuesta |
|
|
CR |
1 (1) |
|
VGPR |
18 (26) |
|
PR |
16 (23) |
IC = intervalo de confianza; CR = respuesta completa; PR = respuesta parcial; VGPR = muy buena respuesta parcial.
a Los pacientes elegibles tuvieron de 1 a 3 líneas de terapia previas y fueron refractarios al último régimen.
b Determinado por parte de un Comité de Revisión Independiente.
c Intervalo de confianza exacto.
CONTRAINDICACIONES: KYPROLIS® está contraindicado en pacientes con hipersensibilidad a carfilzomib o sus derivados, o cualquier excipiente en la formulación.
RESTRICCIONES DE USO DURANTE EL EMBARAZO Y LA LACTANCIA:
Embarazo:
Resumen del riesgo: KYPROLIS® puede causar daño fetal en base a los hallazgos en estudios con animales y su mecanismo de acción (ver Farmacocinética). No hay datos disponibles sobre el uso de KYPROLIS® en mujeres embarazadas para evaluar los riesgos asociados con los medicamentos. KYPROLIS® causó letalidad fetal embrionaria en conejos a dosis más bajas que la dosis clínica (ver Precauciones en relación con efectos de carcinogénesis, mutagénesis y teratogénesis, y sobre la fertilidad). Aconseje a las mujeres embarazadas sobre el riesgo potencial para el feto. Se desconoce el riesgo estimado de fondo de defectos mayores al nacimiento y de abortos espontáneos para la población indicada.
Todos los embarazos tienen un riesgo de fondo de defectos de nacimiento, pérdida y otros resultados adversos.
Lactancia:
Resumen de riesgo: No hay información en relación con la presencia de KYPROLIS® en la leche materna humana, los efectos sobre el niño en lactancia, o los efectos sobre la producción de leche. Debido a la posibilidad de reacciones adversas graves en el niño amamantado, se aconseja a las mujeres lactando que no amamanten durante el tratamiento con KYPROLIS® y durante las 2 semanas posteriores al tratamiento.
Mujeres y hombres con potencial reproductivo:
Basándose en su mecanismo de acción y los hallazgos en animales, KYPROLIS® puede causar daño fetal cuando se administra a una mujer embarazada.
Pruebas de embarazo: Realice pruebas de embarazo en mujeres con potencial reproductivo antes de iniciar el tratamiento con KYPROLIS®.
Anticoncepción:
Mujeres: Informar a las mujeres con potencial reproductivo que usen un método anticonceptivo eficaz durante el tratamiento con KYPROLIS® y durante al menos 6 meses después de la dosis final.
Hombres: Advertir a los hombres con parejas sexuales femeninas con potencial reproductivo, que usen un anticonceptivo eficaz durante el tratamiento con KYPROLIS® y durante al menos 3 meses después de la dosis final.
Infertilidad: Con base en el mecanismo de acción, KYPROLIS® puede tener un efecto sobre la fertilidad del hombre o la mujer (ver Farmaconética y Precauciones en relación con efectos de carcinogénesis, mutagénesis y teratogénesis, y sobre la fertilidad). No hay datos sobre el efecto de KYPROLIS® sobre la fertilidad en humanos.
REACCIONES SECUNDARIAS Y ADVERSAS: Las siguientes reacciones adversas se describen en otra parte, en la sección Precauciones generales de la información para prescribir:
• Toxicidades cardiacas.
• Falla renal aguda.
• Síndrome de lisis tumoral.
• Toxicidad pulmonar.
• Hipertensión pulmonar.
• Disnea.
• Hipertensión.
• Trombosis venosa.
• Reacciones a la infusión.
• Hemorragia.
• Trombocitopenia.
• Toxicidad hepática y falla hepática.
• Microangiopatía trombótica.
• Síndrome de encefalopatía posterior reversible.
• Leucoencefalopatía multifocal progresiva.
Experiencia de estudios clínicos: Dado que los estudios clínicos son conducidos bajo condiciones ampliamente variables, los índices de eventos adversos observados en los estudios clínicos de un fármaco no pueden ser directamente comparados con índices en los estudios clínicos de otro fármaco, y pueden no reflejar los índices observados en la práctica. La población de seguridad agrupada descrita en Precauciones generales refleja la exposición a KYPROLIS® en 1789 pacientes, administrado en combinación con otros medicamentos en ASPIRE, ENDEAVOR, A.R.R.O.W. y CANDOR. Las reacciones adversas más comunes que se produjeron en al menos un 20% de los pacientes que recibieron KYPROLIS® combinado con otros fármacos fueron anemia, diarrea, fatiga, hipertensión, pirexia, infección del tracto respiratorio superior, trombocitopenia, tos, disnea e insomnio.
KYPROLIS® en combinación con lenalidomida y dexametasona: La seguridad de KYPROLIS® 20/27 mg/m2 dos veces a la semana en combinación con lenalidomida y dexametasona (KRd) fue evaluada en ASPIRE (ver Estudios clínicos). La mediana del número de ciclos iniciados fue 22 ciclos para el brazo KRd y 14 ciclos para el brazo Rd.
Ocurrieron muertes debidas a reacciones adversas dentro de los 30 días desde la última dosis de cualquier terapia en el brazo KRd en 45/392 (12%) pacientes comparado con 42/389 (11%) pacientes que murieron debido a eventos adversos dentro de los 30 días desde la última dosis de cualquier terapia Rd. La causa más frecuente de muerte en pacientes (%) en los dos brazos (KRd versus Rd) incluyó infección 12 (3%) versus 11 (3%), cardiaca 10 (3%) versus 9 (2%), y otras reacciones adversas 23 (6%) versus 22 (6%). Se reportaron reacciones adversas graves en 65% de los pacientes en el brazo de KRd y 57% de los pacientes en el brazo de Rd. Las reacciones adversas graves más frecuentes reportadas en el brazo de KRd en comparación con el brazo de Rd fueron neumonía (17% versus 13%), infección del tracto respiratorio (4% versus 2%), pirexia (4% versus 3%), y embolismo pulmonar (3% versus 2%). La interrupción debida a cualquier reacción adversa ocurrió en el 33% en el brazo KRd versus el 30% en el brazo Rd. Las reacciones adversas que llevaron a la interrupción de KYPROLIS® ocurrieron en el 12% de los pacientes y las reacciones más comunes incluyeron neumonía (1%), infarto de miocardio (0.8%) e infección del tracto respiratorio superior (0.8%). La incidencia de eventos de insuficiencia cardiaca fue del 7% en el brazo KRd versus el 4% en el brazo Rd.
La Tabla 17 resume las reacciones adversas en los primeros 12 ciclos en ASPIRE.
Tabla 17: Reacciones adversas que ocurrieron en los ciclos 1 a 12 (2: 10%) en pacientes que recibieron KRd (Régimen de 20/27 mg/m2) en ASPIRE
|
Reacciones adversas |
KRd (N = 392) n (%) |
Rd (N = 389) n (%) |
||
|
Cualquier grado |
≥ Grado 3 |
Cualquier grado |
≥ Grado 3 |
|
|
Trastornos de la sangre y del sistema linfático |
||||
|
Anemia |
138 (35) |
53 (14) |
127 (33) |
47 (12) |
|
Neutropenia |
124 (32) |
104 (27) |
115 (30) |
89 (23) |
|
Trombocitopenia |
100 (26) |
58 (15) |
75 (19) |
39 (10) |
|
Trastornos gastrointestinales |
||||
|
Diarrea |
119 (30) |
8 (2) |
106 (27) |
12 (3) |
|
Estreñimiento |
68 (17) |
0 (0) |
55 (14) |
1 (0) |
|
Náusea |
63 (16) |
1 (0) |
43 (11) |
3 (1) |
|
Trastornos generales y Alteraciones en el lugar de administración |
||||
|
Fatiga |
113 (29) |
23 (6) |
107 (28) |
20 (5) |
|
Pirexia |
93 (24) |
5 (1) |
64 (17) |
1 (0) |
|
Edema periférico |
59 (15) |
3 (1) |
48 (12) |
2 (1) |
|
Astenia |
54 (14) |
11 (3) |
49 (13) |
7 (2) |
|
Infecciones |
||||
|
Infección del tracto respiratorio superior |
87 (22) |
7 (2) |
54 (14) |
4 (1) |
|
Bronquitis |
55 (14) |
5 (1) |
40 (10) |
2 (1) |
|
Infección viral del tracto respiratorio superior |
55 (14) |
0 (0) |
44 (11) |
0 (0) |
|
Neumoníaa |
54 (14) |
35 (9) |
43 (11) |
27 (7) |
|
Trastornos del Metabolismo y de la Nutrición |
||||
|
Hipocalemia |
78 (20) |
22 (6) |
35 (9) |
12 (3) |
|
Hipocalcemia |
55 (14) |
10 (3) |
39 (10) |
5 (1) |
|
Hiperglucemia |
43 (11) |
18 (5) |
33 (9) |
15 (4) |
|
Trastornos musculoesqueléticos y del tejido conjuntivo |
||||
|
Espasmos musculares |
92 (24) |
3 (1) |
75 (19) |
3 (1) |
|
Dolor de espalda |
41 (11) |
4 (1) |
54 (14) |
6 (2) |
|
Trastornos del sistema nervioso |
||||
|
Neuropatías periféricas b |
43 (11) |
7 (2) |
39 (10) |
4 (1) |
|
Trastornos psiquiátricos |
||||
|
Insomnio |
64 (16) |
6 (2) |
51 (13) |
8 (2) |
|
Trastornos respiratorios, torácicos y mediastínicos |
||||
|
Tosc |
93 (24) |
2 (1) |
54 (14) |
0 (0) |
|
Disnead |
71 (18) |
8 (2) |
61 (16) |
6 (2) |
|
Trastornos de la piel y del tejido subcutáneo |
||||
|
Erupción |
45 (12) |
5 (1) |
54 (14) |
5 (1) |
|
Trastornos vasculares |
||||
|
Eventos trombóticos y embólicose |
49 (13) |
16 (4) |
23 (6) |
9 (2) |
|
Hipertensiónf |
41 (11) |
12 (3) |
15 (4) |
4 (1) |
KRd = KYPROLIS®, lenalidomida y dexametasona; Rd = lenalidomida y dexametasona.
a Neumonía incluye neumonía y bronconeumonía.
b Neuropatías periféricas incluye neuropatía periférica, neuropatía periférica sensorial y neuropatía periférica motora.
c Tos incluye tos y tos productiva.
d Disnea incluye disnea y disnea por esfuerzo.
e Eventos venosos, trombóticos y embólicos, incluyen trombosis venosa profunda, embolismo pulmonar, tromboflebitis superficial, tromboflebitis, trombosis venosa de extremidad, síndrome post-trombótico, trombosis venosa.
f Hipertensión incluye hipertensión, crisis hipertensiva.
Hubo 274 (70%) pacientes en el brazo KRd que recibieron tratamiento más allá del Ciclo 12. No hubo nuevas reacciones adversas clínicamente relevantes que emergieran en los ciclos de tratamiento posteriores.
Reacciones adversas que ocurren a una frecuencia de < 10%:
Trastornos de la sangre y del sistema linfático: Neutropenia febril, linfopenia.
Trastornos cardiacos: Paro cardiaco, insuficiencia cardiaca, insuficiencia cardiaca congestiva, infarto al miocardio, isquemia de miocardio, efusión pericárdica.
Trastornos del oído y laberínticos: Sordera, tinnitus. Trastornos oculares: Catarata, visión borrosa.
Trastornos gastrointestinales: Dolor abdominal, dolor abdominal superior, dispepsia, hemorragia gastrointestinal, dolor de muelas.
Trastornos generales y alteraciones en el lugar de administración: Escalofríos, reacción en el sitio de infusión, falla orgánica múltiple, dolor.
Infecciones: Colitis por Clostridium difficiele, influenza, infección pulmonar, rinitis, septicemia, infección del tracto urinario, infección viral.
Trastornos del metabolismo y de la nutrición: Deshidratación, hipercalemia, hiperuricemia, hipoalbuminemia, hiponatremia, síndrome de lisis tumoral.
Trastornos musculoesqueléticos y del tejido conjuntivo: debilidad muscular, mialgia.
Trastornos del sistema nervioso: Hipoestesia, hemorragia intracraneal, parestesia.
Trastornos psiquiátricos: Ansiedad, delirio.
Trastornos renales y urinarios: Insuficiencia renal, insuficiencia renal aguda, daño renal.
Trastornos respiratorios, torácicos y mediastínicos: Disfonia, epistaxis, dolor orofaríngeo, embolismo pulmonar, edema pulmonar, hemorragia pulmonar.
Trastornos de la piel y del tejido subcutáneo: Eritema, hiperhidrosis, prurito.
Trastornos vasculares: Trombosis de vena profunda, hemorragia, hipotensión.
Las reacciones adversas Grado 3 o mayor que ocurrieron durante los Ciclos 1 a 12 con una diferencia sustancial (≥ 2%) entre los dos brazos fueron neutropenia, trombocitopenia, hipocalemia e hipofosfatemia.
La Tabla 18 describe las anormalidades de laboratorio Grado 3 a 4 reportadas en ASPIRE.
Tabla 18: Anormalidades de laboratorio grado 3 a 4 (≥ 10%) durante los Ciclos 1 a 12 en pacientes que reciberon KRd (Régimen de 20/27 mg/m2) en ASPIRE
|
Anormalidad de Laboratorio |
KRd (N = 392) n (%) |
Rd (N = 389) n (%) |
|
Disminución de linfocitos |
182 (46) |
119 (31) |
|
Disminución de la cuenta absoluta de neutrófilos |
152 (39) |
141 (36) |
|
Disminución de fósforo |
122 (31) |
106 (27) |
|
Disminución de plaquetas |
101 (26) |
59 (15) |
|
Disminución de la cuenta total de glóbulos blancos |
97 (25) |
71 (18) |
|
Disminución de hemoglobina |
58 (15) |
68 (18) |
|
Aumento de glucosa |
53 (14) |
30 (8) |
|
Disminución de potasio |
41 (11) |
23 (6) |
KRd = KYPROLIS®, Lenalidomida y Dexametasona; Rd = Lenalidomida y Dexametasona.
KYPROLIS® en combinación con dexametasona: La seguridad de KYPROLIS® en combinación con dexametasona fue evaluada en dos estudios abiertos, aleatorizados (ENDEAVOR y A.R.R.O.W.) (ver Estudios clínicos).
ENDEAVOR: La seguridad de KYPROLIS® 20/56 mg/m2 dos veces a la semana en combinación con dexametasona (Kd) se evaluó en ENDEAVOR (ver Estudios clínicos). Los pacientes recibieron tratamiento durante una mediana de 48 semanas en el grupo de Kd y 27 semanas en el brazo de bortezomib/dexametasona (Vd).
Ocurrieron muertes debidas a reacciones adversas dentro de los 30 días del último tratamiento de estudio en 32/463 (7%) pacientes en el brazo de Kd y 21/456 (5%) pacientes en el brazo de Vd. Las causas de muerte que ocurrieron en pacientes (%) en los dos brazos (Kd versus Vd) incluyeron cardiaca 4 (1%) versus 5 (1%), infecciones 8 (2%) versus 8 (2%), progresión de la enfermedad 7 (2%) versus 4 (1%), pulmonar 3 (1%) versus 2 (< 1%), renal 1 (< 1%) versus 0 (0%), y otros eventos adversos 9 (2%) versus 2 (< 1%).
Se reportaron reacciones adversas graves en 59% de los pacientes en el brazo de Kd y 40% de los pacientes en el brazo de Vd. En ambos brazos, la neumonía fue la reacción adversa grave más frecuentemente reportada (8% versus 9%). La descontinuación debida a cualquier reacción adversa ocurrió en 29% en el brazo de Kd versus 26% en el brazo de Vd. La reacción adversa más frecuente que llevó a la descontinuación fue insuficiencia cardiaca en el brazo de Kd (n = 8, 2%) y neuropatía periférica en el brazo de Vd (n = 22, 5%). La incidencia de eventos de falla cardiaca fue de 11% en el brazo Kd versus 3% en el brazo Vd.
Las reacciones adversas en los primeros 6 meses de la terapia que ocurrieron en una tasa de 10% o más en el brazo Kd se presentan en la Tabla 19.
Tabla 19: Reacciones adversas (≥ 10%) ocurriendo en los meses 1 a 6 en Pacientes que Recibieron Kd (Régimen de 20/56 mg/m2) en ENDEAVOR
|
Reacción adversa |
Kd (N = 463) n (%) |
Vd (N = 456) n (%) |
|||
|
Cualquier grado |
Grado ≥ 3 |
Cualquier grado |
Grado ≥ 3 |
||
|
Trastornos de la sangre y del sistema linfático |
|||||
|
Anemia |
161 (35) |
57 (12) |
112 (25) |
43 (9) |
|
|
Trombocitopeniaa |
125 (27) |
45 (10) |
112 (25) |
64 (14) |
|
|
Trastornos gastrointestinales |
|||||
|
Diarrea |
117 (25) |
14 (3) |
149 (33) |
27 (6) |
|
|
Náusea |
70 (15) |
4 (1) |
68 (15) |
3 (1) |
|
|
Constipación |
60 (13) |
1 (0) |
113 (25) |
6 (1) |
|
|
Vómito |
45 (10) |
5 (1) |
33 (7) |
3 (1) |
|
|
Trastornos generales y Alteraciones en el lugar de administración |
|||||
|
Fatiga |
116 (25) |
14 (3) |
126 (28) |
25 (6) |
|
|
Pirexia |
102 (22) |
9 (2) |
52 (11) |
3 (1) |
|
|
Astenia |
73 (16) |
9 (2) |
65 (14) |
13 (3) |
|
|
Edema periférico |
62 (13) |
3 (1) |
62 (14) |
3 (1) |
|
|
Infecciones |
|||||
|
Infección del tracto respiratorio superior |
67 (15) |
4 (1) |
55 (12) |
3 (1) |
|
|
Bronquitis |
54 (12) |
5 (1) |
25 (6) |
2 (0) |
|
|
Trastornos musculoesqueléticos y del tejido conjuntivo |
|||||
|
Espasmos musculares |
70 (15) |
1 (0) |
23 (5) |
3 (1) |
|
|
Dolor de espalda |
64 (14) |
8 (2) |
61 (13) |
10 (2) |
|
|
Trastornos del sistema nervioso |
|||||
|
Dolor de cabeza |
67 (15) |
4 (1) |
39 (9) |
2 (0) |
|
|
Neuropatías periféricasb,c |
56 (12) |
7 (2) |
170 (37) |
23 (5) |
|
|
Trastornos psiquiátricos |
|||||
|
Insomnio |
105 (23) |
5 (1) |
116 (25) |
10 (2) |
|
|
Trastornos respiratorios, torácicos y mediastínicos |
|||||
|
Disnead |
128 (28) |
23 (5) |
69 (15) |
8 (2) |
|
|
Tose |
97 (21) |
0 (0) |
61 (13) |
2 (0) |
|
|
Trastornos vasculares |
|||||
|
Hipertensiónf |
83 (18) |
30 (7) |
33 (7) |
12 (3) |
|
Kd = KYPROLIS® y dexametasona; Vd = bortezomib y dexametasona.
a La trombocitopenia incluye disminución de cuenta de plaquetas y trombocitopenia.
b Neuropatías periféricas incluyen neuropatía periférica, neuropatía periférica sensorial, neuropatía periférica motora.
c Ver Estudios clínicos.
d Disnea incluye disnea y disnea por esfuerzo.
e Tos incluye tos y tos productiva.
f Hipertensión incluye hipertensión, crisis hipertensiva, y emergencia hipertensiva.
La tasa de evento de neuropatía periférica ≥ Grado 2 en el brazo de Kd fue de 7% (IC 95%: 5, 9) versus 35% (IC 95%: 31, 39) en el brazo Vd.
Reacciones adversas que ocurren a una frecuencia de < 10%:
Trastornos de la sangre y del sistema linfático: Neutropenia febril, leucopenia, linfopenia, neutropenia, microangiopatía trombótica, púrpura trombocitopénica trombótica.
Trastornos cardiacos: Fibrilación atrial, paro cardiaco, falla cardiaca, insuficiencia cardiaca congestiva, infarto al miocardio, isquemia de miocardio, palpitaciones, taquicardia.
Trastornos del oído y laberínticos: Tinnitus.
Trastornos oculares: Catarata, visión borrosa.
Trastornos gastrointestinales: Dolor abdominal, dolor abdominal superior, dispepsia, hemorragia gastrointestinal, dolor de muelas.
Trastornos generales y alteraciones en el sitio de administración: Dolor de pecho, escalofríos, enfermedad parecida a la influenza, reacciones en el sitio de infusión (incluyendo inflamación, dolor y eritema), malestar, dolor.
Trastornos hepatobiliares: Colestasis, insuficiencia hepática, hiperbilirrubinemia.
Trastornos del sistema inmune: Hipersensibilidad al fármaco.
Infecciones: Bronconeumonía, gastroenteritis, influenza, infección pulmonar, nasofaringitis, neumonía, rinitis, septicemia, infección del tracto urinario, infección viral.
Trastornos del metabolismo y de la nutrición: Disminución de apetito, deshidratación, hipercalcemia, hipercalemia, hiperuricemia, hipoalbuminemia, hipocalcemia, hipomagnesemia, hiponatremia, hipofosfatemia, síndrome de lisis tumoral.
Trastornos musculoesqueléticos y del tejido conjuntivo: Debilidad muscular, dolor musculoesquelético de pecho, dolor musculoesquelético, mialgia.
Trastornos del sistema nervioso: Accidente cerebrovascular, mareo, hipoestesia, parestesia, síndrome de encefalopatía posterior reversible.
Trastornos psiquiátricos: Ansiedad.
Trastornos renales y urinarios: Insuficiencia renal, insuficiencia renal aguda, daño renal.
Trastornos respiratorios, torácicos, y mediastínicos: Síndrome de dificultad respiratoria aguda, disfonía, epistaxis, enfermedad pulmonar intersticial, dolor orofaríngeo, neumonitis, embolismo pulmonar, edema pulmonar, hipertensión pulmonar, sibilancias.
Trastornos de la piel y del tejido subcutáneo: Eritema, hiperhidrosis, prurito, erupciones.
Trastornos vasculares: Trombosis venosa profunda, enrojecimiento, hipotensión.
La Tabla 20 describe anormalidades de laboratorio Grado 3 a 4 reportadas en una tasa de ≥ 10% en el brazo Kd.
Tabla 20: Anormalidades de Laboratorio Grado 3 a 4 (≥ 10%) en los Meses 1 a 6 en Pacientes que recibieron Kd (Régimen de 20/56 mg/m2) en ENDEAVOR
|
Anormalidad de laboratorio |
Kd (N = 463) n (%) |
Vd (N = 456) n (%) |
|
Disminución de linfocitos |
249 (54) |
180 (40) |
|
Incremento del ácido úrico |
244 (53) |
198 (43) |
|
Disminución de hemoglobina |
79 (17) |
68 (15) |
|
Disminución de plaquetas |
85 (18) |
77 (17) |
|
Disminución de fósforo |
74 (16) |
61 (13) |
|
Disminución en la depuración de creatininaa |
65 (14) |
49 (11) |
|
Incremento de potasio |
55 (12) |
21 (5) |
Kd = KYPROLIS® y Dexametasona; Vd=Bortezomib y Dexametasona.
a Calculado usando la fórmula de Cockcroft-Gault.
A.R.R.O.W.:
La seguridad de KYPROLIS® en combinación con dexametasona se evaluó en A.R.R.O.W. (ver Estudios clínicos). Los pacientes de A.R.R.O.W. recibieron tratamiento durante una mediana de 38 semanas en el brazo de Kd 20/70 mg/m2 una vez a la semana y de 29, 1 semanas en el brazo de Kd 20/27 mg/m2 dos veces por semana. El perfil de seguridad para el régimen de Kd 20/70 mg/m2 una vez a la semana fue similar al de Kd 20/27 mg/m2 dos veces a la semana.
Las muertes debidas a reacciones adversas dentro de los 30 días posteriores al último tratamiento del estudio ocurrieron en 22/238 (9%) pacientes en el brazo de Kd 20/70 mg/m2 y en 18/235 (8%) pacientes en el brazo Kd 20/27 mg/m2. Las reacciones adversas fatales más frecuentes que ocurren en pacientes (%) en los dos brazos (una vez por semana Kd 20/70 mg/m2 versus dos veces por semana Kd 20/27 mg/m2) fueron sepsis 2 (< 1%) versus 2 (< 1%), shock séptico 2 (< 1%) versus 1 (< 1%) e infección 2 (< 1%) versus 0 (0%).
Se notificaron reacciones adversas graves en el 43% de los pacientes en el brazo de Kd 20/70 mg/m2 y en el 41% de los pacientes en el brazo de Kd 20/27 mg/m2. En ambos grupos, la neumonía fue la reacción adversa grave más frecuentemente reportada (8% versus 7%).
La interrupción debida a cualquier reacción adversa ocurrió en el 13% en el brazo de Kd 20/70 mg/m2 frente al 12% en el brazo de Kd 20/27 mg/m2. La reacción adversa más frecuente que llevó a la interrupción fue la lesión renal aguda (2% versus 2%). La incidencia de eventos de falla cardiaca fue del 3.8% en el brazo de Kd 20/70 mg/m2 una vez a la semana versus el 5.1% en el brazo de Kd 20/27 mg/m2 dos veces a la semana.
Las reacciones adversas que ocurrieron a una tasa del 10% o más en cualquiera de los brazos Kd se presentan en la Tabla 21.
Tabla 21: Reacciones adversas en pacientes que recibieron Kd (≥ 10% en cualquier brazo Kd) en A.R.R.O.W.)
|
Reacción adversa |
Una vez por semana Kd 20/70 mg/m2 (N = 238) n (%) |
Dos veces por semana Kd 20/27 mg/m2 (N = 235) n (%) |
||
|
Cualquier grado |
Grado ≥ 3 |
Cualquier grado |
Grado ≥ 3 |
|
|
Trastornos de la sangre y del sistema linfático |
||||
|
Anemiaa |
64 (26.9) |
42 (17.6) |
76 (32.3) |
42 (17.9) |
|
Trombocitopeniab |
53 (22.3) |
26 (10.9) |
41 (17.4) |
27 (11.5) |
|
Neutropeniac |
30 (12.6) |
21 (8.8) |
27 (11.5) |
17 (7.2) |
|
Trastornos gastrointestinales |
||||
|
Diarrea |
44 (18.5) |
2 (0.8) |
47 (20.0) |
3 (1.3) |
|
Náusea |
34 (14.3) |
1 (0.4) |
26 (11.1) |
2 (0.9) |
|
Trastornos generales y Alteraciones en el lugar de administración |
||||
|
Pirexia |
55 (23.1) |
2 (0.8) |
38 (16.2) |
4 (1.7) |
|
Fatiga |
48 (20.2) |
11 (4.6) |
47 (20.0) |
5 (2.1) |
|
Astenia |
24 (10.1) |
3 (1.3) |
25 (10.6) |
2 (0.9) |
|
Edema periférico |
18 (7.6) |
0 (0.0) |
25 (10.6) |
2 (0.9) |
|
Infecciones |
||||
|
Infección del tracto respiratoriod |
70 (29.4) |
7 (2.9) |
79 (33.6) |
7 (3.0) |
|
Neumonía |
28 (11.8) |
24 (10.1) |
20 (8.5) |
16 (6.8) |
|
Bronquitis |
27 (11.3) |
2 (0.8) |
25 (10.6) |
5 (2.1) |
|
Trastornos musculoesqueléticos y del tejido conjuntivo |
||||
|
Dolor de espalda |
28 (11.8) |
2 (0.8) |
28 (11.9) |
4 (1.7) |
|
Trastornos del sistema nervioso |
||||
|
Dolor de cabeza |
25 (10.5) |
1 (0.4) |
23 (9.8) |
1 (0.4) |
|
Transtornos psiquiátricos |
||||
|
Insomnio |
35 (14.7) |
2 (0.8) |
47 (20.0) |
0 (0.0) |
|
Trastornos respiratorios, torácicos y mediastínicos |
||||
|
Tose |
37 (15.5) |
2 (0.8) |
31 (13.2) |
0 (0.0) |
|
Disneaf |
28 (11.8) |
1 (0.4) |
26 (11.1) |
2 (0.9) |
|
Trastornos vasculares |
||||
|
Hipertensióng |
51 (21.4) |
13 (5.5) |
48 (20.4) |
12 (5.1) |
Kd = KYPROLIS® y dexametasona.
a La anemia incluye anemia, disminución del hematocrito y disminución de la hemoglobina.
b La trombocitopenia incluye disminución del recuento de plaquetas y trombocitopenia.
c La neutropenia incluye el recuento de neutrófilos disminuido y neutropenia.
d La infección del tracto respiratorio incluye infección del tracto respiratorio, infección del tracto respiratorio inferior, infección del tracto respiratorio superior e infección del tracto respiratorio superior virald.
e La tos incluye tos y tos productiva.
f La disnea incluye disnea y disnea de esfuerzo.
g La hipertensión incluye hipertensión y crisis hipertensiva.
Reacciones adversas que ocurren a una frecuencia de <10%.
Trastornos sanguíneos y del sistema linfático: Neutropenia febril, leucopenia, linfopenia, neutropenia, microangiopatía trombótica.
Trastornos cardiacos: Fibrilación auricular, paro cardiaco, insuficiencia cardiaca, insuficiencia cardiaca congestiva, infarto de miocardio, isquemia miocárdica, palpitaciones, derrame pericárdico, taquicardia.
Trastornos del oído y laberínticos: Tinnitus.
Trastornos oculares: Cataratas, visión borrosa.
Trastornos gastrointestinales: Dolor abdominal, dolor abdominal superior, estreñimiento, dispepsia, dolor de muelas, vómitos.
Trastornos generales y afecciones en el lugar de administración: Dolor en el pecho, escalofríos, enfermedades similares a la influenza, reacciones en el lugar de la infusión (incluida inflamación, dolor y eritema), malestar general, dolor.
Trastornos hepatobiliares: Colestasis, insuficiencia hepática, hiperbilirrubinemia.
Infecciones: Colitis por Clostridium difficile, gastroenteritis, influenza, infección pulmonar, nasofaringitis, rinitis, sepsis, shock séptico, infección del tracto urinario, infección viral.
Trastornos del metabolismo y de la nutrición: Disminución del apetito, deshidratación, hipercalcemia, hiperglucemia, hipercalemia, hiperuricemia, hipoalbuminemia, hipocalcemia, hipomagnesemia, hiponatremia, hipofosfatemia, síndrome de lisis tumoral.
Trastornos musculoesqueléticos y del tejido conjuntivo: Espasmos musculares, debilidad muscular, dolor torácico musculoesquelético, dolor musculoesquelético, mialgia.
Trastornos del sistema nervioso: Accidente cerebrovascular, mareos, parestesia, neuropatía periférica.
Trastornos psiquiátricos: Ansiedad, delirio.
Trastornos renales y urinarios: Lesión renal aguda, insuficiencia renal, daño renal.
Trastornos respiratorios, torácicos y mediastínicos: Síndrome de dificultad respiratoria aguda, disfonía, epistaxis, enfermedad pulmonar intersticial, dolor orofaríngeo, neumonitis, hemorragia pulmonar, embolia pulmonar, hipertensión pulmonar, edema pulmonar, sibilancias.
Trastornos de la piel y del tejido subcutáneo: Eritema, hiperhidrosis, prurito, erupción cutánea.
Trastornos vasculares: Trombosis venosa profunda, sofocos, hipotensión.
KYPROLIS® en Combinación con daratumumab intravenoso y dexametasona:
La La seguridad de KYPROLIS® en combinación con daratumumab intravenoso y dexametasona se evaluó en dos estudios (CANDOR y EQUULEUS).
CANDOR: La seguridad de KYPROLIS® 20/56 mg/m2 dos veces a la semana en combinación con daratumumab intravenoso y dexametasona (DKd) se evaluó en CANDOR (Ver Estudios clínicos). Pacientes recibieron KYPROLIS® durante una mediana de 58 semanas en el brazo de DKd y de 40 semanas en el brazo de Kd.
Se reportaron reacciones adversas graves en el 56% de los pacientes en el brazo de DKd y en el 46% de los pacientes en el brazo de Kd. Las reacciones adversas graves más frecuentes informadas en el brazo de DKd en comparación con el brazo de Kd fueron neumonía (14% versus 9%), pirexia (4,2% versus 2,0%), influenza (3.9% versus 1.3%), septicemia (3.9% versus 1.3%), anemia (2.3% versus 0.7%), bronquitis (1.9% versus 0%) y diarrea (1.6% versus 0%). Se produjeron reacciones adversas fatales dentro de los 30 días posteriores a la última dosis de cualquier tratamiento del estudio en un 10% de los 308 pacientes en el brazo de DKd en comparación con un 5% de los 153 pacientes en el brazo de Kd. La reacción adversa fatal más frecuente (DKd versus Kd) fue infección, 4.5% versus 2.6%.
La interrupción permanente del fármaco debido a una reacción adversa en pacientes que recibieron KYPROLIS® se produjo en un 21% de los pacientes del brazo de DKd versus un 22% en el brazo de Kd. Las reacciones adversas más frecuentes que llevaron a la interrupción del tratamiento con KYPROLIS® fueron insuficiencia cardiaca (1.9%) y fatiga (1.9%) en el brazo de DKd e insuficiencia cardiaca (2.0%), hipertensión (2.0%) y lesión renal aguda (2.0%) en el brazo de Kd. Se interrumpió el tratamiento con KYPROLIS® debido a reacciones adversas en el 71% de los pacientes en el brazo de DKd versus un 63% en el brazo de Kd. Se redujo la dosis de KYPROLIS® debido a 25% de los pacientes del brazo de DKd versus un 20% en el brazo de Kd.
Se produjeron reacciones relacionadas con la infusión después de la primera dosis de KYPROLIS® en un 13% en el brazo de DKd versus un 1% en el brazo de Kd.
La Tabla 22 resume las reacciones adversas en CANDOR.
Tabla 22: Reacciones adversas (≥ 15%) en pacientes que recibieron DKd o Kd (Régimen de 20/56 mg/m2) en CANDOR
|
Reacción adversa |
DKd dos veces a la semana (N = 308) |
2Kd dos veces a la semana (N = 153) |
||
|
Todos los grados (%) |
Grados 3 a 4 |
Todos los grados (%) |
Grados 3 a 4 |
|
|
Trastornos generales y Alteraciones en el lugar de administración |
||||
|
Reacción relacionada con la infusiónª |
41 |
12 |
28 |
5 |
|
Fatigab |
32 |
11 |
28 |
8 |
|
Pirexia |
20 |
1.9 |
15 |
0.7 |
|
Infecciones |
||||
|
Infección del tracto respiratorioc |
40g |
7 |
29 |
3.3 |
|
Neumonía |
18g |
13 |
12 |
9 |
|
Bronquitis |
17 |
2.6 |
12 |
1.3 |
|
Trastornos de la sangre y del sistema linfático |
||||
|
Trombocitopeniad |
37 |
25 |
30 |
16 |
|
Anemiae |
33 |
17 |
31 |
14 |
|
Trastornos gastrointestinales |
||||
|
Diarrea |
32 |
3.9 |
14 |
0.7 |
|
Náuseas |
18 |
0 |
13 |
0.7 |
|
Trastornos vasculares |
||||
|
Hipertensión |
31 |
18 |
28 |
13 |
|
Trastornos respiratorios, torácicos y mediastínicos |
||||
|
Tosf |
21 |
0 |
21 |
0 |
|
Disnea |
20 |
3.9 |
22 |
2.6 |
|
Trastornos psiquiátricos |
||||
|
Insomnio |
18 |
3.9 |
11 |
2.0 |
|
Trastornos musculoesqueléticos y del tejido conjuntivo |
||||
|
Dolor de espalda |
16 |
1.9 |
10 |
1.3 |
DKd = KYPROLIS®, daratumumab y dexametasona; Kd = KYPROLIS® y dexametasona.
ª La incidencia de reacciones relacionadas con la infusión se basa en un conjunto de síntomas (como hipertensión, pirexia, erupción, mialgia, hipotensión, aumento de la presión arterial, urticaria, lesión renal aguda, broncoespasmo, edema de cara, hipersensibilidad, síncope, sibilancia, prurito en el ojo, edema palpebral, insuficiencia renal, hinchazón facial) relacionados con la infusión que se produjeron dentro de 1 día después de la administración de DKd o Kd.
b La fatiga incluye fatiga y astenia.
e La infección del tracto respiratorio incluye infección del tracto respiratorio, infección del tracto respiratorio inferior, infección del tracto respiratorio superior e infección del tracto respiratorio superior viral.
d La trombocitopenia incluye disminución del recuento de plaquetas y trombocitopenia.
e La anemia incluye anemia, disminución del hematocrito y disminución de la hemoglobina.
f La tos incluye tos y tos productiva.
g Incluye reacciones adversas mortales.
Reacciones adversas que ocurren a una frecuencia de < 15%:
Trastornos de la sangre y del sistema linfático: Neutropenia febril, púrpura trombocitopénica trombótica.
Trastornos cardiacos: Fibrilación auricular, paro cardiaco, falla cardiaca, cardiomiopatía, infarto al miocardio, isquemia de miocardio, taquicardia.
Trastornos oculares: Cataratas.
Trastornos gastrointestinales: Dolor abdominal, hemorragia gastrointestinal.
Trastornos generales y alteraciones en el lugar de administración: Dolor torácico, malestar.
Infecciones: Gastroenteritis, influenza, infección pulmonar, nasofaringitis, septicemia, shock séptico, infección del tracto urinario, infección viral.
Exploraciones complementarias: Alanina aminotransferasa elevada, creatinina en sangre elevada, proteína C reactiva elevada, fracción de eyección disminuida.
Trastornos del metabolismo y de la nutrición: Deshidratación, hiperglucemia, hipercalemia, hipocalemia, hiponatremia, síndrome de lisis tumoral.
Trastornos musculoesqueléticos y del tejido conjuntivo: Dolor en una extremidad.
Trastornos del sistema nervioso: Accidente cerebrovascular, hemorragia intracraneal, síndrome de encefalopatía posterior reversible, neuropatía periférica.
Trastornos psiquiátricos: Ansiedad.
Trastornos renales y urinarios: Lesión renal aguda, insuficiencia renal, daño renal.
Trastornos respiratorios, torácicos y mediastínicos: Insuficiencia respiratoria aguda, epistaxis, enfermedad pulmonar intersticial, neumonitis, embolismo pulmonar, hipertensión pulmonar, edema pulmonar.
Trastornos de la piel y del tejido subcutáneo: Erupción.
Trastornos vasculares: Trombosis venosa profunda, crisis hipertensiva.
EQUULEUS
La seguridad de KYPROLIS® 20/70 mg/m2 una vez a la semana en combinación con daratumumab y dexametasona (DKd) se evaluó en EQUULEUS (ver Estudios clínicos). Los pacientes recibieron KYPROLIS® por una mediana de duración de 66 semanas.
Se informaron reacciones adversas graves en un 48% de los pacientes. Las reacciones adversas graves más frecuentes fueron neumonía (4.7%), infección del tracto respiratorio superior (4.7%), carcinoma basocelular (4.7%), influenza (3.5%), deterioro general del estado físico (3.5%) e hipercalcemia (3.5%). Se produjeron reacciones adversas mortales dentro de los 30 días de la última dosis de cualquier tratamiento del estudio en un 3.5% de los pacientes, que murieron de deterioro general del estado físico, falla orgánica múltiple después de aspergilosis pulmonar y progresión de la enfermedad.
Se produjo discontinuación del tratamiento con KYPROLIS® en un 19% de los pacientes. La reacción adversa más frecuente que llevó a la interrupción del tratamiento fue la astenia (2%). Se produjo la interrupción del tratamiento de Kyprolis debido a reacciones adversas en un 77% de los pacientes. Se produjo la reducción de la dosis de KYPROLIS® debido reacciones adversas en un 31% de los pacientes en DKd.
Las reacciones relacionadas con la infusión que se produjeron después de la primera dosis de KYPROLIS® fueron de un 11%. Se informó de reacciones adversas de hipertensión pulmonar en un 4.7% de los pacientes en EQUULEUS.
La Tabla 23 resume las reacciones adversas en EQUULEUS.
Tabla 23: Reacciones adversas (≥ 15%) en pacientes que recibieron DKd o Kd (Régimen de 20/70 mg/m2) en EQUULEUS
|
Reacciones adversas |
DKd una vez a la semana (N = 85) |
|
|
Todos los grados (%) |
Grados 3 a 4 (%) |
|
|
Trastornos de la sangre y del sistema linfático |
||
|
Trombocitopeniaª |
68 |
32 |
|
Anemiab |
52 |
21 |
|
Neutropeniac |
31 |
21 |
|
Linfopeniad |
29 |
25 |
|
Trastornos generales y alteraciones en el lugar de administración |
||
|
Fatigae |
54 |
18 |
|
Reacción relacionada con la infusiónf |
53 |
12 |
|
Pirexia |
37 |
1.2 |
|
Infecciones |
||
|
Infección del tracto respiratoriog |
53 |
3.5 |
|
Bronquitis |
19 |
0 |
|
Nasofaringitis |
18 |
0 |
|
Influenza |
17 |
3.5 |
|
Trastornos gastrointestinales |
||
|
Náuseas |
42 |
1.2 |
|
Vómito |
40 |
1.2 |
|
Diarrea |
38 |
2.4 |
|
Constipación |
17 |
0 |
|
Trastornos respiratorios, torácicos y mediastínicos |
||
|
Disnea |
35 |
3.5 |
|
Tosh |
33 |
0 |
|
Trastornos vasculares |
||
|
Hipertensión |
33 |
20 |
|
Trastornos psiquiátricos |
||
|
Insomnio |
33 |
4.7 |
|
Trastornos del sistema nervioso |
||
|
Dolor de cabeza |
27 |
1.2 |
|
Trastornos musculoesqueléticos y del tejido conjuntivo |
||
|
Dolor de espalda |
25 |
0 |
|
Dolor en una extremidad |
15 |
0 |
DKd = KYPROLIS® , daratumumab y dexametasona; Kd = KYPROLIS® y dexametasona.
ª La trombocitopenia incluye disminución de recuento de plaquetas y trombocitopenia.
b La anemia incluye anemia, disminución del hematocrito y disminución de la hemoglobina.
c La neutropenia incluye el recuento de neutrófilos disminuido y neutropenia.
d La linfopenia incluye recuento de linfocitos disminuido y linfopenia.
e La fatiga incluye fatiga y astenia.
f La incidencia de reacciones relacionadas con la infusión se basa en un conjunto de síntomas (como hipertensión, pirexia, erupción, mialgia, hipotensión, aumento de la presión arterial, urticaria, lesión renal aguda, broncoespasmo, edema de cara, hipersensibilidad, síncope, sibilancia, prurito en el ojo, edema palpebral, insuficiencia renal, edema facial) relacionados con la infusión que se produjeron dentro de 1 día después de la administración de DKd.
g La infección del tracto respiratorio incluye infección del tracto respiratorio, infección del tracto respiratorio inferior, infección del tracto respiratorio superior e infección del tracto respiratorio superior viral.
h La tos incluye tos y tos productiva.
Reacciones adversas que ocurren a una frecuencia de < 15%:
Trastornos de la sangre y del sistema linfático: Neutropenia febril, microangiopatía trombótica.
Trastornos cardiacos: Insuficiencia cardiaca, isquemia de miocardio.
Trastornos gastrointestinales: Dolor abdominal.
Trastornos generales y alteraciones en el lugar de administración: Síndrome de disfunción multiorgánica.
Infecciones: Neumonía, septicemia, shock séptico.
Trastornos del metabolismo y de la nutrición: Deshidratación, hipercalcemia.
Trastornos renales y urinarios: Lesión renal aguda, insuficiencia renal, daño renal.
Trastornos respiratorios, torácicos y mediastínicos: Embolismo pulmonar, hipertensión pulmonar.
Trastornos vasculares: Hipotensión.
KYPROLIS® en pacientes que recibieron monoterapia:
La seguridad de KYPROLIS® 20/27 mg/m2 como una infusión de 10 minutos fue evaluada en estudios clínicos en 598 pacientes con mieloma en recaída y/o refractario (ver Estudios clínicos). Se requirió pre-medicar dexametasona 4 mg antes de cada dosis en el Ciclo 1 y fue opcional para subsecuentes ciclos. La mediana de edad fue 64 años (rango 32 a 87), y aproximadamente 57% fueron hombres. Los pacientes recibieron una mediana de 5 (rango 1 a 20) regímenes previos. La mediana del número de ciclos iniciados fue 4 (rango 1 a 35).
Ocurrieron muertes debidas a reacciones adversas dentro de los 30 días desde la última dosis de KYPROLIS® en 30/598 (5%) pacientes que recibieron monoterapia con KYPROLIS®. Estas reacciones adversas estuvieron relacionadas a trastornos cardiacos en 10 (2%) pacientes, infecciones en 8 (1%) pacientes, trastornos renales en 4 (< 1%) pacientes, y otras reacciones adversas en 8 (1 %) pacientes.
Se reportaron reacciones adversas graves, sin importar la causalidad, en el 50% de los pacientes en los estudios agrupados de monoterapia con KYPROLIS® (N = 598). Las reacciones adversas graves más frecuentes fueron: neumonía (8%), insuficiencia renal aguda (5%), progresión de la enfermedad (4%), pirexia (3%), hipercalcemia (3%), insuficiencia cardíaca congestiva (3%), mieloma múltiple (3%), anemia (2%) y disnea (2%).
En FOCUS, un estudio aleatorizado que comparó Kyprolis como agente único versus corticoides con ciclofosfamida oral opcional para pacientes con mieloma múltiple en recaída o refractario, la mortalidad fue mayor en los pacientes tratados con KYPROLIS® en comparación al brazo de control en el subgrupo de 48 pacientes de ≥ 75 años de edad. La causa más común de discontinuación debida a una reacción adversa fue insuficiencia renal aguda (2%).
La seguridad de la monoterapia con KYPROLIS® dosificado a 20/56 mg/m2 mediante una infusión de 30 minutos fue evaluada en un estudio multicéntrico y abierto en pacientes con mieloma múltiple en recaída y/o refractario (ver Estudios clínicos). Los pacientes recibieron una mediana de 4 (rango 1 a 10) regímenes previos.
Las reacciones adversas que ocurren con la monoterapia con KYPROLIS® se presentan en la Tabla 24.
Tabla 24: Reacciones adversas (≥ 20%) con monoterapia de KYPROLIS®
|
Reacción adversa |
20/56 mg/m2 mediante infusión de 30 minutos (N = 24) |
20/27 mg/m2 mediante infusión de 2-10 minutos (N = 598) |
||
|
Todos los grados n (%) |
Grados 3 a 5 n (%) |
Todos los grados n (%) |
Grados 3 a 5 n (%) |
|
|
Fatiga |
14 (58) |
2 (8) |
238 (40) |
25 (4) |
|
Disneaa |
14 (58) |
2 (8) |
202 (34) |
21 (4) |
|
Pirexia |
14 (58) |
0 |
177 (30) |
11 (2) |
|
Trombocitopenia |
13 (54) |
13 (54) |
220 (37) |
152 (25) |
|
Náusea |
13 (54) |
0 |
211 (35) |
7 (1) |
|
Anemia |
10 (42) |
7 (29) |
291 (49) |
141 (24) |
|
Hipertensiónb |
10 (42) |
3 (13) |
90 (15) |
22 (4) |
|
Escalofrío |
9 (38) |
0 |
73 (12) |
1 (<1) |
|
Cefalea |
8 (33) |
0 |
141 (24) |
7 (1) |
|
Tosc |
8 (33) |
0 |
134 (22) |
2 (< 1) |
|
Vómito |
8 (33) |
0 |
104 (17) |
4 (1) |
|
Linfopenia |
8 (33) |
8 (33) |
85 (14) |
73 (12) |
|
Insomnio |
7 (29) |
0 |
75 (13) |
0 |
|
Mareo |
7 (29) |
0 |
64 (11) |
5 (1) |
|
Diarrea |
6 (25) |
1 (4) |
160 (27) |
8 (1) |
|
Incremento de creatinina en sangre |
6 (25) |
1 (4) |
103 (17) |
15 (3) |
|
Edema periférico |
5 (21) |
0 |
118 (20) |
1 (< 1) |
|
Dolor de espalda |
5 (21) |
1 (4) |
115 (19) |
19 (3) |
|
Infección del tracto respiratorio superior |
5 (21) |
1 (4) |
112 (19) |
15 (3) |
|
Disminución de apetito |
5 (21) |
0 |
89 (15) |
2 (< 1) |
|
Espasmos musculares |
5 (21) |
0 |
62 (10) |
2 (< 1) |
|
Dolor de pecho |
5 (21) |
0 |
20 (3) |
1 (< 1) |
a Disnea incluye disnea y disnea por esfuerzo.
b Hipertensión incluye hipertensión, crisis hipertensiva y emergencia hipertensiva.
c Tos incluye tos y tos productiva.
Reacciones adversas que ocurren a una frecuencia de < 20%:
Trastornos de la sangre y del sistema linfático: Neutropenia febril, leucopenia, neutropenia.
Trastornos cardiacos: Paro cardiaco, falla cardiaca, insuficiencia cardiaca congestiva, infarto al miocardio, isquemia de miocardio.
Trastornos del oído y laberínticos: Tinnitus.
Trastornos oculares: Catarata, visión borrosa.
Trastornos gastrointestinales: Dolor abdominal, dolor abdominal superior, estreñimiento, dispepsia, hemorragia gastrointestinal, dolor de muelas.
Trastornos generales y alteraciones en el lugar de administración: Astenia, reacción en el sitio de infusión, falla orgánica múltiple, dolor.
Trastornos hepatobiliares: Falla hepática.
Infecciones: Bronquitis, bronconeumonía, influenza, infección pulmonar, neumonía, nasofaringitis, infección del tracto respiratorio, rinitis, septicemia, infección del tracto urinario.
Trastornos del metabolismo y de la nutrición: Hipercalcemia, hiperglucemia, hipercalemia, hiperuricemia, hipoalbuminemia, hipocalcemia, hipocalemia, hipomagnesemia, hiponatremia, hipofosfatemia, síndrome de lisis tumoral.
Trastornos musculoesqueléticos y de tejido conjuntivo: Artralgia, dolor musculoesquelético, dolor musculoesquelético de pecho, mialgia, dolor en extremidades.
Trastornos del sistema nervioso: Hipoestesia, hemorragia intracraneal, parestesia, neuropatía periférica motora, neuropatía periférica, neuropatía periférica sensorial.
Trastornos psiquiátricos: Ansiedad.
Trastornos renales y urinarios: Insuficiencia renal aguda, insuficiencia renal, daño renal.
Trastornos respiratorios, torácicos y mediastínicos: Disfonía, epistaxis, dolor orofaríngeo, edema pulmonar, hemorragia pulmonar.
Trastornos de la piel y del tejido subcutáneo: Eritema, hiperhidrosis, prurito, erupción.
Trastornos vasculares: Eventos embólicos y trombóticos, venosos (incluyendo trombosis venosa profunda y embolismo pulmonar), hemorragia, hipotensión.
Reacciones adversas Grado 3 o mayor ocurriendo con una incidencia de > 1% incluyen neutropenia febril, paro cardiaco, insuficiencia cardiaca congestiva, dolor, sepsis, infección del tracto urinario, hiperglucemia, hipercalemia, hiperuricemia, hipoalbuminemia, hipocalcemia, hiponatremia, hipofosfatemia, insuficiencia renal, insuficiencia renal aguda, daño renal, edema pulmonar, e hipotensión.
La Tabla 25 describe las anormalidades de laboratorio Grado 3 a 4 reportadas en una tasa de > 10% para pacientes que recibieron monoterapia de KYPROLIS®.
Tabla 25: Anormalidades de laboratorio Grado 3 a 4 (> 10%) con monoterapia de KYPROLIS®
|
Anormalidad de laboratorio |
KYPROLIS® 20/56 mg/m2 (N = 24) |
KYPROLIS® 20/27 mg/m2 (N = 598) |
|
Disminución de linfocitos |
15 (63) |
151 (25) |
|
Disminución de plaquetas |
11 (46) |
184 (31) |
|
Disminución de hemoglobina |
7 (29) |
132 (22) |
|
Disminución de la cuenta total de glóbulos blancos |
3 (13) |
71 (12) |
|
Disminución de sodio |
2 (8) |
69 (12) |
|
Disminución de la cuenta absoluta de neutrófilos |
2 (8) |
67 (11) |
Experiencia Post-comercialización: Las siguientes reacciones adversas se identificaron durante el uso posterior a la aprobación de KYPROLIS®. Dado que estas reacciones son reportadas de forma voluntaria en una población de tamaño incierto, no siempre es posible estimar de forma confiable su frecuencia o establecer una relación causal con la exposición al fármaco: síndrome hemolítico urémico (HUS, por sus siglas en inglés), reactivación de virus de la hepatitis B, perforación gastrointestinal, pericarditis e infección por citomegalovirus, incluyendo coriorretinitis, neumonitis, enterocolitis, viremia, obstrucción intestinal y pancreatitis aguda.
Uso en poblaciones específicas:
Uso pediátrico: La seguridad y efectividad de KYPROLIS® en pacientes pediátricos no ha sido establecida.
Uso geriátrico: De los 2387 pacientes en estudios clínicos de KYPROLIS®, el 51% tenía 65 años o más, mientras que el 14% tenía 75 años o más. La incidencia de eventos adversos graves fue del 49% en pacientes de < 65 años de edad, del 58% en pacientes de 65 a 7 4 años de edad, y del 63% en pacientes de ≥ 75 años de edad. De los 308 pacientes en CANDOR que recibieron DKd, un 47% tenía 65 años o una edad superior, mientras que un 9% tenía 75 años o una edad superior. Se produjeron reacciones adversas fatales en el brazo de DKd de CANDOR en un 6% de los pacientes de< 65 años, un 14% de los pacientes entre 65 y 74 años y un 14% de los pacientes de ≥ 75 años (ver Reacciones secundarias y adversas). No se observaron diferencias generales en la efectividad entre los pacientes mayores y los más jóvenes.
Daño hepático: Reducir la dosis de KYPROLIS® en un 25% en pacientes con daño hepático leve (bilirrubina total 1 a 1.5 × ULN o cualquier AST o bilirrubina total ≤ ULN y AST > ULN) o moderada (bilirrubina total > 1.5 a 3 × ULN y cualquier AST). No se ha establecido una dosis recomendada de KYPROLIS® para pacientes con insuficiencia hepática severa (bilirrubina total > 3 x ULN y cualquier AST) (ver Dosis y vía de administración y Farmacocinética).
La incidencia de eventos adversos graves fue mayor en pacientes con insuficiencia hepática leve, moderada y grave combinados (22/35 o 63%) que en pacientes con función hepática normal (3/11 o 27%) (ver Precauciones generales y Farmacocinética).
PRECAUCIONES EN RELACIÓN CON EFECTOS DE CARCINOGÉNESIS, MUTAGÉNESIS, TERATOGÉNESIS Y SOBRE LA FERTILIDAD:
Carcinogénesis, mutagénesis, y daño a la fertilidad:
No se realizaron estudios de carcinogénesis con carfilzomib.
Carfilzomib fue clastogénico en la prueba de aberración cromosómica in vitro en linfocitos de sangre periférica. Carfilzomib no fue mutagénico en la prueba in vitro de mutación inversa en bacterias (prueba de Ames) y no fue clastogénico en la prueba in vivo de micronúcleos de médula ósea de ratón.
No se han conducido estudios de fertilidad con carfilzomib. No se observaron efectos sobre los tejidos del sistema reproductor durante los estudios de toxicidad de dosis repetidas de 28 días en ratas y monos o en estudios de toxicidad crónica de 6 meses en ratas y 9 meses en monos.
Toxicología y/o farmacología animal:
Toxicidad cardiovascular: Los monos a los cuales se les administró una dosis única por bolo intravenoso de carfilzomib a 3 mg/kg (aproximadamente 1.3 veces la dosis recomendada en humanos de 27 mg/m2 con base al área de la superficie corporal) presentaron hipotensión, frecuencia cardiaca elevada y niveles séricos de troponina T elevados.
Administración crónica: La administración repetida de carfilzomib por bolo intravenoso de ≥2 mg/kg/dosis en ratas y 2 mg/kg/dosis en monos utilizando esquemas de dosificación similares a aquellos utilizados clínicamente resultó en muerte a causa de toxicidad en el sistema cardiovascular (insuficiencia cardiaca, fibrosis cardiaca, acumulación de líquido pericárdico, hemorragia/degeneración cardiaca), gastrointestinal (necrosis/hemorragia), renal (glomerulopatía, necrosis tubular, disfunción) y pulmonar (hemorragia/inflamación). La dosis de 2 mg/kg/dosis en ratas es aproximadamente la mitad de la dosis recomendada en humanos de 27 mg/m2 con base en el área de la superficie corporal. La dosis de 2 mg/kg/dosis en monos es aproximadamente equivalente a la dosis recomendada en humanos con base al área de superficie corporal.
Carfilzomib administrado por vía intravenosa a ratas preñadas y conejas durante el periodo de organogénesis no fue teratogénico en dosis de hasta 2 mg/kg/día en ratas y 0.8 mg/kg/día en conejos. En conejos, hubo un aumento en la pérdida previa al implante a ≥ 0.4 mg/kg/día y un aumento en las reabsorciones tempranas y la pérdida posterior al implante y una disminución en el peso fetal a la dosis materna tóxica de 0.8 mg/kg/día. Las dosis de 0.4 mg/kg/día y 0.8 mg/kg/día en conejos son aproximadamente 20% y 40%, respectivamente, de la dosis recomendada en humanos de 27 mg/m2 según el área de superficie corporal.
INTERACCIONES MEDICAMENTOSAS Y DE OTRO GÉNERO: Carfilzomib es metabolizado principalmente a través de la actividad de las peptidasas y epóxido hidrolasas y, como resultado, es poco probable que el perfil farmacocinético de carfilzomib se vea afectado por la administración concomitante de inhibidores e inductores del citocromo P450. No se espera que carfilzomib pueda influenciar la exposición a otros fármacos.
ALTERACIONES EN LOS RESULTADOS DE PRUEBAS DE LABORATORIO: Ninguna.
PRECAUCIONES GENERALES:
Toxicidades cardiacas: Se han producido nuevos casos o empeoramiento de insuficiencia cardiaca pre-existente (por ejemplo, insuficiencia cardiaca congestiva, edema pulmonar, disminución en la fracción de eyección), cardiomiopatía, isquemia e infarto al miocardio incluyendo fatalidades, posteriores a la administración de KYPROLIS®. Algunos eventos ocurrieron en pacientes con función ventricular basal normal. En estudios clínicos con KYPROLIS®, estos eventos ocurrieron durante el curso de la terapia con KYPROLIS®. Ha ocurrido muerte por paro cardiaco a un día de la administración de KYPROLIS®. En estudios aleatorizados, abiertos, multicéntricos para terapias de combinación, la incidencia de eventos por falla cardiaca fue 8% y la de las arritmias fue 8% (la mayoría de las cuales fueron por fibrilación auricular y taquicardia sinusal) (ver Reacciones secundarias y adversas).
Monitorizar a los pacientes para detectar signos o síntomas clínicos de insuficiencia cardiaca o isquemia cardiaca. Evaluar inmediatamente si se sospecha de toxicidad cardiaca. Interrumpir el tratamiento con KYPROLIS® en caso de eventos adversos cardiacos Grado 3 o 4, hasta que se alcance la recuperación y evaluar si reiniciar el tratamiento con KYPROLIS® con una reducción de dosis con base en una evaluación de beneficio/riesgo (ver Dosis y vía de administración).
Mientras que por un lado se requiere hidratación adecuada previa a cada dosificación en el Ciclo 1, por otro lado, se debe monitorear a todos los pacientes para detectar evidencia de sobrecarga de volumen, especialmente pacientes en riesgo de insuficiencia cardiaca. Ajustar la ingesta total de fluidos según sea lo apropiado clínicamente, en pacientes con insuficiencia cardiaca basal o quienes estén en riesgo de insuficiencia cardiaca (ver Dosis y vía de administración).
El riesgo de falla cardiaca se incrementa en pacientes ≥ 75 años de edad en comparación con los pacientes más jóvenes. En los ensayos clínicos no participaron pacientes con insuficiencia cardiaca Clase III y IV conforme a la NYHA, infarto al miocardio reciente, y aquellos con alteraciones en la conducción, angina o arritmias no controladas con fármacos. Estos pacientes pueden estar en mayor riesgo de complicaciones cardiacas; para estos pacientes, complete una evaluación médica integral (incluyendo el control de la presión sanguínea y el manejo de líquidos) antes de empezar el tratamiento con KYPROLIS® o bien permanecer bajo un seguimiento cercano [ver Uso en poblaciones específicas (Uso geriátrico)].
Falla renal aguda: Se han producido casos de falla renal aguda en pacientes que recibieron KYPROLIS®. Algunos de estos eventos han sido fatales. Los eventos adversos de insuficiencia renal (incluyendo falla renal) han ocurrido en aproximadamente 9% de los pacientes que recibieron KYPROLIS®. La falla renal aguda fue reportada con mayor frecuencia en pacientes con mieloma múltiple avanzado en recaída y refractario que recibieron monoterapia con KYPROLIS®. El riesgo de falla renal fatal fue mayor en pacientes con una depuración de creatinina estimada basal reducida (calculada utilizado la ecuación de Cockcroft y Gault).
Monitorizar la función renal con mediciones regulares de creatinina sérica y/o estimado de depuración de creatinina. Reducir o interrumpir la dosis según sea apropiado (ver Dosis y vía de administración).
Síndrome de Lisis Tumoral: Se han reportado casos de síndrome de lisis tumoral (TLS por sus siglas en inglés), incluyendo resultados fatales, en pacientes que recibieron KYPROLIS®. Pacientes con mieloma múltiple y elevada carga tumoral deben ser considerados con mayor riesgo de TLS.
Se sugiere administrar fluidos orales e intravenosos antes de la administración de KYPROLIS® en el Ciclo 1 y en los ciclos subsecuentes según se requiera. Considerar el uso de fármacos para reducir el ácido úrico en pacientes con alto riesgo para TLS. Monitorizar TLS durante el tratamiento y de presentarse resolverlo de inmediato incluyendo la interrupción del tratamiento con KYPROLIS® hasta la resolución del TLS (ver Dosis y vía de administración).
Toxicidad pulmonar: Han ocurrido casos de Síndrome de Dificultad Respiratoria Aguda (ARDS, por sus siglas en Inglés) e insuficiencia respiratoria aguda en aproximadamente un 2% de los pacientes que recibieron KYPROLIS®. Además, se produjo enfermedad pulmonar infiltrativa difusa aguda, tal como neumonitis y enfermedad pulmonar intersticial en aproximadamente el 2% de los pacientes que recibieron KYPROLIS®. Algunos eventos fueron fatales. En la eventualidad de toxicidad pulmonar inducida por fármaco, discontinuar KYPROLIS®.
Hipertensión pulmonar: Se reportó hipertensión arterial pulmonar en aproximadamente 2% de los pacientes que recibieron KYPROLIS® con Grado 3 o mayor en menos del 1%.
Evaluar con escaneo cardiaco y/u otras pruebas según lo indicado. Interrumpir el uso de KYPROLIS® en caso de hipertensión pulmonar hasta que esta condición se haya resuelto o haya regresado a condiciones basales y considerar si se reinicia KYPROLIS® con base a una valoración de beneficio/riesgo.
Disnea: La disnea fue reportada en el 25% de los pacientes tratados con KYPROLIS® con Grado 3 o mayor en el 4%.
Evaluar los casos de disnea para excluir condiciones cardiopulmonares incluyendo falla cardiaca y síndromes pulmonares. Interrumpir el uso de KYPROLIS® en los casos de disnea Grado 3 y 4 hasta que se resuelvan o hayan regresado a condiciones basales. Considerar si se reinicia KYPROLIS® con base a una valoración del riesgo/beneficio (ver Dosis y vía de administración y Reacciones secundarias y adversas [Experiencia en estudios clínicos]).
Hipertensión: La hipertensión, incluyendo crisis y emergencia hipertensiva, ha sido observada con KYPROLIS®. En ASPIRE, la incidencia de eventos de hipertensión fue del 17% en el brazo KRd versus 9% en el brazo Rd. En ENDEAVOR, la incidencia de eventos de hipertensión fue 34% en el brazo Kd versus 11 % en el brazo Vd. En CANDOR, la incidencia de eventos de hipertensión fue 31 % en el brazo DKd versus 27% en el brazo Kd. Algunos de estos eventos han sido fatales.
Se sugiere optimizar la presión sanguínea antes de comenzar con KYPROLIS®. Monitorizar la presión sanguínea regularmente en todos los pacientes en tratamiento con KYPROLIS®. Si la hipertensión no puede ser controlada adecuadamente, se deberá interrumpir KYPROLIS® y evaluar. Considerar si se reinicia KYPROLIS® con base en evaluación de beneficio/riesgo.
Trombosis venosa: Eventos tromboembólicos venosos (incluyendo trombosis venosa profunda y embolismo pulmonar) han sido observados con KYPROLIS®. En ASPIRE, con tromboprofilaxis usada en ambos brazos, la incidencia de eventos tromboembólicos venosos en los primeros 12 ciclos fue de 13% en el brazo KRd versus 6% en el brazo Rd. En ENDEAVOR, la incidencia de eventos tromboembólicos venosos en los meses 1 a 6 fue de 9% en el brazo Kd versus 2% en el brazo Vd. Con la monoterapia de KYPROLIS®, la incidencia de eventos tromboembólicos venosos fue 2%.
Proporcione tromboprofilaxis para pacientes que están siendo tratados con KYPROLIS® en combinación con lenalidomida y dexametasona, con dexametasona; o con daratumumab intravenoso y dexametasona. Seleccione el régimen de tromboprofilaxis basado en los riesgos subyacentes de los pacientes.
Para los pacientes que usan anticonceptivos orales o anticoncepción hormonal asociado con un riesgo de trombosis, considere anticoncepción no hormonal durante el tratamiento cuando KYPROLIS® se administra en combinación (ver Restricciones de uso durante el embarazo y la lactancia).
Reacciones a la infusión: Han ocurrido reacciones relacionadas con la infusión incluyendo reacciones potencialmente mortales en pacientes recibiendo KYPROLIS®. Los síntomas incluyen fiebre, escalofríos, artralgia, mialgia, enrojecimiento facial, edema facial, edema laríngeo, vómito, debilidad, dificultad para respirar, hipotensión, síncope, dolor torácico o angina. Estas reacciones pueden ocurrir inmediatamente después o dentro de las 24 horas posteriores a la administración de KYPROLIS®. Administrar dexametasona previo a la administración de KYPROLIS® puede reducir la incidencia e intensidad de las reacciones relacionadas con la infusión (ver Dosis y vía de administración y Reacciones secundarias y advesas).
Hemorragia: Se han reportado casos de hemorragia grave o fatal en pacientes tratados con KYPROLIS® (ver Reacciones secundarias y adversas). Los eventos hemorrágicos han incluido hemorragia gastrointestinal, pulmonar e intracraneal y epistaxis. La hemorragia puede ser espontánea, y la hemorragia intracraneal ha ocurrido sin trauma. Se ha reportado hemorragia en pacientes con recuentos plaquetarios bajos o normales. La hemorragia también se ha reportado en pacientes que no estaban en terapia antiplaquetaria o anticoagulación.
Evaluar inmediatamente los signos y síntomas de pérdida de sangre. Reducir o suspender la dosis según sea apropiado (ver Dosis y vía de administración y Reacciones secundarias y adversas).
Trombocitopenia: KYPROLIS® causa trombocitopenia con nadir plaquetario que se observa entre el día 8 y el día 15 de cada ciclo de 28 días, con una recuperación en la cuenta de plaquetas a cifras basales que ocurre usualmente al inicio del ciclo subsecuente (ver Reacciones secundarias). Se reportó trombocitopenia en aproximadamente 32% de los pacientes en los estudios clínicos con KYPROLIS®. Puede ocurir hemorragia (ver Reacciones secundarias y adversas.
Monitorizar frecuentemente el recuento de plaquetas durante el tratamiento con KYPROLIS®. Reducir o suspender la dosis según sea apropiado (ver Dosis y vía de administración).
Toxicidad hepática y falla hepática: Se han reportado casos de insuficiencia hepática, incluyendo casos mortales (2%) durante el tratamiento con KYPROLIS®. KYPROLIS® puede causar incremento en los niveles de transaminasas séricas (ver Reacciones secundarias y adversas).
Monitorizar regularmente las enzimas hepáticas independientemente de los valores basales. Reducir o suspender la dosis según sea apropiado (ver Dosis y vía de administración).
Microangiopatía Trombótica: Se han reportado casos de microangiopatía trombótica, incluyendo púrpura trombocitopénica trombótica/síndrome hemolítico urémico (TTP/HUS, por sus siglas en Inglés) en pacientes que han recibido KYPROLIS. Algunos de estos eventos han sido fatales.
Monitorizar signos y síntomas de TTP/HUS. Si se sospechan estos diagnósticos, se deberá suspender KYPROLIS® y evaluar . Si se excluye el diagnóstico de TTP/HUS, se puede reiniciar el tratamiento con KYPROLIS®. Se desconoce la seguridad en pacientes con TTP/HUS al reiniciar la terapia con KYPROLIS®.
Síndrome de encefalopatía posterior reversible:
Se han reportado casos de síndrome de encefalopatía posterior reversible (PRES, por sus siglas en Inglés) en pacientes recibiendo KYPROLIS®. PRES, previamente denominado Síndrome de Leucoencefalopatía Posterior Reversible (RPLS), es un trastorno neurológico, el cual puede presentarse con convulsiones, cefalea, letargia, confusión, ceguera, alteración de la conciencia, y otras alteraciones visuales y neurológicas, además de hipertensión, y el diagnóstico es confirmado por medio de imágenes neuro-radiológicas (MRI). Interrumpir KYPROLIS® si se sospecha de PRES y evaluar. Se desconoce la seguridad al reiniciar la terapia con KYPROLIS® en pacientes que previamente experimentaron PRES.
Leucoencefalopatía multifocal progresiva: Se ha informado de leucoencefalopatía multifocal progresiva (PML, por sus siglas en inglés), que puede ser mortal, con KYPROLIS®. Además de KYPROLIS®, existen otros posibles factores que pueden contribuir a su ocurrencia, como terapia inmunosupresiva anterior o concurrente que puede causar inmunosupresión.
Considere la existencia de PML en cualquier paciente con nuevos signos o síntomas neurológicos o cambios en los preexistentes. Si se sospecha de PML, interrumpa la administración de KYPROLIS® e inicie la evaluación de PML incluida una consulta neurológica.
Incremento en las toxicidades fatales y graves en combinación con melfalán y prednisona en pacientes recientemente diagnosticados no elegibles para trasplante: En CLARION un estudio clínico de 955 pacientes no elegibles para trasplante con mieloma múltiple de reciente diagnóstico aleatorizados a KYPROLIS® (20/36 mg/m2 con infusión de 30 minutos dos veces a la semana por cuatro de cada ciclo de seis semanas), melfalán y prednisona (KMP) o bortezomib, melfalán y prednisona (VMP), se observó una mayor incidencia de reacciones adversas fatales (7% versus 4%) y reacciones adversas graves (50% versus 42%) en el brazo KMP comparado con los pacientes en el brazo VMP, respectivamente. Se observó que los pacientes en el brazo KMP tuvieron una mayor incidencia de reacciones adversas de cualquier grado incluyendo falla cardiaca (11% versus 4%), hipertensión (25% versus 8%), falla renal aguda (14% versus 6%), y disnea (18% versus 9%). Este estudio no alcanzó su medición primaria de superioridad en la supervivencia libre de progresión (PFS) para el brazo KMP. KYPROLIS®, en combinación con melfalán y prednisona no está indicado para pacientes no elegibles para trasplante con mieloma múltiple de reciente diagnóstico.
Toxicidad embrio-fetal: Según el mecanismo de acción y los hallazgos en animales, KYPROLIS® puede causar daño fetal cuando se administra a una mujer embarazada. Carfilzomib administrado por vía intravenosa a conejas embarazadas durante la organogénesis a una dosis de aproximadamente el 40% de la dosis clínica de 27 mg/m2 basada en el área de superficie corporal causó pérdida post-implantación y una disminución en el peso fetal.
Advierta a las mujeres embarazadas sobre el riesgo potencial para el feto. Advierta a las mujeres con potencial reproductivo que usen anticonceptivos eficaces durante el tratamiento con KYPROLIS® y durante los 6 meses posteriores a la dosis final. Advierta a los hombres con parejas femeninas con potencial reproductivo que usen anticonceptivos eficaces durante el tratamiento con KYPROLIS® y durante los 3 meses posteriores a la dosis final (ver Uso en poblaclones especificas y Precauciones en relaclón con efectos de carcinogénesis, mutagénesis y teratogénesis, y sobre la fertilidad).
Efectos en la habilidad para manejar u operar maquinaria: Informe a los pacientes que KYPROLIS® puede causar fatiga, mareo, desmayos y/o reducción de la presión arterial. Advierta a los pacientes de no manejar ni operar maquinaria si presentan alguno de estos síntomas.
DOSIS Y VÍA DE ADMINISTRACIÓN:
Cálculo de dosis: Para pacientes con un área de superficie corporal (BSA, por sus siglas en inglés) de 2.2 m2 o menos, calcule la dosis de KYPROLIS® con el BSA real. No se necesita hacer ajustes de dosis para cambios de peso menor o iguales al 20%.
En pacientes con un BSA superior a 2.2 m2, calcule la dosis de KYPROLIS® con un BSA de 22 m2.
Dosis recomendada: KYPROLIS® en combinación con lenalidomida y dexametasona: Administrar KYPROLIS® vía intravenosa mediante una infusión de 10 minutos en los días 1, 2, 8, 9, 15 y 16 de cada ciclo de 28 días en combinación con lenalidomida y dexametasona hasta el ciclo 12, como se muestra en la Tabla 26 (ver Estudios clínicos). La dosis inicial recomendada de KYPROLIS® es de 20 mg/m2 en el ciclo 1, días 1 y 2. De ser tolerado, la dosis deberá escalarse a 27 mg/m2 en el ciclo 1, día 8. Desde el ciclo 13, administrar KYPROLIS® en los días 1, 2, 15, 16 hasta el Ciclo 18. Suspender KYPROLIS® después del ciclo 18. Continuar con la lenalidomida y la dexametasona hasta que se produzca la progresión de la enfermedad o una toxicidad inaceptable. Ver la Información para Prescribir de la lenalidomida y la dexametasona para obtener información adicional sobre la dosis.
Tabla 26: KYPROLIS® 20/27 mg/m2 dos veces por semana (Infusión de 10 minutos) en combinación con lenalidomida y dexamentasona
|
Ciclo 1 |
|||||||||||
|
Semana 1 |
Semana 2 |
Semana 3 |
Semana 4 |
||||||||
|
Día 1 |
Día 2 |
Días 3 a 7 |
Día 8 |
Día 9 |
Días 10 a 14 |
Día 15 |
Día 16 |
Días 17 a 21 |
Día 22 |
Días 23 a 28 |
|
|
KYPROLIS® (mg/m2) |
20 |
20 |
- |
27 |
27 |
- |
27 |
27 |
- |
- |
- |
|
Dexametasona (mg) |
40 |
- |
- |
40 |
- |
- |
40 |
- |
- |
40 |
- |
|
Lenalidomida |
25 mg diariamente en los Días 1 a 21 |
- |
- |
||||||||
|
Ciclos 2 a 12 |
|||||||||||
|
Semana 1 |
Semana 2 |
Semana 3 |
Semana 4 |
||||||||
|
Día 1 |
Día 2 |
Días 3 a 7 |
Día 8 |
Día 9 |
Días 10 a 14 |
Día 15 |
Día 16 |
Días 17 a 21 |
Día 22 |
Días 23 a 28 |
|
|
KYPROLIS® (mg/m2) |
27 |
27 |
- |
27 |
27 |
- |
27 |
27 |
- |
- |
- |
|
Dexametasona (mg) |
40 |
- |
- |
40 |
- |
- |
40 |
- |
- |
40 |
- |
|
Lenalidomida |
25 mg diariamente en los Días 1 a 21 |
- |
- |
||||||||
|
Ciclos 13 y posterioresa |
|||||||||||
|
Semana 1 |
Semana 2 |
Semana 3 |
Semana 4 |
||||||||
|
Día 1 |
Día 2 |
Días 3 a 7 |
Día 8 |
Día 9 |
Días 10 a 14 |
Día 15 |
Día 16 |
Días 17 a 21 |
Día 22 |
Días 23 a 28 |
|
|
KYPROLIS® (mg/m2) |
27 |
27 |
- |
- |
- |
- |
27 |
27 |
- |
- |
- |
|
Dexametasona (mg) |
40 |
- |
- |
40 |
- |
- |
40 |
- |
- |
40 |
- |
|
Lenalidomida |
25 mg diariamente en los Días 1 a 21 |
- |
- |
||||||||
a KYPROLIS® es administrado hasta el Ciclo 18, la Lenalidomida y la Dexametasona continúan a partir de esa semana
KYPROLIS® en combinación con dexametasona:
Régimen de 20/56 mg/m2 dos veces por semana mediante infusión de 30 minutos: Administrar KYPROLIS® vía intravenosa como una infusión de 30 minutos en los Días 1, 2, 8, 9, 15 y 16 de cada ciclo de 28 días en combinación con dexametasona hasta la progresión de la enfermedad o hasta una toxicidad inaceptable, como se muestra en la Tabla 27 (ver Estudios clínicos). La dosis inicial recomendada de KYPROLIS® es 20 mg/m2 en el Ciclo 1, Días 1 y 2. Si se tolera, escalar la dosis a 56 mg/m2 en el Ciclo 1, Día 8. Administrar dexametasona de 30 minutos a 4 horas antes de KYPROLIS®. Referir a la Información para Prescribir de dexametasona a fin de obtener información adicional sobre la dosis.
Tabla 27: KYPROLIS® 20/56 mg/m2 dos veces a la semana (Infusión de 30 minutos) en combinación con dexametasona
|
Ciclo 1 |
||||||||||||
|
Semana 1 |
Semana 2 |
Semana 3 |
Semana 4 |
|||||||||
|
Día 1 |
Día 2 |
Días 3 a 7 |
Día 8 |
Día 9 |
Días 10 a 14 |
Día 15 |
Día 16 |
Días 17 a 21 |
Día 22 |
Día 23 |
Días 24 a 28 |
|
|
KYPROLIS® (mg/m2) |
20 |
20 |
- |
56 |
56 |
- |
56 |
56 |
- |
- |
- |
- |
|
Dexametasona (mg) |
20 |
20 |
- |
20 |
20 |
- |
20 |
20 |
- |
20 |
20 |
- |
|
Ciclos 2 y posteriores |
||||||||||||
|
Semana 1 |
Semana 2 |
Semana 3 |
Semana 4 |
|||||||||
|
Día 1 |
Día 2 |
Días 3 a 7 |
Día 8 |
Día 9 |
Días 10 a 14 |
Día 15 |
Día 16 |
Días 17 a 21 |
Día 22 |
Día 23 |
Días 24 a 28 |
|
|
KYPROLIS (mg/m2) |
56 |
56 |
- |
56 |
56 |
- |
56 |
56 |
- |
- |
- |
- |
|
Dexametasona (mg) |
20 |
20 |
- |
20 |
20 |
- |
20 |
20 |
- |
20 |
20 |
- |
Una vez por semana un régimen de 20/70 mg/m2 mediante infusión de 30 minutos: Administrar KYPROLIS® vía intravenosa como una infusión de 30 minutos en los días 1, 8 y 15 de cada ciclo de 28 días en combinación con dexametasona hasta la progresión de la enfermedad o hasta una toxicidad inaceptable, como se muestra en la Tabla 28 (ver Estudios clínicos). La dosis inicial recomendada de Kyprolis es 20 mg/m2 en el Ciclo 1, día 1. Si se tolera, escalar la dosis a 70 mg/m2 en el Ciclo 1, día 8. Administrar dexametasona de 30 minutos a 4 horas antes de KYPROLIS®. Ver la Información para Prescribir de dexametasona a fin de obtener información adicional sobre la dosis.
Tabla 28: KYPROLIS® 20/70 mg/m2 una vez a la semana (Infusión de 30 minutos) en combinación con dexametasona
|
Ciclo 1 |
||||||||||||
|
Semana 1 |
Semana 2 |
Semana 3 |
Semana 4 |
|||||||||
|
Día 1 |
Día 2 |
Días 3 a 7 |
Día 8 |
Día 9 |
Días 10 a 14 |
Día 15 |
Día 16 |
Días 17 a 21 |
Día 22 |
Día 23 |
Días 24 a 28 |
|
|
KYPROLIS® (mg/m2) |
20 |
- |
- |
70 |
- |
- |
70 |
- |
- |
- |
- |
- |
|
Dexametasona (mg) |
40 |
- |
- |
40 |
- |
- |
40 |
- |
- |
40 |
- |
- |
|
Ciclo 2 a 9 |
||||||||||||
|
Semana 1 |
Semana 2 |
Semana 3 |
Semana 4 |
|||||||||
|
Día 1 |
Día 2 |
Días 3 a 7 |
Día 8 |
Día 9 |
Días 10 a 14 |
Día 15 |
Día 16 |
Días 17 a 21 |
Día 22 |
Día 23 |
Días 24 a 28 |
|
|
KYPROLIS® (mg/m2) |
70 |
- |
- |
70 |
- |
- |
70 |
- |
- |
- |
- |
- |
|
Dexametasona (mg) |
40 |
- |
- |
40 |
- |
- |
40 |
- |
- |
40 |
- |
|
|
Ciclos 10 y posteriores |
||||||||||||
|
Semana 1 |
Semana 2 |
Semana 3 |
Semana 4 |
|||||||||
|
Día 1 |
Día 2 |
Días 3 a 7 |
Día 8 |
Día 9 |
Días 10 a 14 |
Día 15 |
Día 16 |
Días 17 a 21 |
Día 22 |
Día 23 |
Días 24 a 28 |
|
|
KYPROLIS® (mg/m2) |
70 |
- |
- |
70 |
- |
- |
70 |
- |
- |
- |
- |
- |
|
Dexametasona (mg) |
40 |
- |
- |
40 |
- |
- |
40 |
- |
- |
- |
- |
- |
KYPROLIS® en combinación con daratumumab intravenoso y dexametasona:
Régimen de 20/56 mglm2 dos veces por semana mediante infusión de 30 minutos: Administrar KYPROLIS® por vía intravenosa como una infusión de 30 minutos los días 1, 2, 8, 9, 15 y 16 de cada ciclo de 28 días en combinación con daratumumab intravenoso y dexametasona hasta la progresión de la enfermedad o una toxicidad inaceptable, como se muestra en la Tabla 29 (ver Estudios clínicos). La dosis inicial recomendada de KYPROLIS® es 20 mg/m2 en el Ciclo 1, días 1 y 2. De ser tolerado, la dosis deberá escalarse a 56 mg/m2 en el Ciclo 1, día 8, y de ahí en adelante. Administrar dexametasona de 30 minutos a 4 horas antes de KYPROLIS® y de 1 a 3 horas antes del daratumumab intravenoso. Ver la Información para Prescribir de daratumumab intravenoso y dexametasona si desea obtener información adicional sobre la dosis.
Tabla 29: KYPROLIS® 20/56 mg/m2 dos veces por semana (Infusión de 30 minutos) en combinación con daratumumab intravenoso y dexametasona
|
Ciclo 1 |
||||||||||||
|
Semana 1 |
Semana 2 |
Semana 3 |
Semana 4 |
|||||||||
|
Día 1 |
Día 2 |
Días 3 a 7 |
Día 8 |
Día 9 |
Días 10 a 14 |
Día 15 |
Día 16 |
Días 17 a 21 |
Día 22 |
Día 23 |
Días 24 a 28 |
|
|
KYPROLIS® (mg/m2) |
20 |
20 |
- |
56 |
56 |
- |
56 |
56 |
- |
- |
- |
- |
|
Dexametasona (mg)a |
20 |
20 |
- |
20 |
20 |
- |
20 |
20 |
- |
40 |
- |
- |
|
Daratumumab (mg/kg) |
8 |
8 |
- |
16 |
- |
- |
16 |
- |
- |
16 |
- |
- |
|
Ciclo 2 |
||||||||||||
|
Semana 1 |
Semana 2 |
Semana 3 |
Semana 4 |
|||||||||
|
Día 1 |
Día 2 |
Días 3 a 7 |
Día 8 |
Día 9 |
Días 10 a 14 |
Día 15 |
Día 16 |
Días 17 a 21 |
Día 22 |
Día 23 |
Días 24 a 28 |
|
|
KYPROLIS® (mg/m2) |
56 |
56 |
- |
56 |
56 |
- |
56 |
56 |
- |
- |
- |
- |
|
Dexametasona (mg)* |
20 |
20 |
- |
20 |
20 |
- |
20 |
20 |
- |
40 |
- |
- |
|
Daratumumab (mg/kg) |
16 |
- |
- |
16 |
- |
- |
16 |
- |
- |
16 |
- |
- |
|
Ciclos 3 a 6 |
||||||||||||
|
Semana 1 |
Semana 2 |
Semana 3 |
Semana 4 |
|||||||||
|
Día 1 |
Día 2 |
Días 3 a 7 |
Día 8 |
Día 9 |
Días 10 a 14 |
Día 15 |
Día 16 |
Días 17 a 21 |
Día 22 |
Día 23 |
Días 24 a 28 |
|
|
KYPROLIS® (mg/m2) |
56 |
56 |
- |
56 |
56 |
- |
56 |
56 |
- |
- |
- |
- |
|
Dexametasona (mg)* |
20 |
20 |
- |
20 |
20 |
- |
20 |
20 |
- |
40 |
- |
- |
|
Daratumumab (mg/kg) |
16 |
- |
- |
- |
- |
- |
16 |
- |
- |
- |
- |
- |
|
Ciclos 7 y posteriores |
||||||||||||
|
Semana 1 |
Semana 2 |
Semana 3 |
Semana 4 |
|||||||||
|
Día 1 |
Día 2 |
Días 3 a 7 |
Día 8 |
Día 9 |
Días 10 a 14 |
Día 15 |
Día 16 |
Días 17 a 21 |
Día 22 |
Día 23 |
Días 24 a 28 |
|
|
KYPROLIS® (mg/m2) |
56 |
56 |
- |
56 |
56 |
- |
56 |
56 |
- |
- |
- |
- |
|
Dexametasona (mg)* |
20 |
20 |
- |
20 |
20 |
- |
20 |
20 |
- |
40 |
- |
- |
|
Daratumumab (mg/kg) |
16 |
- |
- |
- |
- |
- |
- |
- |
- |
- |
- |
- |
* En pacientes mayores de 75 años, administre 20 mg de Dexametasona por vía oral o intravenosa una vez a la a la semana después de la primera semana.
Régimen de 20/70 mg/m2 una vez por semana mediante infusión de 30 minutos:
Administrar KYPROLIS® por vía intravenosa como una infusión de 30 minutos los días 1, 8 y 15 de cada ciclo de 28 días en combinación con daratumumab intravenoso y dexametasona hasta la progresión de la enfermedad o una toxicidad inaceptable, como se muestra en la Tabla 30 (ver Estudios clínicos). La dosis inicial recomendada de KYPROLIS® es 20 mg/m2 en el Ciclo 1, día 1. De ser tolerado, la dosis deberá escalarse a 70 mg/m2 en el Ciclo 1, Día 8, y de ahí en adelante. Administrar dexametasona de 30 minutos a 4 horas antes de KYPROLIS® y de 1 a 3 horas antes del daratumumab intravenoso. Ver la Información para Prescribir de daratumumab intravenoso y dexametasona si desea obtener información adicional sobre la dosis.
Tabla 30: KYPROLIS® 20/70 mg/m2 una vez a la semana (Infusión de 30 minutos) en combinación con daratumumab intravenoso y dexametasona
|
Ciclo 1 |
||||||||||||
|
Semana 1 |
Semana 2 |
Semana 3 |
Semana 4 |
|||||||||
|
Día 1 |
Día 2 |
Días 3 a 7 |
Día 8 |
Día 9 |
Días 10 a 14 |
Día 15 |
Día 16 |
Días 17 a 21 |
Día 22 |
Día 23 |
Días 24 a 28 |
|
|
KYPROLIS® (mg/m2) |
20 |
- |
- |
70 |
- |
- |
70 |
- |
- |
- |
- |
- |
|
Dexametasona (mg)* |
20 |
20 |
- |
20 |
20 |
- |
20 |
20 |
- |
20 |
20 |
- |
|
Daratumumab (mg/kg) |
8 |
8 |
- |
16 |
- |
- |
16 |
- |
- |
16 |
- |
- |
|
Ciclo 2 |
||||||||||||
|
Semana 1 |
Semana 2 |
Semana 3 |
Semana 4 |
|||||||||
|
Día 1 |
Día 2 |
Días 3 a 7 |
Día 8 |
Día 9 |
Días 10 a 14 |
Día 15 |
Día 16 |
Días 17 a 21 |
Día 22 |
Día 23 |
Días 24 a 28 |
|
|
KYPROLIS® (mg/m2) |
70 |
- |
- |
70 |
- |
- |
70 |
- |
- |
- |
- |
- |
|
Dexametasona (mg)* |
20 |
20 |
- |
20 |
20 |
- |
20 |
20 |
- |
20 |
20 |
- |
|
Daratumumab (mg/kg) |
16 |
- |
- |
16 |
- |
- |
16 |
- |
- |
16 |
- |
- |
|
Ciclos 3 a 6 |
||||||||||||
|
Semana 1 |
Semana 2 |
Semana 3 |
Semana 4 |
|||||||||
|
Día 1 |
Día 2 |
Días 3 a 7 |
Día 8 |
Día 9 |
Días 10 a 14 |
Día 15 |
Día 16 |
Días 17 a 21 |
Día 22 |
Día 23 |
Días 24 a 28 |
|
|
KYPROLIS® (mg/m2) |
70 |
- |
- |
70 |
- |
- |
70 |
- |
- |
- |
- |
- |
|
Dexametasona (mg)* |
20 |
20 |
- |
40 |
- |
- |
20 |
20 |
- |
40 |
- |
- |
|
Daratumumab (mg/kg) |
16 |
- |
- |
- |
- |
- |
16 |
- |
- |
- |
- |
- |
|
Ciclos 7 y posteriores |
||||||||||||
|
Semana 1 |
Semana 2 |
Semana 3 |
Semana 4 |
|||||||||
|
Día 1 |
Día 2 |
Días 3 a 7 |
Día 8 |
Día 9 |
Días 10 a 14 |
Día 15 |
Día 16 |
Días 17 a 21 |
Día 22 |
Día 23 |
Días 24 a 28 |
|
|
KYPROLIS® (mg/m2) |
70 |
- |
70 |
56 |
- |
70 |
- |
- |
- |
- |
||
|
Dexametasona (mg)* |
20 |
20 |
- |
40 |
- |
- |
40 |
- |
- |
40 |
- |
- |
|
Daratumumab (mg/kg) |
16 |
- |
- |
- |
- |
- |
- |
- |
- |
- |
- |
- |
* En pacientes mayores de 75 años, administre 20 mg de Dexametasona por vía oral o intravenosa una vez a la semana después de la primera semana.
Monoterapia con KYPROLIS®:
Régimen de 20/27 mg/m2 dos veces por semana mediante infusión de 10 minutos: Administrar KYPROLIS® vía intravenosa mediante una infusión de 10 minutos (ver Estudios clínicos). En los ciclos 1 hasta el 12, administrar KYPROLIS® en los días 1, 2, 8, 9, 15 y 16 de cada ciclo de 28 días como se muestra en la Tabla 31 . A partir del Ciclo 13, administrar KYPROLIS® en los días 1, 2, 15 y 16 de cada ciclo de 28 días. Pre-medicar con dexametasona 4 mg oral o intravenoso 30 minutos a 4 horas antes de cada dosis de KYPROLIS® en el Ciclo 1, posteriormente según se requiera para minimizar las reacciones de la infusión. La dosis inicial recomendada de KYPROLIS® es 20 mg/m2 en el Ciclo 1 en los días 1 y 2. Si se tolera, escalar la dosis a 27 mg/m2 en el día 8 del Ciclo 1 y de ahí en adelante. Continuar con KYPROLIS® hasta la progresión de la enfermedad o una toxicidad inaceptable.
Tabla 31: Monoterapia con KYPROLIS® 20/27 mg/m2 dos veces por semana (Infusión de 10 minutos)
|
Ciclo 1 |
||||||||||
|
Semana 1 |
Semana 2 |
Semana 3 |
Semana 4 |
|||||||
|
Día 1 |
Día 2 |
Días 3 a 7 |
Día 8 |
Día 9 |
Días 10 a 14 |
Día 15 |
Día 16 |
Días 17 a 21 |
Día 22 a 28 |
|
|
KYPROLIS® (mg/m2)a |
20 |
20 |
- |
27 |
27 |
- |
27 |
27 |
- |
- |
|
Ciclos 2 a 12 |
||||||||||
|
Semana 1 |
Semana 2 |
Semana 3 |
Semana 4 |
|||||||
|
Día 1 |
Día 2 |
Días 3 a 7 |
Día 8 |
Día 9 |
Días 10 a 14 |
Día 15 |
Día 16 |
Días 17 a 21 |
Día 22 a 28 |
|
|
KYPROLIS® (mg/m2) |
27 |
27 |
- |
27 |
27 |
- |
27 |
27 |
- |
- |
|
Ciclos 13 y posteriores |
||||||||||
|
Semana 1 |
Semana 2 |
Semana 3 |
Semana 4 |
|||||||
|
Día 1 |
Día 2 |
Días 3 a 7 |
Día 8 |
Día 9 |
Días 10 a 14 |
Día 15 |
Día 16 |
Días 17 a 21 |
Día 22 a 28 |
|
|
KYPROLIS® (mg/m2) |
27 |
27 |
- |
- |
- |
- |
27 |
27 |
- |
- |
ª Se requiere pre-medicación con dexametasona para cada dosis de KYPROLIS® en el ciclo 1.
Régimen de 20/56 mg/m2 dos veces por semana mediante infusión de 30 minutos: Administrar KYPROLIS® vía intravenosa mediante una infusión de 30 minutos (ver Estudios clínicos). En los Ciclos 1 hasta el 12, administrar KYPROLIS® en los días 1, 2, 8, 9, 15 y 16 de cada ciclo de 28 días como lo muestra la Tabla 32. Desde el Ciclo 13, administrar KYPROLIS® en los días 1, 2, 15 y 16 de cada ciclo de 28 días. Pre-medicar con dexametasona 8 mg oral o intravenosa 30 minutos a 4 horas antes de cada dosis de KYPROLIS® en el Ciclo 1, posteriormente según se requiera para minimizar las reacciones de la infusión. La dosis de arranque recomendada de KYPROLIS® es 20 mg/m2 en el Ciclo 1 en los días 1 y 2. Si se tolera, escalar la dosis a 56 mg/m2 en el día 8 del Ciclo 1.
Continuar con KYPROLIS® hasta la progresión de la enfermedad o una toxicidad inaceptable.
Tabla 32: Monoterapia con KYPROLIS® 20/56 mg/m2 Dos veces por semana (Infusión de 30 minutos)
|
Ciclo 1 |
||||||||||
|
Semana 1 |
Semana 2 |
Semana 3 |
Semana 4 |
|||||||
|
Día 1 |
Día 2 |
Días 3 a 7 |
Día 8 |
Día 9 |
Días 10 a 14 |
Día 15 |
Día 16 |
Días 17 a 21 |
Día 22 a 28 |
|
|
KYPROLIS® (mg/m2)a |
20 |
20 |
- |
56 |
56 |
- |
56 |
56 |
- |
- |
|
Ciclos 2 a 12 |
||||||||||
|
Semana 1 |
Semana 2 |
Semana 3 |
Semana 4 |
|||||||
|
Día 1 |
Día 2 |
Días 3 a 7 |
Día 8 |
Día 9 |
Días 10 a 14 |
Día 15 |
Día 16 |
Días 17 a 21 |
Día 22 a 28 |
|
|
KYPROLIS® (mg/m2) |
56 |
56 |
- |
56 |
56 |
- |
56 |
56 |
- |
- |
|
Ciclos 13 y posteriores |
||||||||||
|
Semana 1 |
Semana 2 |
Semana 3 |
Semana 4 |
|||||||
|
Día 1 |
Día 2 |
Días 3 a 7 |
Día 8 |
Día 9 |
Días 10 a 14 |
Día 15 |
Día 16 |
Días 17 a 21 |
Día 22 a 28 |
|
|
KYPROLIS® (mg/m2) |
56 |
56 |
- |
- |
- |
- |
56 |
56 |
- |
- |
ª Se requiere pre-medicación con Dexametasona para cada dosis de KYPROLIS® en el Ciclo 1.
Precauciones de administración:
Hidratación: Se requiere una hidratación adecuada antes de la dosificación en el Ciclo 1, especialmente en pacientes con alto riesgo de síndrome de lisis tumoral o toxicidad renal. Considere la hidratación tanto con líquidos orales (30 mL por kg por lo menos 48 horas antes del Ciclo 1, Día 1) como líquidos por vía intravenosa (250 mL a 500 mL de solución intravenosa apropiada antes de cada una de las dosis del Ciclo 1). Si se requiere, administre adicionalmente de 250 mL a 500 mL de solución por vía intravenosa después de la administración de KYPROLIS®. Continuar con la hidratación por vía intravenosa y/o oral, según sea necesario en los ciclos posteriores.
Monitorizar a los pacientes por evidencia de sobrecarga de volumen y ajustar la hidratación a las necesidades individuales del paciente, especialmente en pacientes con o en riesgo de insuficiencia cardiaca (consulte Precauciones generales).
Monitorización de electrólitos: Durante el tratamiento con KYPROLIS® se deberá monitorizar regularmente los niveles de potasio sérico (ver Reacciones secundarias y adversas).
Premedicaciones y medicamentos concomitantes: Pre medicar con la dosis recomendada de dexametasona para monoterapia o dexametasona administrada como parte de la terapia en combinación. Administrar dexametasona vía oral o intravenosa por lo menos 30 minutos, pero no más de 4 horas antes de todas las dosis de KYPROLIS® durante el Ciclo 1 para reducir la incidencia y gravedad de las reacciones a la infusión (ver Precauciones generales). Reinicie la premedicación con dexametasona si se presentan estos síntomas durante los ciclos subsecuentes.
Proporcione tromboprofilaxis para los pacientes tratados con KYPROLIS® en combinación con otras terapias (ver Precauciones generales).
Se debe considerar la profilaxis antiviral para reducir el riesgo de reactivación del herpes zoster (ver Reacciones secundarias y adversas).
Modificaciones de la dosis para reacciones adversas: En la Tabla 33 se muestran las medidas y modificaciones de dosis recomendadas para KYPROLIS®. Las reducciones en el nivel de dosis se presentan en la Tabla 34. Ver la Información para Prescribir de lenalidomida, daratumumab intravenoso y dexametasona, respectivamente, para ver las modificaciones recomendadas de dosificación asociadas con cada producto.
Tabla 33: Modificaciones de la dosis para Reacciones adversas
|
Toxicidad hematológica (ver Precauciones generales y Reacciones secundarias y adversas) |
Medidas recomendadas |
|
CAN menos de 0.5 x 109/L |
Detener la dosis Si se observa recuperación mayor o igual que 0.5 x 109/L continuar con el mismo nivel de dosis Para reducciones subsecuentes menores de 0.5 x 109/L, seguir las recomendaciones antes mencionadas y considerar 1 nivel en reducción de dosis al reiniciar con KYPROLISa |
|
Neutropenia febril: CAN menor de 0.5 x 109/L y temperatura oral mayor que 38.5°C o dos lecturas consecutivas mayores a 38.0°C por 2 horas |
Detener la dosis Si la CAN regresa a valores basales y la fiebre desaparece, reanudar en el mismo nivel de dosis |
|
Plaquetas menores a 10 x 109/L o evidencia de sangrado con trombocitopenia |
Detener la dosis Si hay recuperación a mayor o igual que 10 x 109/L y/o el sangrado está controlado, continuar con el mismo nivel de dosis En reducciones subsecuentes en plaquetas, menor que 10 x 109/L, seguir las mismas recomendaciones indicadas arriba y considerar 1 reducción en nivel de dosis cuando se reinicie KYPROLIS®a |
|
Toxicidad Renal (ver Precauciones generales) |
Medidas recomendadas |
|
Creatinina sérica igual o mayor a 2 veces la basal o Depuración de creatinina menor a 15 mL/min o disminuciones en la depuración de creatinina menor o igual que 50% sobre el valor basal o necesidad de hemodiálisis |
Detener la dosis y monitorizar continuamente la función renal (creatinina sérica o depuración de creatinina) Si es atribuible a KYPROLIS®, reanudar cuando la función renal se haya recuperado al 25% de la capacidad basal, iniciando con 1 dosis reducida en un nivela Si no es atribuible a KYPROLIS®, la dosis puede ser reanudada a discreción del proveedor de atención médica Para pacientes en hemodiálisis recibiendo KYPROLIS®, la dosis se administrará después del procedimiento de hemodiálisis |
|
Otra toxicidad no-hematológica (ver Reacciones secundarias y adversas) |
Medida recomendada |
|
Para cualquier otro tipo de toxicidad no hematológica, severas o que ponen en riesgo la vidab |
Detener la dosis hasta su resolución o hasta que se retome a la situación basal. Considere la posibilidad de reiniciar el siguiente tratamiento programado a una reducción en el nivel de dosisa |
CAN = cuenta absoluta de neutrófilos.
a Ver Tabla 34 para reducciones del nivel de la dosis.
b Grados 3 y 4.
Tabla 34: Reducciones del nivel de la dosis para Reacciones adversas
|
Régimen |
Dosis |
Primera reducción de la dosis |
Segunda reducción de la dosis |
Tercera reducción de la dosis |
|
KYPROLIS® y Dexametasona O KYPROLIS®, Daratumumab y Dexametasona (una vez por semana) |
70 mg/m2 |
56 mg/m2 |
45 mg/m2a |
36 mg/m2a |
|
KYPROLIS® y Dexametasona O KYPROLIS®, Daratumumab y Dexametasona O Monoterapia con KYPROLIS® (dos veces por semana) |
56 mg/m2 |
45 mg/m2 |
36 mg/m2 |
27 mg/m2a |
|
KYPROLIS®, Lenalidomida y Dexametasona O Monoterapia con KYPROLIS® (dos veces por semana) |
27 mg/m2 |
20 mg/m2 |
15 mg/m2a |
- |
Nota: Los tiempos de infusión permanecen sin cambio durante la(s) reducción(es) en la dosis.
a Si la toxicidad persiste, descontinuar el tratamiento con KYPROLIS®.
Modificación de dosis para daño hepático: Para pacientes con daño hepático leve (bilirrubina total 1 a 1.5 x ULN o cualquier AST o bilirrubina total ≤ ULN y AST > ULN) o moderado (bilirrubina total > 1.5 a 3 x ULN y cualquier AST), reduzca la dosis de KYPROLIS® en 25% (ver Uso en poblaciones específicas y farmacocinética).
Dosificación recomendada para enfermedad renal en etapa terminal: Para pacientes con enfermedad renal en etapa final quienes están en hemodiálisis, administrar KYPROLIS® después del procedimiento de hemodiálisis.
Preparación y administración: Los frascos ámpula de Kyprolis no contienen conservadores antimicrobianos y están previstos para un solo uso. La solución reconstituida contiene carfilzomib a una concentración de 2 mg/mL.
Lea las instrucciones de preparación completas antes de la reconstitución. Los medicamentos parenterales deben inspeccionarse visualmente en busca de partículas y decoloración antes de la administración, siempre que la solución y el envase lo permita.
Reconstitución/Pasos de Preparación:
1. Retirar el frasco ámpula del refrigerador justo antes de usarse.
2. Calcule la dosis (mg/m2) y el número de frascos ámpula de KYPROLIS® requeridos utilizando el área de la superficie corporal del paciente (BSA) basal.
3. Utilice una aguja de calibre 21 o mayor (aguja con diámetro externo de 0.8 mm o menor) para reconstituir de manera aséptica cada frasco ámpula inyectando lentamente 29 mL (para un frasco ámpula de 60 mg) de Agua Estéril para Inyección, a través del tapón y dirigiendo la solución HACIA LA PARED INTERIOR DEL FRASCO ÁMPULA para minimizar la formación de espuma. (No reconstituir KYPROLIS® con solución salina normal). No hay datos que respalden el uso de dispositivos de transferencia de sistema cerrado con KYPROLIS®.
4. Gire y/o invierta suavemente el frasco ámpula despacio por cerca de 1 minuto, o hasta la disolución completa. NO AGITE para evitar generar espuma. Si se ve espuma, permita que la solución se asiente en el frasco ámpula hasta que la espuma desaparezca (aproximadamente 5 minutos) y la solución esté clara.
5. Inspeccione visualmente para detección de partículas o cualquier cambio de color previo a la administración. El producto reconstituido debe ser una solución clara e incolora, y no debe administrarse si se observa cualquier cambio de coloración o partículas.
6. Deseche cualquier porción no utilizada que quede en el frasco ámpula. NO combine las porciones no utilizadas de los frascos ámpula. NO administre más de una dosis del frasco ámpula.
7. Administre KYPROLIS® directamente por infusión intravenosa o en una bolsa intravenosa de 50 mL o 100 mL con Dextrosa al 5% Inyectable. No debe ser administrado como bolo intravenoso.
8. Cuando se administre en una bolsa intravenosa, utilice una aguja de calibre 21 o mayor (aguja con diámetro externo de 0.8 mm o menor) para retirar del frasco ámpula la dosis calculada y diluya en una bolsa intravenosa de 50 mL o 100 mL con Dextrosa al 5% Inyectable, (con base en la dosis total calculada y el tiempo de infusión). No diluya KYPROLIS® con solución salina normal.
9. Purgue la vía de administración intravenosa con solución salina normal o con Dextrosa al 5%, inmediatamente antes y después de la administración de KYPROLIS®.
10. No mezcle KYPROLIS® ni lo administre mediante una infusión con otros medicamentos.
Las estabilidades de KYPROLIS® reconstituido bajo varias condiciones de temperatura y contenedor se muestran en la Tabla 35.
Tabla 35: Estabilidad de KYPROLIS® reconstituido
|
Condiciones de almacenaje de KYPROLIS® reconstituido |
Estabilidadª por contenedor |
||
|
Vial |
Jeringa |
Bolsa intravenosa (D5Wb) |
|
|
Refrigerado (2°C a 8°C) |
24 horas |
24 horas |
24 horas |
|
Temperatura ambiente (15°C a 30°C) |
4 horas |
4 horas |
4 horas |
a Tiempo total desde la reconstitución a la administración no debe exceder 24 horas.
b 5% Dextrosa inyectable.
MANIFESTACIONES Y MANEJO DE LA SOBREDOSIFICACIÓN O INGESTA ACCIDENTAL: Posterior a una dosis de 200 mg de KYPROLIS® administrada por error, se ha reportado inicio agudo de escalofríos, hipotensión, insuficiencia renal, trombocitopenia, y linfopenia.
Se desconoce el antídoto específico para la sobredosificación con carfilzomib. En caso de sobredosificación, monitoree a los pacientes por reacciones adversas y proporcione tratamiento de apoyo según sea apropiado.
PRESENTACIÓN:
Para inyección: 60 mg como polvo liofilizado en un frasco ámpula de una sola dosis para reconstitución.
RECOMENDACIONES SOBRE ALMACENAMIENTO: KYPROLIS®
Los frascos ámpula no abiertos deberán mantenerse en refrigeración (2°C a 8°C). Conserve el frasco ámpula en su caja para protegerlo de la luz.
Solución reconstituida:
El tiempo transcurrido desde la reconstitución a la administración no debe exceder 24 horas. Almacene la solución reconstituida en el frasco ámpula, jeringa o bolsa de infusión en refrigeración (2°C a 8°C) hasta por 24 horas o a temperatura ambiente (15°C a 30°C) durante un máximo de 4 horas.
Los frascos ámpula de KYPROLIS® no contienen conservantes antimicrobianos y están destinados a un solo uso. Se deben observar las medidas asépticas apropiadas.
Cualquier producto no usado o material de desperdicio deberá ser desechado de acuerdo con los requerimientos o normas locales.
LEYENDAS DE PROTECCIÓN:
Este medicamento deberá ser prescrito por médicos especialistas en oncología y hematología. Manténgase fuera del alcance de los niños. Su venta requiere receta médica. No se use durante el embarazo y lactancia. No se administre a menores de 18 años. Literatura exclusiva para el médico.
Reporte las sospechas de reacción adversa al correo: farmacovigilancia@cofepris.gob.mx o
farmacovigilanciamx@amgen.com
Representante Legal:
AMGEN MÉXICO S.A. de C.V.
Av. Vasco de Quiroga No. 3000, Piso 4,
Col. Santa Fe, C.P. 01210, Álvaro Obregón,
Ciudad de México, México.
®Marca Registrada


