
KUVAN - Tabletas
Sustancia(s):
- Dihidrocloruro De Sapropterina
Presentaciones:
- 1 Caja , 1 Frasco(s) , 30 Tabletas
- 1 Caja , 1 Frasco(s) , 120 Tabletas
- 1 Caja , 1 Frasco(s) , 240 Tabletas
FORMA FARMACÉUTICA Y FORMULACIÓN:
Cada TABLETA contiene:
Dihidrocloruro de sapropterina 100 mg
Excipientes, c.b.p. 1 tableta.
INDICACIONES TERAPÉUTICAS: KUVAN® está indicado para el tratamiento de la hiperfenilalaninemia (HPA) en pacientes adultos y pediátricos mayores a 4 años con fenilcetonuria.
FARMACOCINÉTICA Y FARMACODINAMIA:
Mecanismo de acción: KUVAN® es una forma sintética de BH4, el cofactor para la enzima fenilalanina hidroxilasa (PAH). La PAH hidroxila a la Phe mediante una reacción oxidativa para formar tirosina. En pacientes con PKU, la actividad de la PAH está ausente o es deficiente. El tratamiento con BH4 puede activar a la enzima PAH residual, mejorar el metabolismo oxidativo normal de la Phe y reducir la concentración de Phe en algunos pacientes.
Farmacodinamia: En pacientes con PKU que responden al tratamiento con BH4, la concentración sanguínea de Phe disminuye dentro de un lapso de 24 horas después de la administración única de dihidrocloruro de sapropterina, aunque el máximo efecto sobre la concentración de Phe puede llevar hasta un mes, dependiendo del paciente. Una dosis única diaria de KUVAN® es adecuada para mantener estable la concentración sanguínea de Phe por un periodo de 24 horas. Se evaluaron 24 pacientes con concentraciones sanguíneas de Phe que variaban de 516 a 986 µmol/L (media 747 ± 153 µmol/L) con monitoreo de la concentración sanguínea de Phe a 24 horas después de una dosis diaria por la mañana de 10 mg/kg/día. La concentración sanguínea de Phe permaneció estable durante un periodo de observación de 24 horas. No se observaron incrementos sustanciales en la concentración sanguínea de Phe después de la ingesta de alimentos durante todo el periodo de 24 horas.
Las dosis superiores a 20 mg/kg/día no han sido evaluadas en estudios clínicos.
Estudios clínicos:
Estudios clínicos en PKU: La eficacia y seguridad del KUVAN® fueron evaluadas en 4 estudios clínicos en pacientes con PKU.
El estudio 1 fue un ensayo clínico multicéntrico, abierto y sin grupo control de 489 pacientes con PKU de 8 a 48 años de edad (promedio de 22 años), quienes presentaron concentraciones iniciales de Phe en sangre > 450 µmol/L y no estaban en dieta restringida en Phe. Todos los pacientes recibieron tratamiento con KUVAN® 10 mg/kg/día por 8 días. Para los propósitos de este estudio, la respuesta al tratamiento de KUVAN® se definió como una disminución > 30% de Phe en sangre desde el inicio. En el día 8, 96 pacientes (20%) fueron identificados como pacientes que responden.
El estudio 2 fue multicéntrico , doble ciego, placebo controlado de 88 pacientes con PKU quienes respondieron a KUVAN® en el estudio 1. Después de un periodo de lavado del estudio 1, los pacientes fueron asignados de manera aleatoria igualmente, ya sea a KUVAN® 10 mg/kg/día (n = 41) o placebo (n = 47) por 6 semanas. La eficacia se evaluó por el cambio promedio en la concentración de Phe en sangre desde el inicio a la semana 6 en el grupo tratado con KUVAN® comparado al cambio promedio en el grupo placebo.
Los resultados mostraron que al inicio la concentración promedio (± SD) de Phe en sangre fue de 843 (± 300) µmol/L en el grupo tratado con KUVAN® y 888 (± 323) µmol/L en el grupo placebo. A la semana 6, el grupo tratado con KUVAN® tuvo una concentración promedio (± SD) de Phe en sangre de 607 (± 377) µmol/L y el grupo placebo tuvo una concentración promedio (± SD) de Phe en sangre de 891 (± 348) µmol/L.
A la semana 6, los grupos tratados con KUVAN® y placebo tuvieron cambios promedio en la concentración de Phe en sangre de -239 y 6 µmol/L, respectivamente (cambios promedio en porcentaje de -29 % (± 32) y 3% (± 33), respectivamente).
La diferencia entre los grupos fue estadísticamente significativa (p < 0.001) (tabla 1).
Tabla 1. Resultados de Phe en sangre en el estudio 2
|
Sapropterina (n = 41) |
Placebo (n = 47) |
|
|
Concentración sanguínea inicial de Phe1 (µmol/L) |
||
|
Promedio (± SD) |
843 (± 300) |
888 (± 323) |
|
Percentiles (25ªVº, 75ªVº) |
620, 990 |
618, 1,141 |
|
Concentración sanguínea de Phe a la semana 6 (µmol/L) |
||
|
Promedio (± SD) |
607 (± 377) |
891 (± 348) |
|
Percentiles (25ªVº, 75ªVº) |
307, 812 |
619, 1,143 |
|
Cambio promedio en Phe en sanare del inicio a la semana 6 (µmol/L) |
||
|
Media ajustada (± SE)2 |
-239 (±± 38) |
6 (± 36) |
|
Percentiles (25ªVº, 75ªVº) |
-397, -92 |
-96, 93 |
|
Cambio promedio en porcentaje en Phe en sangre del inicio a la semana 6 |
||
|
Promedio (± SD) |
-29 (± 32) |
3 (± 33) |
|
Percentiles (25ªVº. 75ªVº) |
-61, -11 |
-13, 12 |
1 Las concentraciones promedio iniciales (BL) mostradas en esta tabla representan el promedio de 3 concentraciones de tratamiento previo (semana 2, semana 1 y semana 0). El tratamiento con KUVAN® o placebo empezó en la semana 0.
2 p < 0.001, media ajustada y error estándar de un modelo de ANCOVA, con el cambio en la concentración sanguínea de Phe del inicio a la semana 6 como la variable de respuesta, así como ambos grupos de tratamiento y la concentración sanguínea inicial de Phe como covariables.
El cambio en Phe en sangre se advirtió en el grupo tratado con KUVAN® en la semana 1 y se mantuvo hasta la semana 6 (figura 1).
Figura 1. Concentración promedio de fenilalanina (Phe) en sangre a través del tiempo1
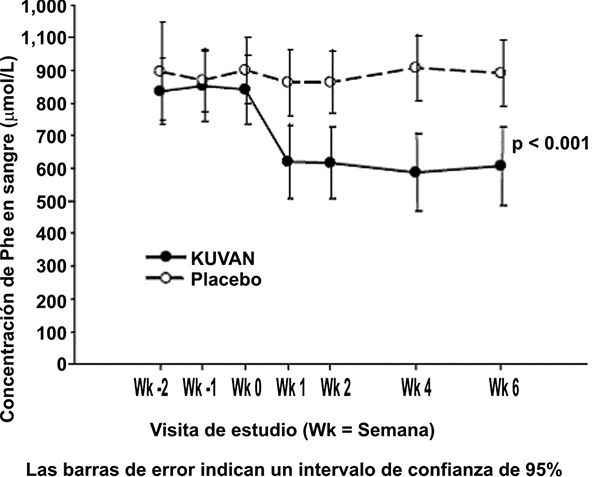
El estudio 3 fue multicéntrico, abierto y de extensión en el cual 80 pacientes quienes respondieron al tratamiento con KUVAN® en el estudio 1 y completaron el estudio 2 experimentaron 6 semanas de titulación de la dosis forzada con 3 dosis diferentes de KUVAN®. Los tratamientos consistieron de 3 cursos consecutivos de 2 semanas de KUVAN® en dosis de 5, luego 20 y luego 10 mg/kg/día. La concentración de Phe en sangre fue monitoreada después de 2 semanas de tratamiento en cada concentración de dosis. Al inicio, la Phe sanguínea promedio (± SD) fue 844 (± 398) µmol/L. Al final del tratamiento con 5, 10 y 20 mg/kg/día, las concentraciones sanguíneas promedio (± SD) de Phe fueron de 744 (± 384) µmol/L, 640 (± 382) µmol/L y 581 (± 399) µmol/L, respectivamente (tabla 2).
Tabla 2. Resultados de Phe en sangre de la titulación de la dosis forzada en el estudio 3
|
Concentración de dosis de KUVAN® (mg/kg/día) |
Núm de pacientes |
Concentración sanguínea promedio (± SD) de Phe (µmol/L) |
Cambios promedio (± SD) en la concentración sanguínea de Phe desde la semana 0 (µmol/L) |
|
Inicio (sin tratamiento) |
80 |
844 (± 398) |
- |
|
5 |
80 |
744 (± 384) |
-100 (± 295) |
|
10 |
80 |
640 (± 382) |
-204 (± 303) |
|
20 |
80 |
581 (± 399) |
-263 (± 318) |
El estudio 4 fue multicéntrico, con 90 niños con PKU de 4 a 12 años de edad, quienes estaban en dietas restringidas en Phe y tenían concentraciones sanguíneas de Phe < 480 µmol/L en la selección. Todos los pacientes fueron tratados de manera abierta con KUVAN® 20 mg/kg/día por 8 días. La respuesta al KUVAN® se definió como una disminución > 30% de Phe en sangre desde el inicio al día 8. En el día 8, 50 pacientes (56%) tuvieron una disminución > 30% de Phe en sangre.
Farmacocinética: Los estudios con voluntarios sanos han mostrado una absorción comparable del dihidrocloruro de sapropterina cuando las tabletas se disuelven en agua o jugo de naranja y se toman en ayunas. La administración de tabletas disueltas después de una comida alta en grasas y calorías produjo incrementos medios en Cmáx. de 84% y un AUC de 87% (disueltos en agua). Sin embargo, se observó una amplia variabilidad en los valores de sujetos individuales para la Cmáx. y AUC en los diferentes modos de administración y condiciones de alimentación. En los estudios clínicos de KUVAN®, se administró el fármaco en la mañana como tableta disuelta sin importar los alimentos.
La vida media de eliminación promedio en pacientes con PKU fue de aproximadamente 6.7 horas (intervalo de 3.9 a 17 horas), comparable con los valores observados en sujetos sanos (intervalo de 3.0 a 5.3 horas).
Un análisis farmacocinético de la población con sapropterina que incluyó a pacientes con edades entre 9 y 49 años no mostró efectos sobre la farmacocinética del dihidrocloruro de sapropterina. No se ha estudiado la farmacocinética en pacientes menores de 9 años y mayores de 49 años.
Absorción: La sapropterina es absorbida después de la administración oral de la tableta disuelta, y la concentración sanguínea máxima (Cmáx.) es alcanzada 3 o 5 horas después de la administración en estado de ayunas .
El grado y la extensión de la absorción de la sapropterina es influenciada por la comida. La absorción de la sapropterina es mayor después de una comida alta en grasas y alta en calorías comparada con la absorción en ayunas , resultando en promedio en concentraciones sanguíneas máximas 40-85% mayores alcanzadas de 4 a 5 horas después de la administración .
La biodisponibilidad absoluta o biodisponibilidad para humanos después de la administración oral no se conoce.
Distribución: En estudios no clínicos, la sapropterina se distribuyó primariamente en los riñones, glándulas adrenales e hígado, medida por los niveles de las concentraciones de biopterina total y reducida.
En ratas, después de la administración de sapropterina marcada radiactivamente, se encontró que radiactividad se distribuye en el feto. La excreción de biopterina total en la leche fue demostrada en ratas por vía intravenosa. No se observó un incremento en las concentraciones totales de biopterina en los fetos o en la leche después de la administración de 10 mg/kg de dihidrocloruro de sapropterina.
Biotransformación: El dihidrocloruro de sapropterina es primariamente metabolizado en el hígado a dihidrobiopterina y biopterina. Ya que el dihidrocloruro de sapropterina es una versión sintética de la 6R-BH4 natural, puede ser razonable anticipar que sufra el mismo metabolismo, incluyendo la 6R-BH4.
Eliminación: Después de la administración intravenosa en ratas, el dihidrocloruro de sapropterina es principalmente excretado en la orina. Después de la administración oral es principalmente eliminado a través de las heces, mientras que una pequeña proporción es excretado en la orina.
CONTRAINDICACIONES: Contraindicaciones a la sapropterina o a cualquiera de los excipientes.
RESTRICCIONES DE USO DURANTE EL EMBARAZO Y LA LACTANCIA:
Embarazo categoría C.
No utilizar en mujeres embarazadas .
A las mujeres que se exponen a KUVAN® durante el embarazo se les pide que ingresen en el registro de pacientes de KUVAN®.
Se han llevado a cabo estudios de teratogenicidad con sapropterina en ratas con dosis orales de hasta 400 mg/kg/día (aproximadamente 3 veces la dosis humana máxima recomendada de 20 mg/kg/día, con base en el área de superficie corporal) y en conejos con dosis orales de hasta 600 mg/kg/día (aproximadamente 10 veces la dosis humana máxima recomendada, con base en el área de superficie corporal). No se encontraron indicios claros de actividad teratogénica en ninguna de las especies. Sin embargo, en el estudio de teratogenicidad en conejos, se observó un incremento (no estadísticamente significativo) en la incidencia de holoprosencefalia en la dosis de 600 mg/kg/día en comparación con los controles.
No existen estudios adecuados y bien controlados de KUVAN® en mujeres embarazadas. Debido a que los estudios de reproducción animal no siempre son un elemento de predicción para la respuesta en humanos, este fármaco deberá usarse durante el embarazo sólo si fuera claramente necesario. Un estudio de 468 embarazos y 331 nacidos vivos en mujeres afectadas por PKU (estudio de colaboración de fenilcetonuria materna, Rouse 1997) demostró que concentraciones de Phe no controladas superiores a 600 µmol/L están asociadas con una muy alta incidencia de dimorfismo neurológico cardiaco y facial, y anormalidades del crecimiento. Un buen control dietario de la concentración de Phe durante el embarazo es esencial para reducir la incidencia de efectos teratogénicos inducidos por la Phe.
No existen datos clínicos sobre exposiciones durante el embarazo. Los estudios en animales no indican efectos peligrosos directos o indirectos con respecto al embarazo, desarrollo fetal/embrionario, parto o desarrollo posnatal.
Los niveles sanguíneos de fenilalanina maternos deben ser estrictamente controlados durante el embarazo. Si los niveles de fenilalanina maternos no son estrictamente controlados antes y durante el embarazo, esto podría ser peligroso para la madre y para el feto. Una dieta restringida en fenilalanina supervisada por el médico previo a y durante el embarazo es el tratamiento de primera elección en este grupo de pacientes. El uso de KUVAN® debe ser considerado solamente si el manejo estricto del régimen dietético no reduce adecuadamente los niveles sanguíneos de fenilalanina.
Parto: Se desconocen los efectos de KUVAN® sobre el parto en mujeres embarazadas.
Madres en lactancia: La sapropterina es excretada en la leche de ratas lactando tratadas por vía intravenosa, pero no por vía oral. No se sabe si la sapropterina se excreta en la leche humana. Debido al potencial de la sapropterina para reacciones adversas serias en lactantes y debido al potencial para la tumorigenicidad mostrada por la sapropterina en el estudio de carcinogenicidad en ratas, se debe tomar la decisión de si se debe suspender la lactancia o suspender la administración del fármaco, tomando en cuenta la importancia del fármaco para la madre.
No se sabe si la sapropterina o sus metabolitos son excretados en la leche humana. KUVAN® no debe ser utilizado durante la lactancia.
REACCIONES SECUNDARIAS Y ADVERSAS:
Experiencia de estudios clínicos con PKU: En estudios clínicos, se administró KUVAN® a 579 pacientes con PKU en dosis que variaron de 5 a 20 mg/kg/día y con duraciones de tratamiento que variaron de 1 a 30 semanas. La edad de los pacientes variaba de 4 a 49 años. La población de pacientes estaba distribuida casi uniformemente en sexo y aproximadamente 95% de los pacientes eran caucásicos.
Las reacciones adversas más serias durante la administración de KUVAN® (independientemente de la relación con el tratamiento) fueron gastritis, lesión en médula espinal, infección por estreptococos, carcinoma testicular e infección en vías urinarias. Se observó neutropenia de leve a moderada durante la administración de KUVAN® en 24 de los 579 pacientes (4%). Las más comunes (³ 4 % de pacientes tratados con KUVAN®) en todos los estudios (n = 579) fueron cefalea, diarrea, dolor abdominal, infección en las vías respiratorias superiores, dolor faringolaríngeo, vómito y náuseas.
Los datos descritos a continuación reflejan la exposición de 74 pacientes con PKU a KUVAN® con dosis de 10 a 20 mg/kg/día durante 6 a 10 semanas en 2 estudios clínicos doble ciego placebo controlados. La incidencia total de las reacciones adversas en pacientes que recibieron KUVAN® fue similar a la registrada con pacientes que recibieron placebo.
Debido a que los estudios fueron conducidos bajo condiciones variadas, las tasas de reacciones adversas pudieran no predecir las tasas observadas en pacientes en la práctica clínica. La tabla 1 enumera las reacciones adversas surgidas durante el tratamiento (independientemente de la relación) que se presentaron en al menos 4% de los pacientes tratados con KUVAN® en los estudios clínicos doble ciego controlados con placebo que se describieron anteriormente. Se clasificó la frecuencia de reacciones adversas registrada en términos del MedDRA (tabla 3).
Tabla 3. Resumen de reacciones adversas por término preferido que se presentaron en £ 4% de los pacientes en estudios clínicos controlados con KUVAN®
|
Tratamiento |
||
|
KUVAN® |
Placebo |
|
|
Pacientes tratados |
n = 74 |
n = 59 |
|
Término preferido |
n (%) |
n (%) |
|
Cualquier reacción adversa |
47 (64) |
42 (71) |
|
Cefalea |
11 (15) |
8 (14) |
|
Infección de las vías respiratorias superiores |
9 (12) |
14 (24) |
|
Rinorrea |
8 (11) |
0 |
|
Dolor faringolaríngeo |
7 (10) |
1 (2) |
|
Diarrea |
6 (8) |
3 (5) |
|
Vómito |
6 (8) |
4 (7) |
|
Tos |
5 (7) |
3 (5) |
|
Pirexia |
5 (7) |
4 (7) |
|
Contusión |
4 (5) |
1 (2) |
|
Dolor abdominal |
4 (5) |
5 (8) |
|
Exantema |
4 (5) |
4 (7) |
|
Congestión nasal |
3 (4) |
0 |
En estudios clínicos abiertos sin grupo control en los cuales los pacientes recibieron KUVAN® en dosis de 5 a 20 mg/kg/día, las reacciones adversas fueron similares en tipo y frecuencia a las registradas en los estudios clínicos doble ciego controlados con placebo.
Otras reacciones adversas dentro de las enfermedades del metabolismo y trastornos de la nutrición es la hipofenilalaninemia.
Rebote, definido como un incremento en los niveles sanguíneos de fenilalanina sobre los niveles pretratamiento, puede ocurrir al cesar el tratamiento.
Experiencia en seguridad de estudios clínicos para indicaciones diferentes a PKU: Aproximadamente 800 voluntarios sanos y pacientes con trastornos diferentes a PKU, algunos de los cuales presentaban trastornos neurológicos subyacentes o enfermedad cardiovascular, recibieron diferentes formulaciones del mismo principio activo (sapropterina) en 19 estudios clínicos controlados y no controlados. En estos estudios clínicos, los sujetos recibieron sapropterina en dosis que variaron de 1 a 20 mg/kg/día por periodos de exposición de 1 día a 2 años. Las reacciones adversas serias y graves (independientemente de la relación) durante la administración de sapropterina fueron convulsiones, exacerbación de las convulsiones (véase Reacciones secundarias y adversas y Precauciones generales), mareo, hemorragia gastrointestinal, hemorragia posterior al procedimiento, cefalea, irritabilidad, infarto del miocardio, sobre estimulación e insuficiencia respiratoria. Las reacciones adversas comunes fueron cefalea, edema periférico, artralgia, poliuria, agitación, mareo e infección de las vías respiratorias superiores.
Experiencia posterior a la comercialización: Se han identificado las siguientes reacciones adversas durante un programa de vigilancia de seguridad en Japón después de 10 años de la aprobación de otra formulación del mismo ingrediente activo (sapropterina). Este programa de vigilancia de seguridad fue realizado en 30 pacientes, 27 de los cuales presentaban trastornos diferentes a PKU y una condición neurológica subyacente. Las reacciones adversas más comunes fueron convulsiones y exacerbación de las convulsiones en 3 de los pacientes sin PKU (véase Reacciones secundarias y adversas y Precauciones generales) y un incremento en la gamma-glutamiltransferasa (GGT) en 2 de los pacientes sin PKU.
PRECAUCIONES EN RELACIÓN CON EFECTOS DE CARCINOGÉNESIS, MUTAGÉNESIS, TERATOGÉNESIS Y SOBRE LA FERTILIDAD: Se realizó un estudio de carcinogenicidad de 2 años con ratas F-344 y un estudio de carcinogenicidad de 78 semanas con ratones CD-1.
En el estudio de carcinogenicidad oral de 104 semanas en ratas se usaron dosis de sapropterina de 25, 80 y 250 mg/kg/día (0.2, 0.7 y 2 veces la dosis humana máxima recomendada de 20 mg/kg/día, respectivamente, con base en el área de superficie corporal). En el estudio de carcinogenicidad oral de 78 semanas en ratones se usaron dosis de sapropterina de 25, 80 y 250 mg/kg/día (0.1, 0.3 y 2 veces la dosis humana máxima recomendada de 20 mg/kg/día, respectivamente, con base en el área de superficie corporal). En el estudio de carcinogenicidad en ratas de 2 años, se observó un incremento estadísticamente significativo en la incidencia de feocromocitoma suprarrenal benigno en ratas macho tratadas con la dosis de 250 mg/kg/día (aproximadamente 2 veces la dosis humana máxima recomendada, con base en el área de superficie corporal) en comparación con las ratas tratadas con el vehículo. El estudio de carcinogenicidad en ratones no mostró indicios de un efecto carcinogénico, pero el estudio no fue ideal debido a que su duración fue de 78 semanas en lugar de 104 semanas.
La sapropterina fue genotóxica en una prueba de Ames in vitro en concentraciones de 625 µg (TA98) y 5,000 µg (TA100) por placa, sin activación metabólica. Sin embargo, no se observó genotoxicidad en la prueba de Ames in vitro con activación metabólica. La sapropterina fue genotóxica en el ensayo de aberración cromosómica in vitro en células pulmonares de hámster chino en concentraciones de 0.25 y 0.5 mM. La sapropterina no fue mutagénica en el ensayo con micronúcleos in vivo en ratones a dosis de hasta 2,000 mg/kg/día (aproximadamente 8 veces la dosis humana máxima recomendada de 20 mg/kg/día, con base en el área de superficie corporal).Se observó que la sapropterina, en dosis orales de hasta 400 mg/kg/día (aproximadamente 3 veces la dosis humana máxima recomendada, con base en el área de superficie corporal) no tuvo efectos sobre la fertilidad ni función reproductiva en ratas macho y hembra.
Se han llevado a cabo estudios de teratogenicidad con sapropterina en ratas con dosis orales de hasta 400 mg/kg/día (aproximadamente 3 veces la dosis humana máxima recomendada de 20 mg/kg/día, con base en el área de superficie corporal) y en conejos con dosis orales de hasta 600 mg/kg/día (aproximadamente 10 veces la dosis humana máxima recomendada, con base en el área de superficie corporal). No se encontraron indicios claros de actividad teratogénica en ninguna de las especies. Sin embargo, en el estudio de teratogenicidad en conejos, se observó un incremento (no estadísticamente significativo) en la incidencia de holoprosencefalia en la dosis de 600 mg/kg/día en comparación con los controles.
No existen estudios adecuados y bien controlados de KUVAN® en mujeres embarazadas. Debido a que los estudios de reproducción animal no siempre son un elemento de predicción para la respuesta en humanos, este fármaco deberá usarse durante el embarazo sólo si fuera claramente necesario. Un estudio de 468 embarazos y 331 nacidos vivos en mujeres afectadas por PKU (estudio de colaboración de fenilcetonuria materna, Rouse 1997) demostró que concentraciones de Phe no controladas superiores a 600 µmol/L están asociadas con una muy alta incidencia de dimorfismo neurológico cardiaco y facial, y anormalidades del crecimiento. Un buen control dietario de la concentración de Phe durante el embarazo es esencial para reducir la incidencia de efectos teratogénicos inducidos por la Phe.
INTERACCIONES MEDICAMENTOSAS Y DE OTRO GÉNERO: No se llevaron a cabo estudios de interacciones medicamentosas. Pacientes masculinos bajo tratamiento con sildenafil pueden tener interacciones con KUVAN®.
ALTERACIONES EN LOS RESULTADOS DE PRUEBAS DE LABORATORIO: Ninguna reportada.
PRECAUCIONES GENERALES:
Monitoreo de la concentración de Phe en sangre durante el tratamiento: El tratamiento con KUVAN® debe ser dirigido por médicos experimentados en el manejo de PKU. Las elevaciones prolongadas en la concentración de Phe sanguínea en pacientes con PKU pueden producir daño neurológico grave, incluido retraso mental grave, microcefalia, lenguaje retardado, convulsiones y anormalidades conductuales. Esto se puede presentar incluso si los pacientes están tomando KUVAN®, pero no están controlando de manera adecuada su concentración de Phe sanguínea dentro del intervalo objetivo recomendado.
No se han realizado estudios a largo plazo con resultados neurocognitivos con tratamiento con KUVAN®. A la inversa, las concentraciones prolongadas de Phe sanguínea demasiado bajas se han asociado con el catabolismo y desdoblamiento de proteínas. Se requiere el manejo activo de la ingesta de Phe en la dieta, mientras se toma KUVAN® para asegurar un adecuado control de la Phe y un equilibrio nutricional.
Se recomienda consultar al médico en caso de una enfermedad concomitante debido a que los niveles de fenilalanina sanguíneos se pueden incrementar.
Identifique a los pacientes que no responden al tratamiento con KUVAN®: No todos los pacientes con PKU responden al tratamiento con KUVAN®. En estudios clínicos, aproximadamente 20 a 56% de los pacientes con PKU responden al tratamiento con KUVAN®. No se puede predeterminar la respuesta al tratamiento mediante pruebas de laboratorio (por ejemplo, pruebas genéticas) y sólo se puede determinar mediante un estudio terapéutico de KUVAN® ( véase Dosis y vía de administración).
Trate a todos los pacientes con una dieta restringida en Phe: Los pacientes con PKU que estén siendo tratados con KUVAN® también deberán llevar una dieta restringida en Phe.El inicio de la terapia con KUVAN® no elimina la necesidad de un monitoreo adecuado por profesionales capacitados para asegurar que el control de la Phe sanguínea se mantiene en el contexto del manejo en curso de la dieta.
Úsese con precaución en pacientes con insuficiencia hepática: No se han evaluado pacientes con insuficiencia hepática en estudios clínicos con KUVAN®. Los pacientes con insuficiencia hepática deberán ser monitoreados cuidadosamente cuando reciban KUVAN® debido a que el daño hepático se ha asociado con un metabolismo de Phe afectado.
Monitoree las reacciones alérgicas: Los pacientes con alguna alergia grave conocida a alguno de los componentes de KUVAN® no deberán tomar KUVAN®.
En estudios clínicos realizados con KUVAN® no se observaron reacciones alérgicas graves. Se deberán considerar los riesgos y beneficios del tratamiento continuo con KUVAN® en pacientes con reacciones alérgicas de leves a moderadas (tal como exantema).
Úsese con precaución cuando se coadministre KUVAN® con medicamentos que se sabe inhiben el metabolismo de folatos: Los fármacos que se sabe afectan el metabolismo de folatos (por ejemplo, metotrexato) y sus derivados deberán usarse con precaución cuando se administre KUVAN® debido a que estos fármacos pueden reducir la concentración de BH4 al inhibir la enzima dihidropteridina reductasa (DHPR).
Úsese con precaución cuando se coadministre KUVAN® con medicamentos que se sabe afectan la vasodilatación mediada por óxido nítrico: Se debe tener precaución con la administración de KUVAN® en pacientes que estén recibiendo fármacos que afecten la vasodilatación mediada por óxido nítrico (por ejemplo, inhibidores de PDE-5 como sildenafil, vardenafil o tadalafil), debido a que el dihidrocloruro de sapropterina y los inhibidores de PDE-5 pueden inducir vasodilatación. El efecto aditivo de la coadministración de sapropterina y el inhibidor de PDE-5 puede llevar a una reducción en la presión arterial. Sin embargo, el uso combinado de estos medicamentos no ha sido evaluado en humanos. En estudios con animales, KUVAN® administrado por vía oral en combinación con un inhibidor de PDE-5 no afectó la presión arterial.
Úsese con precaución cuando se coadministre KUVAN® y levodopa: Se deberá tener precaución con la administración de KUVAN® a pacientes que estén recibiendo levodopa. En un programa de vigilancia de seguridad después de 10 años de la comercialización para una indicación diferente a la PKU que usó otra formulación del mismo ingrediente activo (sapropterina), 3 pacientes con trastornos neurológicos subyacentes presentaron convulsiones, exacerbación de las convulsiones, sobre estimulación o irritabilidad durante la coadministración de levodopa y sapropterina .
Pacientes con insuficiencia renal: No han sido evaluados pacientes con insuficiencia renal en los estudios clínicos. Los pacientes que presentan insuficiencia renal deben ser cuidadosamente monitoreados cuando reciban KUVAN®.
Se recomienda precaución cuando se utilice KUVAN® en pacientes con predisposición a convulsiones: En estudios clínicos con deficiencia de BH4 tratados con una preparación de sapropterina, se observaron convulsiones y exacerbación de las convulsiones. Esto no fue observado en los ensayos clínicos en pacientes con PKU.
Efectos sobre la capacidad para conducir y para el uso de maquinaria: No se han realizado estudios sobre los efectos sobre la capacidad para conducir y para el uso de maquinaria.
DOSIS Y VÍA DE ADMINISTRACIÓN:
Dosificación: La dosis inicial recomendada de KUVAN® es 10 mg/kg/día una vez al día.
La respuesta a la terapia se determina por el cambio en la Phe sanguínea después del tratamiento con KUVAN® a 10 mg/kg/día por un periodo de hasta 1 mes. Se deberá revisar la concentración de Phe sanguínea después de 1 semana de tratamiento con KUVAN® y de manera periódica hasta por 1 mes. Si la Phe sanguínea no se reduce desde el inicio con 10 mg/kg/día, se podrá incrementar la dosis a 20 mg/kg/día. Los pacientes cuya Phe sanguínea no se reduzca después de 1 mes con tratamiento de 20 mg/kg/día son pacientes que no responden, y se deberá suspender el tratamiento con KUVAN® en esos pacientes.
Una vez que se ha establecido el grado de respuesta a KUVAN®, se podrá ajustar la dosificación dentro del intervalo de 5 a 20 mg/kg/día de acuerdo con la respuesta a la terapia. Las dosis de KUVAN® superiores a 20 mg/kg/día no han sido evaluadas en estudios clínicos.
Uso pediátrico: Se han tratado con KUVAN® pacientes pediátricos con PKU con edades de 4 a 16 años en estudios clínicos (véase Estudios clínicos). No se han evaluado en estudios clínicos la seguridad y eficacia de KUVAN® en pacientes pediátricos menores de 4 años de edad. Se recomienda el monitoreo sanguíneo frecuente en la población pediátrica para asegurar un control adecuado de la concentración de Phe sanguínea.
La administración a pacientes menores de 4 años es bajo la responsabilidad del prescriptor.
Uso geriátrico: Los estudios clínicos de KUVAN® en pacientes con PKU no incluyen pacientes con 65 años de edad o mayores. Se desconoce si estos pacientes responden de manera diferente que los pacientes más jóvenes.
Administración: KUVAN® (dihidrocloruro de sapropterina) tabletas debe ser administrado por vía oral con los alimentos para incrementar la absorción, de preferencia a la misma hora cada día. Las tabletas de KUVAN® deben disolverse en 120-240 ml de agua o jugo de manzana y tomarse dentro de 15 minutos de la disolución. Las tabletas pueden tardar algunos minutos en disolverse. Para hacer que se disuelvan más rápido, revuelva o aplástelos. Las tabletas pudieran no disolverse completamente. Los pacientes podrán ver pequeños grumos flotando en la parte superior del agua o jugo de manzana. Esto es normal y los pacientes pueden tragarlos sin problema. Si después de beber la medicina los pacientes siguen viendo grumos, pueden agregar más agua o jugo de manzana para asegurarse de que están tomando toda la medicina. Una dosis perdida debe reponerse lo más pronto posible, pero no se deben tomar 2 dosis en el mismo día.
El método de administración de KUVAN® se podría adaptar a los niños y a los pacientes jóvenes con peso inferior a 20 kilogramos y especialmente al volumen de solución que estos niños pueden consumir. La necesidad de líquidos de un niño es de 100 a 120 ml/kg, que representa cerca de 400 ml/día para un recién nacido de 3.5 kilogramos y por lo tanto de no más de 70 ml por comida. Hasta la edad de 6 meses, la ingesta adecuada recomendada es de 0.7 L/día1.
La posibilidad de administrar las tabletas en pequeños volúmenes de agua se ha analizado en estudios de disolución in vitro para apoyar la administración de las tabletas del dihidrocloruro de sapropterina a los niños y a los pacientes jóvenes que pueden consumir solamente pequeñas cantidades de la solución con el medicamento. Estos datos demuestran una liberación adecuada de 93-96% de la dosis prevista cuando hasta 2 tabletas fueron machacadas en 5 ml de agua.
Además, se han estudiado la liberación y la estabilidad del principio activo de las tabletas aisladas disueltas en 20 o 80 ml de agua. Los resultados demuestran que el dihidrocloruro de sapropterina está disuelto casi totalmente después de 5 minutos en agua y un minuto de revolvimiento y son estables en ambos volúmenes de agua por lo menos dos horas.
En las tablas siguientes se presenta la dosis total de KUVAN® (según el peso corporal y la dosis prescritos, ya sea 10 o 20 mg/kg/día), el número de tabletas que se disolverán en los volúmenes específicos de agua, en rangos de 20 a 80 ml y el volumen exacto de la solución preparada que se administrará, usando un dispositivo de medición exacto.
Estas recomendaciones de administración son para asegurarse de que el volumen total de solución consumido es apropiado a los pacientes muy jóvenes, para asegurar que la dosis exacta de KUVAN® es administrada a los pacientes y para limitar el riesgo de errores durante la administración por los padres o los cuidadores.
La administración para los niños que pesen más de 20 kg es de acuerdo a lo mencionado en el primer párrafo.
1 Campbell SM. Hydratation Needs throughout the Lifespan. J Am Coll Nutr. 2007 Oct; 26(5 Suppl): 585S-587S.
Tabla 4.1. Cálculo y disolución de la dosis de KUVAN®, 10 mg/kg/día
|
Peso (kg) |
Dosis (mg/kg/día) |
Dosis total (mg/día) |
Volumen de disolución (ml) |
Número de tabletas a ser disueltas |
Volumen de solución a ser administrado (ml) |
|
3 |
10 |
30 |
20 |
1 |
6 |
|
3.5 |
10 |
35 |
20 |
1 |
7 |
|
4 |
10 |
40 |
20 |
1 |
8 |
|
4.5 |
10 |
45 |
20 |
1 |
9 |
|
5 |
10 |
50 |
20 |
1 |
10 |
|
5.5 |
10 |
55 |
20 |
1 |
11 |
|
6 |
10 |
60 |
20 |
1 |
12 |
|
6.5 |
10 |
65 |
20 |
1 |
13 |
|
7 |
10 |
70 |
20 |
1 |
14 |
|
7.5 |
10 |
75 |
20 |
1 |
15 |
|
8 |
10 |
80 |
20 |
1 |
16 |
|
8.5 |
10 |
85 |
20 |
1 |
17 |
|
9 |
10 |
90 |
20 |
1 |
18 |
|
9.5 |
10 |
95 |
20 |
1 |
19 |
|
10 |
10 |
100 |
20 |
1 |
20 |
|
11 |
10 |
110 |
40 |
2 |
22 |
|
12 |
10 |
120 |
40 |
2 |
24 |
|
13 |
10 |
130 |
40 |
2 |
26 |
|
14 |
10 |
140 |
40 |
2 |
28 |
|
15 |
10 |
150 |
40 |
2 |
30 |
|
16 |
10 |
160 |
40 |
2 |
32 |
|
17 |
10 |
170 |
40 |
2 |
34 |
|
18 |
10 |
180 |
40 |
2 |
36 |
|
19 |
10 |
190 |
40 |
2 |
38 |
|
20 |
10 |
200 |
40 |
2 |
40 |
Tabla 4.2. Cálculo y disolución de la dosis de KUVAN®, 20 mg/kg/día
|
Peso (kg) |
Dosis (mg/kg/día) |
Dosis total (mg/día) |
Volumen de disolución (ml) |
Número de tabletas a ser disueltas |
Volumen de solución a ser administrado (ml) |
|
3 |
20 |
60 |
20 |
1 |
12 |
|
3.5 |
20 |
70 |
20 |
1 |
14 |
|
4 |
20 |
80 |
20 |
1 |
16 |
|
4.5 |
20 |
90 |
20 |
1 |
18 |
|
5 |
20 |
100 |
20 |
1 |
20 |
|
5.5 |
20 |
110 |
40 |
2 |
22 |
|
6 |
20 |
120 |
40 |
2 |
24 |
|
6.5 |
20 |
130 |
40 |
2 |
26 |
|
7 |
20 |
140 |
40 |
2 |
28 |
|
7.5 |
20 |
150 |
40 |
2 |
30 |
|
8 |
20 |
160 |
40 |
2 |
32 |
|
8.5 |
20 |
170 |
40 |
2 |
34 |
|
9 |
20 |
180 |
40 |
2 |
36 |
|
9.5 |
20 |
190 |
40 |
2 |
38 |
|
10 |
20 |
200 |
40 |
2 |
40 |
|
11 |
20 |
220 |
60 |
3 |
44 |
|
12 |
20 |
240 |
60 |
3 |
48 |
|
13 |
20 |
260 |
60 |
3 |
52 |
|
14 |
20 |
280 |
60 |
3 |
56 |
|
15 |
20 |
300 |
60 |
3 |
60 |
|
16 |
20 |
320 |
80 |
4 |
64 |
|
17 |
20 |
340 |
80 |
4 |
68 |
|
18 |
20 |
360 |
80 |
4 |
72 |
|
19 |
20 |
380 |
80 |
4 |
76 |
|
20 |
20 |
400 |
80 |
4 |
80 |
MANIFESTACIONES Y MANEJO DE LA SOBREDOSIFICACIÓN O INGESTA ACCIDENTAL: En la única sobredosis registrada con KUVAN®, un paciente que participaba en un estudio de 26 semanas recibió una dosis única de 4,500 mg (36 mg/kg) en vez de 2,600 mg (20 mg/kg) en la semana 16.
El paciente registró cefalea leve y mareo leve inmediatamente después de tomar la dosis; ambos síntomas se resolvieron dentro de un lapso de 1 hora sin intervención de tratamiento. Los resultados de las pruebas de laboratorio de función hepática obtenidos inmediatamente después del acontecimiento estuvieron dentro de los límites normales. El paciente suspendió la terapia durante 24 horas y luego reinició la administración de KUVAN® sin registros de signos o síntomas anormales.
Manejo: El tratamiento de la sobredosis debe ser dirigida a los síntomas.
PRESENTACIONES:
Cajas con:
Frasco con 30 tabletas.
Frasco con 120 tabletas.
Frasco con 240 tabletas.
RECOMENDACIONES SOBRE ALMACENAMIENTO: Consérvese a temperatura no mayor de 25°C y en lugar seco. Mantener el frasco perfectamente cerrado para protegerlo de la humedad.
LEYENDAS DE PROTECCIÓN:
Su venta requiere receta médica. Literatura exclusiva para médicos. Mantenga KUVAN® fuera del alcance de los niños. No se use durante el embarazo y la lactancia. Este medicamento sólo podrá ser administrado por médicos especialistas pediatras, pediatrasendocrinólogos, genetistas o neurólogos.
Hecho en Alemania por:
Excella GmbH
Nürnberger Str. 12
90537 Feucht, Alemania
Acondicionado en Uruguay por:
Ares Trading S.A (ATUSA)
Ruta 8 Km 17.500, Zona América
Montevideo, Uruguay
Acondicionado en Alemania por:
Merck KGaA
Frankfurter Strasse 250
D-64293 Darmstadt
Acondicionado en Austria por:
Merck KGaA & Co Werk Spittal
Hösslgasse 20
9800 Spittal/Drau, Austria
Distribuido en México por:
MERCK, S. A. de C. V.
Calle 5 No. 7
Fracc. Industrial Alce Blanco, C.P. 53370
Naucalpan de Juárez, Edo. de México
No. de Oficio: 123300EL870031

