

JEVTANA
CABAZIXATEL
Solución inyectable
1 Caja , 1 Frasco(s) ámpula , 60/1.5 mg/ml
FORMA FARMACÉUTICA Y FORMULACIÓN:
El frasco ámpula contiene:
Cabazitaxel acetona solvato 60 mg
Vehículo cbp 1.5 mL
El frasco ámpula con diluyente contiene:
Etanol al 96% en agua inyectable 4.5 mL
INDICACIONES TERAPÉUTICAS: JEVTANA® en combinación con prednisona o prednisolona, está indicado para el tratamiento de pacientes con cáncer de próstata metastásico refractario a terapia hormonal, previamente tratados con un esquema que contiene docetaxel.
FARMACOCINÉTICA Y FARMACODINAMIA:
Farmacocinética: Se realizó un análisis en la población que participó en el estudio de farmacocinética, que incluyó a 170 pacientes con tumores sólidos avanzados (n = 69), cáncer de mama metastásico (n = 34) y cáncer de próstata metastásico (n = 67). Estos pacientes recibieron dosis de cabazitaxel en el rango de 10 a 30 mg/m2 semanalmente o cada 3 semanas.
Absorción: Después de la administración de cabazitaxel en dosis de 25 mg/m2 por vía intravenosa durante 1 hora a pacientes con cáncer de próstata metastásico (n = 67), el promedio de la Cmáx. fue de 226 ng/ml (coeficiente de variación, CV 107%) y fue alcanzado al final de la infusión de 1 hora (Tmáx). El promedio del ABC fue de 991 ng• h/mL (CV: 34%).
No se observaron desviaciones mayores a la proporcional de la dosis en el rango de 10 a 30 mg/m2 en pacientes con tumores sólidos avanzados (n = 126).
Distribución: El volumen de distribución (Vss) fue de 4870 L (2640 L/m2 en pacientes con una media de área de superficie corporal de 1.84 m2) en el estado estable.
In vitro, la unión de cabazitaxel a las proteínas del suero humano fue del 89 al 92% y no fue saturable hasta 50,000 ng/mL, lo cual cubre la concentración máxima observada en los estudios clínicos. Cabazitaxel se une principalmente a la albúmina sérica humana (82.1%) y lipoproteínas (87.9% a las HDL, 69.8% a las LDL y 55.8% a las VLDL). La relación de la concentración de sangre/plasma in vitro en sangre humana, estuvo en el rango desde 0.90 hasta 0.99, indicando que cabazitaxel es igualmente distribuido entre la sangre y el plasma.
Metabolismo: Cabazitaxel es metabolizado extensamente en el hígado (≥ 95%), principalmente por la isoenzima CYP3A4 (80 a 90%). Cabazitaxel es el principal compuesto circulante en el plasma humano. Se detectaron siete metabolitos en plasma (incluyendo 3 metabolitos activos derivados de la O-desmetilación), contribuyendo el principal metabolito al 5% de la detección del compuesto de origen. Alrededor de 20 metabolitos de cabazitaxel son excretados en la orina y heces humanas.
El riesgo potencial de inhibición (i/Ki) del cabazitaxel in vitro, en concentraciones clínicamente relevantes, es posible hacia fármacos que son un sustrato principalmente para CYP3A. Además, cabazitaxel no indujo isoenzimas CYP (CYP1A, CYP2C y CYP3A) in vitro. No obstante, se debe evitar el uso concomitante de inductores potentes de CYP3A así como de inhibidores potentes de CYP3A junto con cabazitaxel. En caso de que sea necesaria la coadministración de cabazitaxel y de inductores/inhibidores potentes de CYP3A, entonces se deberá considerar disminuir la dosis de cabazitaxel a 25% de la dosis recomendada.
Los estudios de interacción en humanos han demostrado que cabazitaxel (25 mg/m2 administrado en una infusión única de 1 hora) no modifica los niveles plasmáticos de midazolam, un sustrato de CYP3A. Por lo tanto, cabazitaxel no es un inhibidor de CYP3A in vivo.
Eliminación: Después de una infusión IV de 1 hora de (14C)-cabazitaxel en dosis de 25 mg/m2 a pacientes, aproximadamente el 80% de la dosis administrada se eliminó dentro de las siguientes 2 semanas. Cabazitaxel es principalmente excretado en las heces como numerosos metabolitos (76% de la dosis); mientras que la excreción renal de cabazitaxel y sus metabolitos representa menos del 3.7% de la dosis (2.3% como fármaco inalterado en la orina). Seguido de una infusión intravenosa para una hora, la concentración plasmática de cabazitaxel se describe como la farmacocinética de tres compartimentos con alfa, beta y gama, vida media de 4 minutos, 2 horas y 95 horas, respectivamente. Cabazitaxel muestra una depuración del plasma de 48.5 L/h (26.4 L/h/m2 en pacientes con un área de superficie corporal mediana de 1.84 m2).
Poblaciones especiales:
Edad avanzada: En el análisis de farmacocinética de la población de pacientes ≤ 65 años de edad (N = 100) y pacientes de > 65 años (N = 70), la edad no afectó la farmacocinética de cabazitaxel.
Pacientes pediátricos: La seguridad y efectividad de JEVTANA® no ha sido establecida en niños.
Insuficiencia hepática: No se han realizado estudios formales en pacientes con insuficiencia hepática; no obstante, dado que cabazitaxel se metaboliza por vía hepática principalmente, es de esperar un incremento en la exposición a cabazitaxel.
Insuficiencia renal: Cabazitaxel es excretado mínimamente a través de los riñones (2.3% de la dosis). No se han realizado estudios formales de farmacocinética con cabazitaxel en pacientes con insuficiencia renal. Sin embargo, el análisis en la población que participó en el estudio de farmacocinética, realizado en 170 pacientes, incluyó a 14 pacientes con insuficiencia renal moderada (depuración de creatinina en el rango de 30 a 50 ml/min) y 59 pacientes con insuficiencia renal leve (depuración de creatinina en el rango de 50 a 80 ml/min), mostró que la insuficiencia renal leve a moderada no tuvo efectos importantes sobre la farmacocinética de cabazitaxel.
Farmacodinamia:
Mecanismo de acción: Cabazitaxel es un agente antineoplásico que actúa interrumpiendo la red de los microtúbulos en las células. Cabazitaxel se une a la tubulina y promueve el ensamble de la tubulina en los microtúbulos, mientras inhibe simultáneamente su desensamble. Esto lleva a la estabilización de los microtúbulos, lo cual resulta en inhibición de las funciones mitóticas y de interfase celular.
Efectos de farmacodinamia: Cabazitaxel demostró un amplio espectro de actividad antitumoral contra tumores humanos avanzados, xenoimplantes en ratones, incluyendo glioblastomas humanos intracraneales. Cabazitaxel es activo en tumores sensibles a docetaxel. Adicionalmente cabazitaxel demostró actividad en modelos tumorales insensibles a quimioterapia, incluyendo docetaxel.
Eficacia clínica/estudios clínicos: La eficacia y seguridad de JEVTANA® en combinación con prednisona o prednisolona, fue evaluada en un estudio abierto, aleatorizado, multicéntrico, internacional, en pacientes con cáncer de próstata metastásico hormono-refractario, previamente tratado con esquemas que incluían docetaxel.
La supervivencia global (SG) fue el objetivo final primario de eficacia del estudio.
Los objetivos finales secundarios incluyeron el periodo de supervivencia libre de progresión (SLP, definido como el tiempo desde la aleatorización hasta la progresión del tumor, progresión del antígeno prostático específico [APE], progresión del dolor o muerte debida a cualquier causa, lo que ocurriera primero), la tasa de respuesta tumoral basada en los criterios de evaluación de respuesta en tumores sólidos (RECIST), la progresión del APE (definida como un incremento ≥ 25% o > 50% en el APE de pacientes que no responden o que responden, respectivamente), la respuesta del APE (declinación en los niveles séricos de APE de por lo menos 50%), progresión del dolor (evaluado utilizando la escala de Intensidad del dolor presente [IDP] a partir del cuestionario de McGill-Melzack y una evaluación analgésica [EA] y respuesta del dolor [definido como 2 puntos de mayor reducción con respecto al valor promedio del registro basal en la escala de IDP, sin aumento concomitante en la EA, o reducción ≥ 50% en el uso de analgésicos con respecto al promedio de EA en el registro basal, sin aumento concomitante del dolor]).
Un total de 755 pacientes fueron aleatorizados a recibir cabazitaxel 25 mg/m2 por vía intravenosa cada 3 semanas durante un máximo de 10 ciclos con prednisona o prednisolona 10 mg por vía oral diariamente (n = 378) o a recibir mitoxantrona 12 mg/m2 por vía intravenosa cada 3 semanas durante un máximo de 10 ciclos con prednisona o prednisolona 10 mg por vía oral diariamente (n = 377).
Este estudio incluyó a pacientes mayores de 18 años con cáncer de próstata metastásico hormono-refractario, ya fuese medible por los criterios de RECIST o con enfermedad no medible con niveles crecientes del APE o aparición de nuevas lesiones, así como el estado de desempeño de la Eastern Cooperative Oncology Group (ECOG) de 0 a 2. Los pacientes deberían tener una cuenta de neutrófilos > 1,500/mm3, plaquetas > 100,000/mm3, hemoglobina > 10 g/dL, creatinina < 1.5 x arriba del límite normal (ALN), bilirrubina total < 1 x ALN, AST/STGO < 1.5 x ALN y ALT/STGP < 1.5 ALN.
Los pacientes con historia de insuficiencia cardiaca congestiva o infarto del miocardio en los últimos 6 meses o pacientes con arritmias cardiacas no controladas, angina de pecho y/o hipertensión, no se incluyeron en el estudio.
Los datos demográficos, incluyendo edad, raza y estado de desempeño de la ECOG (0 a 2), estuvieron balanceados entre los grupos de tratamiento. En el grupo de JEVTANA®, la edad promedio fue de 68 años, con rango de 46 a 92, y la distribución racial fue del 83.9% caucásicos, 6.9% asiáticos, 5.3% negros y (4%) otros.
La mediana de ciclos en el grupo JEVTANA® fue de 6 ciclos y en el grupo de mitoxantrona fue de 4 ciclos. El porcentaje de pacientes que completaron el tratamiento en el estudio (10 ciclos) fue de 29.4% y 13.5% respectivamente.
La supervivencia global fue más prolongada en el brazo de cabazitaxel, teniendo los pacientes del grupo tratado con cabazitaxel una disminución del 30% en el riesgo de muerte comparada con el brazo de mitoxantrona (índice de riesgo = 0.70, IC 95% (0.59-0.83) (véase la Tabla 1. Eficacia y la Gráfica 1. Supervivencia global).
Tabla 1. Eficacia: Eficacia de JEVTANA® en el tratamiento de pacientes con cáncer de próstata metastásico hormono-refractario (análisis de la población con intención a tratar) – Objetivo Final Primario.
|
JEVTANA® + prednisona n = 378 |
Mitoxantrona + prednisona n = 377 |
|
|
Supervivencia gobal |
||
|
Número de pacientes muertos (%) |
234 (61.9%) |
279 (74%) |
|
Media de supervivencia (meses) (IC 95%) |
15.1 (14.1-16.3) |
12.7 (11.6-13.7) |
|
Índice de riesgo (IR) (IC 95%) |
0.70 (0.59-0.83) |
|
|
Valor de p |
< 0.0001 |
|
IR estimado utilizando el modelo de Cox: un índice de riesgo de menos de 1, favorece a JEVTANA®.
Gráfica 1. de Supervivencia global
Curvas de Kaplan-Meier para la supervivencia Global
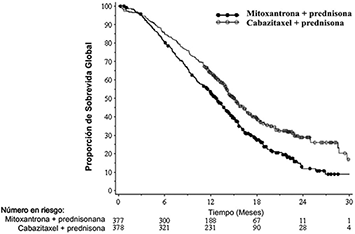
Hubo mejoría en la SLP en el brazo de JEVTANA® en comparación con el brazo de mitoxantrona, 2.8 (2.4-3.0) meses versus 1.4 (12.4-1.7) respectivamente, IR (IC95%) 0.74 (0.65-0.86), p < 0.0001.
Hubo una tasa significativamente más alta de respuesta tumoral, del 14.4% (IC95%: 9.6-19.3) en los pacientes del brazo de JEVTANA®, en comparación con el 4.4% (IC95%: 1.6-7.2) en los pacientes del brazo de mitoxantrona, p = 0.0005.
Los objetivos finales secundarios del APE fueron positivos en el brazo de JEVTANA®. Hubo una media de progresión del APE a los 6.4 meses (IC 95% 5.1-7.3) en los pacientes del grupo de JEVTANA®, en comparación con 3.1 meses (IC95%: 2.2-4.4) en el brazo de mitoxantrona, IR 0.75 meses (IC 95%: 0.63-0.90), p = 0.0010. La respuesta del APE fue del 39.2% en los pacientes del grupo de JEVTANA® (IC 95%: 33.9-44.5) versus 17.8% en los pacientes del grupo de mitoxantrona (IC 95%: 13.7-22.0), p = 0.0002.
Datos de seguridad no clínicos:
Toxicología general:
Efectos sobre el hígado: Se observó hiperplasia de conductos biliares, necrosis arteriolar/periarteriolar y/o necrosis hepatocelular en perros que recibieron la administración de una dosis única (0.25 mg/kg [5 mg/m2]), durante 5 días (0.2 mg/kg [4 mg/m2]) y semanal (0.325 mg/kg [6.5 mg/m2]). En ratas se observó pigmentación de las células de Kupffer y degeneración/regeneración de los conductos biliares, con la dosis letal más alta de 10 mg/kg (60 mg/m2) en un estudio de 10 ciclos en ratas.
Neurotoxicidad: La neurotoxicidad periférica no reversible caracterizada histopatológicamente por degeneración del nervio ciático y de las raíces nerviosas lumbosacras, se observó en ratones después de 10 o 20 semanas de recibir la administración de una dosis única. El nivel de efectos no observables fue de 15 mg/kg (45 mg/m2) después de la administración intravenosa de una dosis única durante 1 hora.
La neurotoxicidad central caracterizada histopatológicamente por necrosis neuronal y/o vacuolización del encéfalo, edema y degeneración axonal en la médula cervical, fue observada en ratones después de recibir la administración de una dosis única por vía intravenosa durante 1 hora, en dosis de 15 mg/kg (45 mg/m2), considerada suficientemente en exceso de la exposición humana máxima. El nivel de efectos no observables fue de 10 mg/kg (30 mg/m2) (aproximadamente 7 veces el ABC de pacientes con cáncer a la dosis recomendada para el humano) después de la administración intravenosa de una dosis única durante 1 hora.
Trastornos oculares: Se observó edema/degeneración de las fibras subcapsulares del cristalino en ratas, durante un estudio de toxicidad de 10 ciclos con 10-20 mg/kg (60-120 mg/m2 [aproximadamente 2 veces el ABC en pacientes con cáncer que reciben la dosis recomendada para el humano]). El nivel de efectos no observables para los hallazgos microscópicos en el cristalino, fue de 5 mg/kg (30 mg/m2) [aproximadamente el ABC de pacientes con cáncer que reciben la dosis recomendada en el humano]). Estos efectos fueron parcialmente reversibles después de 8 semanas.
Evaluación de riesgos ambientales: Los estudios para evaluar riesgos ambientales, indican que JEVTANA®, no tiene efectos ambientales significativos (ver sección DOSIS Y VÍA DE ADMINISTRACIÓN).
CONTRAINDICACIONES:
En pacientes con:
• Historia de reacciones severas de hipersensibilidad a cabazitaxel o a otros fármacos formulados con polisorbato 80.
• Cuenta de neutrófilos < 1,500/mm3.
• Insuficiencia hepática (bilirrubina ≥ 1 x ALN, o AST/SGOT y/o ALT/SGTP ≥ 1.5 x ALN).
RESTRICCIONES DE USO DURANTE EL EMBARAZO Y LA LACTANCIA:
Embarazo: Debido a la potencial exposición vía líquido seminal, los hombres con parejas que tienen potencial para embarazarse, deben utilizar métodos anticonceptivos confiables durante todo el tratamiento y se recomienda continuarlos hasta por 6 meses después de la última dosis de JEVTANA®.
No existen datos sobre el uso de cabazitaxel en mujeres embarazadas. Los estudios en animales han demostrado embriotoxicidad, fetotoxicidad y abortos al ser expuestos a dosis incluso mucho más bajas que las recomendadas para uso humano. Cabazitaxel cruza la barrera placentaria.
JEVTANA® no está recomendado durante el embarazo.
Lactancia: La información de farmacocinética disponible en animales ha demostrado que cabazitaxel es excretado en la leche, al igual que sus metabolitos (ver sección TERATOGENICIDAD). JEVTANA® no se debe administrar durante el periodo de lactancia.
REACCIONES SECUNDARIAS Y ADVERSAS:
Se utilizó la siguiente frecuencia de la CIOMS, cuando fue aplicable: Muy frecuentes ≥ 10%; frecuentes ≥ 1 y < 10%; no frecuentes ≥ 0.1 y < 1%; raros ≥ 0.01 y < 0.1%; muy raros < 0.01, no conocidos (no pueden ser estimados de los datos disponibles).
La seguridad de JEVTANA® en combinación con prednisona o prednisolona, fue evaluada en 371 pacientes con cáncer de próstata metastásico hormono-refractario, en un estudio Fase III, abierto, aleatorizado, controlado. Los pacientes recibieron una media de 6 ciclos de JEVTANA® o 4 de mitoxantrona.
Las reacciones adversas muy comunes de grado 1-4 (≥ 10%) fueron anemia, leucopenia, trombocitopenia, diarrea, fatiga, náusea, vómito, estreñimiento, astenia, dolor abdominal, hematuria, dolor de espalda, anorexia, neuropatía periférica (incluyendo neuropatía sensorial y motora), pirexia, disnea, disgeusia, tos, artralgias y alopecia.
Las reacciones adversas comunes de grado ≥ 3 (≥ 5%) en el grupo de cabazitaxel fueron la neutropenia, leucopenia, anemia, neutropenia febril, diarrea, fatiga y astenia.
La suspensión del tratamiento debido a reacciones adversas al medicamento ocurrió en 68 pacientes (18.3%) en el grupo de cabazitaxel y en 31 pacientes (8.4%) en el grupo de mitoxantrona. La reacción adversa más frecuente que llevó a la suspensión del tratamiento en el grupo de JEVTANA®, fue la neutropenia y la insuficiencia renal.
La mortalidad no relacionada con progresión de la enfermedad dentro de los 30 días después de la última dosis de cabazitaxel fue de 18 (4.9%) pacientes tratados con cabazitaxel y 3 (< 1%) en los tratados con mitoxantrona. La reacción adversa fatal más común en el grupo tratado con cabazitaxel fue la infección (n = 5). La mayoría (4 de 5 pacientes) con infección fatal relacionada a evento adverso en el estudio TROPIC ocurrió después de una sola dosis de cabazitaxel.
Tabla 2. Reacciones adversas: Incidencia de reacciones adversas y anormalidades hematológicas reportadas en pacientes que recibieron JEVTANA® en combinación con prednisona y en pacientes que recibieron mitoxantrona en combinación con prednisona (con una incidencia de por lo menos 2% más elevada en el grupo JEVTANA® en comparación con el grupo mitoxantrona).
|
Sistema corporal |
JEVTANA® 25 mg/m2 cada 3 semanas en combinación con prednisona 10 mg/día N = 371 |
Mitoxantrona 12 mg/m2 cada 3 semanas en combinación con prednisona 10 mg/día n = 371 |
||
|
Todos los grados n (%) |
Grado ¾ n (%) |
Todos los grados n (%) |
Grado ¾ n (%) |
|
|
Trastornos de la sangre y sistema linfático |
||||
|
Neutropeniaa |
347 (93.5%) |
303 (81.7%) |
325 (87.6%) |
215 (58.0%) |
|
Anemiaa |
361 (97.3%) |
39 (10.5%) |
302 (81.4%) |
18 (4.9%) |
|
Leucopeniaa |
355 (95.7%) |
253 (68.2%) |
343 (92.5%) |
157 (42.3%) |
|
Trombocitopeniaa |
176 (47.4%) |
15 (4%) |
160 (43.1%) |
6 (1.6%) |
|
Neutropenia febril |
--- |
28 (7.5%) |
--- |
5 (1.3%) |
|
Trastornos gastrointestinales |
||||
|
Diarrea |
173 (46.6%) |
23 (6.2%) |
39 (10.5%) |
1 (0.3%) |
|
Náusea |
127 (34.2%) |
7 (1.9%) |
85 (22.9%) |
1 (0.3%) |
|
Vómito |
84 (22.6%) |
7(1.9%) |
38 (10.2%) |
0 |
|
Estreñimiento |
76 (20.5%) |
4 (1.1%) |
57(15.4%) |
2 (0.5%) |
|
Dolor abdominal |
43(11.6%) |
7(1.9%) |
13 (3.5%) |
0 |
|
Dispepsia |
25 (6.7%) |
0 |
6 (1.6%) |
0 |
|
Dolor abdominal alto |
20 (5.4%) |
0 |
5(1.3%) |
0 |
|
Hemorroides |
14 (3.8%) |
0 |
3 (0.8%) |
0 |
|
Enfermedad por reflujo gastroesofágico |
12 (3.2%) |
0 |
3 (0.8%) |
0 |
|
Trastornos generales y reacciones en el sitio de administración |
||||
|
Fatiga |
136 (36.7%) |
18 (4.9%) |
102 (27.5%) |
11 (3.0%) |
|
Astenia |
76 (20.5%) |
17 (4.6%) |
46 (12.4%) |
9 (2.4%) |
|
Pirexia |
45 (12.1%) |
4 (1.1%) |
23 (6.2%) |
1 (0.3%) |
|
Inflamación de mucosas |
22 (5.9%) |
1 (0.3%) |
10 (2.7%) |
1 (0.3%) |
|
Trastornos músculo-esqueléticos y del tejido conectivo |
||||
|
Dolor de espalda |
60 (16.2%) |
14(3.8%) |
45 (12.1%) |
11 (3.0%) |
|
Artralgias |
39 (10.5%) |
4 (1.1%) |
31 (8.4%) |
4 (1.1%) |
|
Espasmos musculares |
27 (7.3%) |
0 |
10 (2.7%) |
0 |
|
Trastornos del metabolismo y nutrición |
||||
|
Anorexia |
59 (15.9%) |
3 (0.8%) |
39 (10.5%) |
3 (0.8%) |
|
Deshidratación |
18 (4.9%) |
8 (2.2%) |
10 (2.7%) |
3 (0.8%) |
|
Trastornos renales y del tracto urinario |
||||
|
Hematuria |
62 (16.7%) |
7 (1.9%) |
14 (3.8%) |
2 (0.5%) |
|
Disuria |
25 (6.7%) |
0 |
5 (1.3%) |
0 |
|
Incontinencia urinaria |
9 (2.4%) |
0 |
1 (0.3%) |
0 |
|
Insuficiencia renal aguda |
8 (2.2%) |
6 (1.6%) |
0 |
0 |
|
Trastornos respiratorios, torácicos y mediastinales |
||||
|
Disnea |
44 (11.9%) |
5 (1.3%) |
17 (4.6%) |
3 (0.8%) |
|
Tos |
40 (10.8%) |
0 |
22 (5.9%) |
0 |
|
Trastornos de la piel y tejido subcutáneo |
||||
|
Alopecia |
37 (10.0%) |
0 |
18 (4.9%) |
0 |
|
Infecciones e infestaciones |
||||
|
Infección del tracto urinario |
27 (7.3%) |
4 (1.1%) |
11 (3.0%) |
3 (0.8%) |
|
Trastornos del sistema nervioso |
||||
|
Disgeusia |
41 (11.1%) |
0 |
15 (4.0%) |
0 |
|
Neuropatía periférica |
30 (8.1%) |
2 (0.5%) |
4 (1.1%) |
1 (0.3%) |
|
Vértigo |
30 (8.1%) |
0 |
21 (5.7%) |
2 (0.5%) |
|
Cefalea |
28 (7.5%) |
0 |
19 (5.1%) |
0 |
|
Neuropatía sensorial periférica |
20 (5.4%) |
1 (0.3%) |
5 (1.3%) |
0 |
|
Trastornos vasculares |
||||
|
Hipotensión |
20 (5.4%) |
2 (0.5%) |
9 (2.4%) |
1 (0.3%) |
a Con base en valores de laboratorio.
Eventos adversos por órganos o sistemas: Alteraciones generales y reacciones en el sitio de la administración.
El edema periférico, observado con una incidencia del 9.2% en todos los grados y una incidencia de 0.5% y 0.3% en grado ≥ 3 en el grupo de cabazitaxel y mitoxantrona respectivamente.
Dolor se observó con una incidencia de 5.4% y 4.9% en todos los grados y fue de 1.1% y 1.9% en grado ≥ 3 en el grupo de cabazitaxel y mitoxantrona, respectivamente.
Neutropenia y eventos clínicos derivados: La incidencia de neutropenia grado ≥ 3 con base en los valores de laboratorio fue del 81.7%. La incidencia de reacciones adversas de neutropenia clínica y neutropenia febril fueron respectivamente del 21.3% y 7.5%. La neutropenia fue la reacción adversa más frecuente que llevó a la suspensión del tratamiento (2.4%). Las complicaciones neutropénicas incluyeron infecciones neutropénicas (0.5%), sepsis neutropénica (0.8%), así como choque séptico (1.1%), el cual en algunos casos tuvo desenlace fatal.
El uso de G-CSF ha demostrado limitar la incidencia y severidad de la neutropenia (ver secciones DOSIS Y VÍA DE ADMINISTRACIÓN y PRECAUCIONES GENERALES).
Trastornos cardiacos y arritmias: Todos los grados de los eventos cardiacos fueron más comunes en el grupo de cabazitaxel, en el cual 6 pacientes (1.6%) presentaron arritmia cardiaca grado ≥ 3. La incidencia de taquicardia en el grupo de cabazitaxel fue de 1.6%, ninguno de los cuales fue grado ≥ 3. La incidencia de fibrilación auricular fue de 1.1% en el grupo de cabazitaxel.
En un estudio multicéntrico, abierto, de brazo único, 94 pacientes con tumores sólidos recibieron cabazitaxel a dosis de 25 mg/m2 cada 3 semanas. Las evaluaciones durante el ciclo 1 en el día 1 hasta las 24 horas siguientes no mostraron cambios > 10 mseg en el intervalo QTc promedio a partir del valor basal. No obstante, debido a las limitantes en el diseño del estudio, no se pueden excluir incrementos pequeños en el intervalo QTc (< 10 mseg) debido a la administración de cabazitaxel.
Trastornos renales y del tracto urinario: Se observó insuficiencia renal en 2.2% en todos los grados y 1.6% en grados ≥ 3 en el grupo de cabazitaxel.
Investigaciones: La incidencia de anemia grado ≥ 3, elevación de AST/SGOT, elevación de ALT/SGTP y elevación de bilirrubina, según los valores de laboratorio fue de 10.6%, 0.9%, 1.1% y 0.6%, respectivamente.
Se observó pérdida de peso en 8.6% y 7.5% en todos los grados y 0% y 0.3% en grados ≥ 3 en el grupo de cabazitaxel y mitoxantrona, respectivamente.
Población de edad avanzada: De los 371 pacientes tratados con JEVTANA® en el estudio de cáncer de próstata, 240 pacientes eran mayores de 65 años, incluyendo a 70 pacientes mayores de 75 años. Se reportaron las siguientes reacciones adversas con tasas ≥ 5% en pacientes mayores de 65 años en comparación con pacientes más jóvenes: fatiga (40.4% versus 29.8%), neutropenia (24.2% versus 17.6%), astenia (23.8% versus 14.5%), pirexia (14.6% versus 7.6%), mareo (10.0 versus 4.6%), infección del tracto urinario (9.6% versus 3.1%) y deshidratación (6.7% versus 1.5%), respectivamente.
La incidencia de las siguientes reacciones adversas de grado ≥ 3 fue más elevada en pacientes ≥ 65 años de edad, en comparación con pacientes más jóvenes: neutropenia según los valores (86.3% versus 73.3%), neutropenia clínica (23.8% versus 16.8%) y neutropenia febril (8.3% versus 6.1%)
Habilidad para conducir o utilizar máquinas: No se han realizado estudios sobre los efectos en la capacidad para conducir o utilizar máquinas. Sin embargo, con base en el perfil de seguridad, JEVTANA® puede tener moderada influencia en la capacidad para conducir y utilizar maquinas, ya que podría causar fatiga y vértigo. Los pacientes deben ser advertidos de no manejar ni utilizar máquinas si experimentan estas reacciones adversas durante el tratamiento.
PRECAUCIONES EN RELACIÓN CON EFECTOS DE CARCINOGÉNESIS, MUTAGÉNESIS, TERATOGÉNESIS Y SOBRE LA FERTILIDAD:
Carcinogenicidad: No se han realizado estudios a largo plazo en animales para evaluar el potencial carcinogénico de cabazitaxel.
Mutagenicidad: Cabazitaxel tuvo resultados negativos en la prueba mutagénica de reversión bacteriana (prueba de AMES).
Genotoxicidad: Cabazitaxel no resultó clastogénico en una prueba in vitro en linfocitos humanos (no produjo inducción de aberraciones cromosómicas estructurales pero aumentó el número de células poliploides) e indujo un aumento de micronúcleos en la prueba in vivo en ratas que recibieron dosis de ≥ 0.5 mg/kg. Sin embargo, estos hallazgos de genotoxicidad son inherentes a la actividad farmacológica del compuesto (inhibición de la despolimerización de la tubulina).
Teratogenicidad: Los estudios preclínicos en ratas y conejos demostraron que cabazitaxel es embriotóxico, fetotóxico y abortivo. Cuando ratas hembra recibieron JEVTANA® por vía intravenosa una vez al día a partir de los 6 a 17 días de la gestación, se observó toxicidad embriofetal con exposiciones más bajas que aquellas utilizadas en humanos que recibieron dosis clínicamente relevantes de cabazitaxel; tal toxicidad consistió en muerte fetal y disminución del peso fetal promedio, asociado con un retraso de la osificación del esqueleto.
Cabazitaxel no produjo anomalías fetales en conejos. Cabazitaxel cruza la barrera placentaria en ratas.
Después de la administración intravenosa de una dosis única de (14C)-cabazitaxel de 0.08 mg/kg a ratas hembra en lactación, menos del 1.5% de la dosis se encontró en la leche materna en las siguientes 24 horas.
Alteración de la fertilidad: Se desconocen los efectos de JEVTANA® sobre la fertilidad. Los estudios en animales mostraron que cabazitaxel afecta al sistema reproductivo de ratas y perros macho.
Cabazitaxel no afectó el desempeño de copulación ni la fertilidad de ratas macho tratadas con dosis de 0.05, 0.1 y 0.2 mg/kg/día. Sin embargo, en estudios de ciclos múltiples, se observó degeneración de vesículas seminales y atrofia de túbulos seminíferos en los testículos de ratas tratadas por vía intravenosa con cabazitaxel en dosis de 5 mg/kg y mínima degeneración testicular en perros (necrosis celular epitelial mínima aislada en el epidídimo) tratados con la dosis de 0.5 mg/kg. La exposición en animales fue similar o más baja que aquellas dosis clínicamente relevantes de cabazitaxel en humanos.
INTERACCIONES MEDICAMENTOSAS Y DE OTRO GÉNERO: Cabazitaxel es metabolizado principalmente por CYP3A.
Los inhibidores potentes de CYP3A (p. ej. ketoconazol, itraconazol, claritromicina, atazanavir, indinavir, nefazodona, nelfinavir, ritonavir, saquinavir, telitromicina, voriconazol) o inductores potentes de CYP3A (p. ej. rifampicina, carbamacepina, fenobarbital o fenitoína) alteran el metabolismo de cabazitaxel. Por ende se debe evitar el uso concomitante de inductores potentes de CYP3A, así como de inhibidores potentes de CYP3A junto con cabazitaxel. En caso de que sea necesaria la coadministración de cabazitaxel y de un inhibidor potente de CYP3A, entonces se deberá realizar un monitoreo estrecho de la toxicidad y considerar disminuir la dosis de cabazitaxel en un 25%, aproximadamente, de la dosis habitual recomendada.
Se debe evitar la coadministración de inductores potentes de CYP3A, ya que pueden disminuir la exposición a cabazitaxel.
La administración de ketoconazol (400 mg una vez/día), un inhibidor potente de CYP3A, resultó en la disminución de 20% del aclaramiento de cabazitaxel correspondiente a 25% de incremento en el ABC (área bajo la curva). La administración concomitante de aprepitant, un inhibidor moderado de CYP3A, no ejerció efecto alguno sobre el aclaramiento o la exposición a cabazitaxel. La administración repetida de rifampicina (600 mg una vez/día), un inductor potente de CYP3A, resultó en el incremento de 21% de aclaramiento de cabazitaxel correspondiente a una disminución de 17% en ABC.
In vitro, cabazitaxel no inhibe las proteínas de resistencia multi-fármacos (por su siglas en inglés, MRP: MRP1 y MRP2; ni a los transportadores de cationes orgánicos (OCT1). Cabazitaxel sí inhibe, in vitro, el transporte de la glucoproteína P (P-gp) (digoxina, vinblastina), así como a las proteínas de resistencia de cáncer de mama (por su siglas en inglés, BCRP (metotrexate) y a los polipéptidos transportadores de aniones orgánicos (OATP1B3) CCK8 a concentraciones de al menos 15 veces respecto a lo observado en la práctica clínica, en tanto que también inhibe al transportador OATP1B1 (Estradiol-17-β-glucurónido) a concentraciones de sólo 5 veces respecto a lo observado en la práctica clínica. Luego entonces, el riesgo de interacción entre cabazitaxel con sustratos de MRP, OCT1, P-gp y OATP1B3 es improbable in vivo a dosis de 25 mg/m2. En tanto que el riesgo de interacción con el transportador OATP1B1 es posible principalmente durante la infusión (1 hora) y hasta 20 minutos después de haber finalizado la infusión.
La prednisona/prednisolona administrada en dosis de 10 mg al día no afecta la farmacocinética de cabazitaxel.
ALTERACIONES EN LOS RESULTADOS DE PRUEBAS DE LABORATORIO: La incidencia de anemia grado ≥ 3, aumento de SGOT, aumento de SGTP y aumento de bilirrubina en base a anormalidades de laboratorio, fue de 10.6%, 0.9%, 1.1% y 0.6%, respectivamente.
En un estudio multicéntrico, abierto, de brazo único, 94 pacientes con tumores sólidos recibieron cabazitaxel a dosis de 25 mg/m2 cada 3 semanas. Las evaluaciones durante el ciclo 1 en el día 1 hasta las 24 horas siguientes no mostraron cambios de > 10 mseg en el intervalo QTc promedio a partir del valor basal. No obstante, debido a las limitantes en el diseño del estudio, no se pueden excluir incrementos pequeños en el intervalo QTc (< 10 mseg) debido a la administración de cabazitaxel.
PRECAUCIONES GENERALES:
Neutropenia: Los pacientes tratados con JEVTANA® pueden recibir profilaxis con G-CSF, de acuerdo con los lineamientos de la Sociedad Americana de Oncología Clínica (ASCO) y/o con los lineamientos institucionales vigentes, para disminuir el riesgo de las complicaciones de la neutropenia (neutropenia febril, neutropenia prolongada o infección neutropénica) o para el manejo de éstas.
El uso de G-CSF ha demostrado que limita la incidencia y severidad de la neutropenia. La neutropenia es la reacción adversa más frecuente a JEVTANA® (ver sección REACCIONES SECUNDARIAS Y ADVERSAS). Es esencial el monitoreo de la biometría hemática completa cada semana durante el 1er. ciclo de tratamiento y posteriormente antes de cada ciclo subsecuente, de tal manera que la dosis pueda ser ajustada en caso de ser necesario (ver sección DOSIS Y VÍA DE ADMINISTRACIÓN).
Reducir la dosis en caso de neutropenia febril o neutropenia prolongada, a pesar del tratamiento apropiado (ver sección DOSIS Y VÍA DE ADMINISTRACIÓN).
Reiniciar el tratamiento únicamente cuando los neutrófilos se recuperen a un nivel de ≥ 1,500/mm3 (ver sección CONTRAINDICACIONES).
Reacciones de hipersensibilidad: Todos los pacientes deben recibir premedicación antes del inicio de la infusión de JEVTANA® (ver sección DOSIS Y VÍA DE ADMINISTRACIÓN). Los pacientes se deben mantener bajo observación estrecha para detectar reacciones de hipersensibilidad, particularmente durante la primera y segunda infusión. Las reacciones de hipersensibilidad pueden ocurrir a los pocos minutos después del inicio de la infusión de cabazitaxel, por lo que deberán estar disponibles los recursos y el equipo para el tratamiento de hipotensión y broncoespasmo. También se pueden presentar reacciones más graves que se acompañen de exantema/eritema generalizado, hipotensión y broncoespasmo. Las reacciones de hipersensibilidad graves requieren la suspensión inmediata de cabazitaxel y el tratamiento adecuado. Los pacientes con historia de reacciones severas de hipersensibilidad no deben recibir nuevamente el medicamento con el fin de verificar la relación causal con cabazitaxel (ver sección CONTRAINDICACIONES).
Síntomas gastrointestinales: Si los pacientes experimentan diarrea después de la administración de JEVTANA®, pueden ser tratados con medicamentos antidiarreicos comunes. Deberán adoptarse medidas apropiadas para rehidratar a los pacientes. Puede ser necesario el retraso del tratamiento o la disminución de la dosis para la diarrea de grado ≥ 3 (ver sección DOSIS Y VÍA DE ADMINISTRACIÓN). Si los pacientes experimentan náuseas o vómito, pueden ser tratados con los antieméticos utilizados habitualmente.
Se ha reportado hemorragia y perforación gastrointestinal, íleo y colitis con desenlaces fatales en pacientes tratados con JEVTANA®. Se recomienda tener precaución con el tratamiento en pacientes de alto riesgo para desarrollar complicaciones gastrointestinales: aquellos con neutropenia, edad avanzada, uso concomitante de antiinflamatorios no esteroideos, tratamiento antiplaquetario o anticoagulante, y pacientes con historia previa de radioterapia pélvica y enfermedades gastrointestinales como ulceración y sangrado gastrointestinal.
Síntomas como dolor abdominal, fiebre, estreñimiento persistente, diarrea con o sin neutropenia pueden ser manifestaciones tempranas de toxicidad grave a nivel gastrointestinal y deben ser evaluadas y tratadas de forma inmediata. Puede ser necesario suspender o retrasar el tratamiento con JEVTANA®.
Trastornos renales: Se han reportado trastornos renales asociados con sepsis, deshidratación grave secundaria a diarrea, vómito y uropatía obstructiva. Se han observado casos de insuficiencia renal con desenlace fatal. Se deben adoptar medidas adecuadas para identificar la causa de manera temprana y dar tratamiento intensivo a los pacientes, si ocurre tal situación. Se debe mantener bajo monitoreo la función renal.
Arritmias cardiacas: Se han reportado arritmias cardiacas, más comúnmente taquicardia y fibrilación auricular (ver sección REACCIONES SECUNDARIAS Y ADVERSAS).
Población de edad avanzada: Los pacientes de edad avanzada (≥ 65 años de edad) son proclives de experimentar ciertas reacciones adversas, incluyendo neutropenia o neutropenia febril (ver sección REACCIONES SECUNDARIAS Y ADVERSAS).
Pacientes con insuficiencia hepática: En pacientes con insuficiencia hepática el tratamiento con JEVTANA® está contraindicado (ver sección DOSIS Y VÍA DE ADMINISTRACIÓN, CONTRAINDICACIONES). Cabazitaxel es ampliamente metabolizado en el hígado, por lo que en presencia de daño hepático es probable que se suscite un incremento de las concentraciones de cabazitaxel (ver FARMACOCINÉTICA, METABOLISMO). Asimismo, los trastornos hepáticos elevan el riesgo de complicaciones graves que ponen en riesgo la vida de pacientes que están bajo tratamiento con otros fármacos pertenecientes a la misma clase de cabazitaxel.
Cabazitaxel no debe ser administrado a pacientes con trastorno hepático (bilirrubina total ≥ ALN o AST y/o ALT ≥ 1.5 veces ALN).
DOSIS Y VÍA DE ADMINISTRACIÓN:
General: El uso de JEVTANA® debe ser exclusivo de unidades especializadas en la administración de agentes citotóxicos y deberá ser administrado bajo la supervisión de un médico oncólogo calificado en el uso de quimioterapia anticáncer.
Premedicación: Previo a cada administración de JEVTANA® se debe llevar a cabo la premedicación para reducir la incidencia y gravedad de una reacción de hipersensibilidad con los siguientes medicamentos por vía intravenosa:
• Antihistamínico (dexaclorfeniramina 5 mg o difenhidramina 25 mg o equivalente).
• Corticosteroide (dexametasona 8 mg o equivalente) y con
• Antagonista H2 (ranitidina o equivalente) (ver sección PRECAUCIONES GENERALES).
Se recomienda la profilaxis antiemética, la cual se puede administrar por vía oral o intravenosa, según sea necesario.
Posología: La dosis recomendada de JEVTANA® es de 25 mg/m2 administrada en infusión intravenosa durante 1 hora cada 3 semanas, en combinación con prednisona (o prednisolona) por vía oral en dosis de 10 mg, administrada diariamente durante todo el tratamiento con JEVTANA®.
Ajustes de dosis: Las modificaciones de dosis deben hacerse si los pacientes experimentan las siguientes reacciones adversas:
Tabla 3. Modificaciones recomendadas a la dosis en caso de reacciones adversas en pacientes tratados con JEVTANA®
|
Reacciones adversas |
Modificación de dosis |
|
Neutropenia grado ≥ 3 (que se prolongue más de 1 semana de duración) a pesar del tratamiento adecuado, incluyendo G-CSF |
Retrasar el tratamiento hasta que la cuenta de neutrófilos sea > 1,500 células/mm3, posteriormente disminuir la dosis de JEVTANA® de 25 mg/m2 a 20 mg/m2. |
|
Neutropenia febril o infección neutropénica |
Retrasar el tratamiento hasta la mejoría o resolución y hasta que la cuenta de neutrófilos sea de > 1,500 células/mm3, posteriormente disminuir la dosis de JEVTANA® de 25 a 20 mg/m2. |
|
Diarrea grado ≥ 3 o diarrea persistente a pesar del tratamiento adecuado, reemplazo de líquidos y electrólitos |
Retrasar el tratamiento hasta mejoría o resolución, posteriormente disminuir la dosis de JEVTANA® de 25 mg/m2 a 20 mg/m2. |
Suspender el tratamiento con JEVTANA® si el paciente continúa experimentando cualquiera de estas reacciones con la dosis de 20 mg/m2.
Poblaciones especiales:
Niños: La seguridad y eficacia de JEVTANA® no se ha establecido en niños.
Pacientes de edad avanzada: No está recomendado un ajuste específico de la dosis para el uso en pacientes de edad avanzada (ver secciones FARMACOCINÉTICA Y FARMACODINAMIA, PRECAUCIONES GENERALES, REACCIONES SECUNDARIAS Y ADVERSAS).
Pacientes con trastorno hepático: JEVTANA® es extensamente metabolizado en hígado. No se han realizado estudios formales en pacientes con daño hepático. Como medida precautoria, JEVTANA® no debe ser administrado a pacientes con daño hepático (nivel de bilirrubina ≥ 1 arriba del límite normal superior [ULN], o AST/TGO y/o ALT/TGP ≥ 1.5 x ULN), (ver secciones PRECAUCIONES GENERALES, CONTRAINDICACIONES).
Pacientes con trastorno renal: JEVTANA® es mínimamente excretado a través de los riñones. No son necesarios los ajustes a la dosis en pacientes con daño renal leve (depuración de creatinina [CLCR]: 50 a 80 mL/min). Es limitada la información de pacientes con insuficiencia renal moderada (CLCR: 30 a 50 mL/min) y grave (CLCR < 30 mL/min) o enfermedad renal en estadio terminal; por lo tanto, estos pacientes deben ser tratados con precaución y vigilados cuidadosamente durante el tratamiento.
Incompatibilidades/compatibilidades: Este medicamento no debe ser mezclado con otros productos, excepto con aquellos mencionados en DOSIS Y VÍA DE ADMINISTRACIÓN.
Siempre diluir el concentrado para solución para infusión de JEVTANA® 60 mg/1.5 ml con el diluyente que se proporciona, antes de adicionar a las soluciones de infusión.
JEVTANA® contiene polisorbato 80, el cual se sabe que incrementa la tasa de extracción del di-(2-etilhexil) ftalato (DEHP) a partir del cloruro de polivinilo (PVC).
No utilizar equipos de infusión de PVC ni equipos de infusión de poliuretano para la preparación y administración de la solución para infusión.
Preparación y manejo: Esta sección se debe leer por completo y de manera cuidadosa antes de iniciar el mezclado y la dilución. JEVTANA® requiere DOS diluciones antes de su administración. Se deben seguir las instrucciones de preparación proporcionadas abajo. NOTA: Tanto los frascos ámpula del concentrado de cabazitaxel como del diluyente contienen un volumen de sobrellenado para compensar la pérdida de líquido durante la preparación. Tal sobrellenado garantiza que después de la dilución utilizando el contenido COMPLETO del líquido diluyente acompañante, la solución inicial diluida contendrá una concentración de 10 mg/ml de JEVTANA®.
Al igual que con cualquier agente antineoplásico, debe tenerse precaución al manejar y preparar las soluciones de JEVTANA®. Se recomienda utilizar guantes.
Si JEVTANA® en cualquier etapa de su manejo, se pone en contacto con la piel, lavar inmediata e intensamente con agua y jabón. Si se pone en contacto con membranas mucosas, lavar inmediata e intensamente con agua.
JEVTANA® debe ser preparado y administrado únicamente por personal entrenado en el manejo de agentes citotóxicos. El personal en estado de embarazo no debe manejar cabazitaxel.
Deberán realizarse los siguientes DOS pasos del proceso de dilución de manera aséptica, para la preparación de la solución para infusión:
Paso 1: Dilución inicial del concentrado para solución para infusión de JEVTANA® 60 mg/1.5 mL con el diluyente proporcionado.
• Coloque por separado el frasco ámpula con el concentrado de JEVTANA® 60 mg/1.5 mL y el diluyente que se proporciona. La solución concentrada debe ser clara si ha sido almacenada apropiadamente (ver sección RECOMENDACIONES SOBRE ALMACENAMIENTO).
• Retirar el contenido completo del diluyente proporcionado utilizando una jeringa, invirtiendo parcialmente el frasco ámpula, e inyectar al frasco ámpula correspondiente del concentrado de JEVTANA® 60 mg/1.5 ml. Para limitar al máximo posible la formación de espuma cuando se inyecte el diluyente, la aguja debe dirigirse a la pared lateral interna del frasco ámpula de la solución concentrada e inyectar lentamente.
• Retirar la jeringa y la aguja y mezclar manualmente y suavemente por medio de inversiones repetidas hasta obtener una solución clara y homogénea. Esto podría tomar aproximadamente 45 segundos.
• Dejar reposar la solución algunos minutos (aproximadamente 5 minutos) y revisar entonces que la solución sea clara y homogénea. Es normal que la espuma persista después de este periodo de tiempo.
• El resultante de la mezcla del concentrado y el diluyente contiene 10 mg/mL de cabazitaxel (por lo menos 6 mL de volumen administrable). Deberá diluirse inmediatamente como se detalla en el paso 2.
Nota importante: El frasco ámpula de la solución concentrada contiene 60 mg de cabazitaxel en 1.5 mL de volumen nominal equivalentes a 73.2 mg de cabazitaxel en 1.83 mL como volumen real de llenado. Tal volumen permite compensar la pérdida de líquido durante la preparación de la premezcla. Asimismo, el frasco ámpula del líquido diluyente contiene 4.5 mL de volumen nominal para dar un volumen real de llenado de 5.67 mL. Tal volumen de sobrellenado garantiza que después de la adición de TODO el contenido del frasco ámpula de diluyente al frasco ámpula de la solución concentrada de 60 mg, se obtenga una concentración final de la premezcla de 10 mg/ml de cabazitaxel.
Paso 2: Preparación de la solución para infusión.
• Retirar la cantidad requerida de la solución inicial diluida de JEVTANA® (10 mg/mL de cabazitaxel), con una jeringa graduada e inyectar en un contenedor estéril sin PVC, que contenga ya sea solución glucosada al 5% o solución de cloruro de sodio al 0.9% para infusión. La concentración de la solución para infusión debe ser entre 0.10 mg/mL y 0.26 mg/mL.
• Como ejemplo, una dosis de 45 mg de JEVTANA® requerirá 4.5 ml (volumen rea de 5.67 mL) de la mezcla del concentrado y diluyente preparada en el paso 1. Puede ser necesario más de un frasco ámpula de la solución inicial diluida, para administrar la dosis prescrita.
• Debido a que la espuma puede persistir sobre la pared del frasco ámpula de esta solución, después de su preparación descrita en el paso 1, es preferible colocar la aguja de la jeringa en la mitad del frasco ámpula cuando se extraiga.
• Retirar la jeringa y mezclar el contenido de la bolsa o botella manualmente, utilizando un movimiento de ondulación.
Al igual que todos los productos parenterales, la solución para infusión resultante debe ser inspeccionada visualmente antes de su administración. La solución que contenga un precipitado, deberá desecharse. Considerar que la solución para infusión puede llegar a sobresaturarse conforme transcurre el tiempo y llegar a cristalizarse, por lo que si llega a darse esta situación, la infusión se debe desechar y no se administrará.
Administración: JEVTANA® se administra por infusión intravenosa.
No utilizar contenedores para infusión de PVC.
No utilizar equipos para infusión de poliuretano.
Utilizar un filtro en la línea de infusión con un diámetro nominal de los poros de 0.22 micrómetros durante la administración.
La solución para infusión de JEVTANA® deberá ser utilizada inmediatamente. Sin embargo, el tiempo de almacenamiento durante su uso puede ser más prolongado bajo las condiciones específicas mencionadas en la sección RECOMENDACIONES SOBRE ALMACENAMIENTO.
Cualquier cantidad del producto no utilizado o materiales utilizados en su preparación, deberán ser desechados de conformidad con los requerimientos locales.
MANIFESTACIONES Y MANEJO DE LA SOBREDOSIFICACIÓN O INGESTA ACCIDENTAL:
Signos y síntomas: Las complicaciones esperadas en caso de sobredosis, podrían ser la exacerbación de las reacciones adversas, tales como supresión de la médula ósea y trastornos gastrointestinales.
Tratamiento: No existe un antídoto conocido para JEVTANA®. En caso de sobredosis, el paciente debe ser internado en una unidad especializada, bajo estrecho monitoreo. Los pacientes deben recibir G-CSF terapéutico tan pronto como se haya identificado la sobredosis. También deben tomarse otras medidas sintomáticas apropiadas.
PRESENTACIÓN: Caja con frasco ámpula, etiquetado con 60 mg/1.5 mL y un frasco ámpula con 4.5 mL de diluyente e instructivo anexo.
RECOMENDACIONES SOBRE ALMACENAMIENTO:
Almacenamiento:
Antes de la dilución: Consérvese a no más de 30ºC. No se refrigere.
Después de la dilución: Para las condiciones de almacenamiento del medicamento diluido, vea a continuación:
Estabilidad:
Estabilidad de la solución inicial diluida en el frasco ámpula: Después de la dilución inicial de JEVTANA® en concentrado con 60 mg/1.5 mL con el diluyente, la mezcla resultante del concentrado-diluyente es estable durante 1 hora si se almacena a temperatura ambiente.
Estabilidad de la solución final diluida en la bolsa de infusión: Después de la dilución final en la bolsa/botella, la solución de infusión puede ser almacenada hasta por 8 horas a temperatura ambiente (incluyendo 1 hora de infusión).
La estabilidad química y física de la solución para infusión ha sido demostrada durante 48 horas bajo condiciones de refrigeración.
Debido a que la solución para infusión está sobresaturada, podría cristalizarse con el paso del tiempo. En este caso la solución no debe utilizarse y deberá desecharse.
LEYENDAS DE PROTECCIÓN:
Literatura exclusiva para médicos. Su venta requiere receta médica. No se deje al alcance de los niños. Consérvese la caja bien cerrada.
Reporte las sospechas de reacción adversa al correo:
farmacovigilancia@cofepris.gob.mx
SANOFI-AVENTIS DE MÉXICO, S.A. de C.V.
Acueducto del Alto Lerma No. 2,
Zona Industrial Ocoyoacac,
C.P. 52740, Ocoyoacac, México, México.
Reg. Núm. 097M2012, SSA IV
Clave interna del documento: MX-(IPPA-Jevtana/V3)-(Cabazitaxel CCDS/V4.1)


