INVOKANA - Tabletas
Sustancia(s):
- Canagliflozina
Presentaciones:
- 1 Caja , 10 Tabletas , 100 Miligramos
- 1 Caja , 14 Tabletas , 100 Miligramos
- 1 Caja , 28 Tabletas , 100 Miligramos
- 1 Caja , 30 Tabletas , 100 Miligramos
- 1 Caja , 56 Tabletas , 100 Miligramos
- 1 Caja , 60 Tabletas , 100 Miligramos
- 1 Caja , 90 Tabletas , 100 Miligramos
- 1 Caja , 100 Tabletas , 100 Miligramos
- 1 Caja , 10 Tabletas , 300 Miligramos
- 1 Caja , 14 Tabletas , 300 Miligramos
- 1 Caja , 28 Tabletas , 300 Miligramos
- 1 Caja , 30 Tabletas , 300 Miligramos
- 1 Caja , 56 Tabletas , 300 Miligramos
- 1 Caja , 60 Tabletas , 300 Miligramos
- 1 Caja , 90 Tabletas , 300 Miligramos
- 1 Caja , 100 Tabletas , 300 Miligramos
FORMA FARMACÉUTICA Y FORMULACIÓN:
INVOKANA® Tabletas, 100 mg y 300 mg
Cada TABLETA contiene:
|
Hemihidrato de Canagliflozina equivalente a de Canagliflozina |
100 mg |
|
Excipiente cbp |
|
Cada TABLETA contiene:
|
Hemihidrato de Canagliflozina equivalente a de Canagliflozina |
300 mg |
|
Excipiente cbp |
|
INDICACIONES TERAPÉUTICAS: INVOKANA® está indicada como adyuvante a la dieta y el ejercicio para mejorar el control de la glucemia en adultos con diabetes mellitus tipo 2.
FARMACOCINÉTICA Y FARMACODINAMIA:
Farmacodinamia:
Grupo farmacoterapéutico: Medicamentos usados en diabetes. Otros medicamentos que disminuyen la glucosa sanguínea, con exclusión de la insulina, código ATC: A10BX11.
Mecanismo de acción: El cotransportador sodio-glucosa tipo 2 (SGLT2), que se expresa en los túbulos renales proximales, se encarga de la mayor parte de la resorción de la glucosa filtrada desde la luz tubular. Se ha demostrado que los pacientes con diabetes tienen resorción elevada de la glucosa renal que pudiese contribuir a las concentraciones persistentemente elevadas de glucosa. La canagliflozina es un inhibidor de SGLT2 activo por vía oral. Al inhibir la SGLT2, la canagliflozina disminuye la resorción de la glucosa filtrada y aminora el umbral renal de la glucosa (TmG) y, por lo tanto, aumenta la excreción urinaria de glucosa, lo que disminuye las cifras elevadas de glucosa plasmática por este mecanismo independiente de la insulina en los pacientes con diabetes tipo 2. La mayor excreción de glucosa urinaria con la inhibición de SGLT2 también se traduce en una diuresis osmótica, con un efecto diurético que lleva a una disminución de la presión arterial sistólica: el aumento de la excreción urinaria de glucosa produce pérdida de calorías y, por lo tanto, disminución del peso corporal, según se ha demostrado en estudios de pacientes con diabetes tipo 2.
La acción de canagliflozina en el incremento de la excreción urinaria de glucosa que disminuye directamente la glucosa plasmática es independiente de insulina. Se ha observado mejoría en el modelo de evaluación de la homeostasis de la función de la célula beta (HOMA B) y mejoría de la célula beta en la secreción de insulina como respuesta a la prueba de tolerancia oral con alimentos mixtos en estudios clínicos con INVOKANA®.
Estudios Fase 3 donde se hizo una prueba de tolerancia de una comida mixta, 300 mg de Canagliflozina aportaron una mayor disminución de la variación de glucosa postprandial que la observada con 100 mg. Este efecto con la dosis de 300 mg de Canagliflozina pudiese, en parte, deberse a la inhibición local del SGLT1 intestinal (un cotransportador importante de la glucosa intestinal) en relación con concentraciones transitorias altas de Canagliflozina en la luz intestinal antes de su absorción.
Efectos farmacodinámicos: Después de dosis orales únicas y múltiples de canagliflozina en pacientes con diabetes tipo 2, se observaron decrementos en TmG dosis dependientes e incremento de la excreción de glucosa urinaria. A partir del valor de inicio de TmG de casi 13 mmol/L, se observó supresión máxima de la medida de TmG de 2 horas con la dosis diaria de 300 mg hasta casi 4 a 5 mmol/L en los pacientes con diabetes tipo 2 en estudios de Fase 1, lo que sugiere un bajo riesgo de hipoglucemia inducida por el tratamiento. Las reducciones en TmG llevaron al incremento en la excreción urinaria de glucosa en sujetos con diabetes tipo 2 tratados ya sea con 100 mg o 300 mg de canagliflozina. A lo largo de los estudios Fase 1 se encontraba en rangos de 77 a 119 g/día. La excreción urinaria de glucosa observada se traduce en la perdida de 308 a 476 kcal/día. Las reducciones en TmG y aumento en la excreción urinaria de glucosa se mantuvieron durante el periodo de dosificación de 26 semanas en pacientes con diabetes tipo 2. Se observaron aumentos moderados en el volumen urinario diario (generalmente < 400 a 500 mL) los cuales diminuyeron tras varios días de uso del tratamiento. La excreción de ácido úrico aumento transitoriamente debido al tratamiento con canagliflozina (de 19% en comparación a la basal, y después disminuyó a 6% en el día 2 y 1% en el día 13). Esto se acompañó de la reducción sustancial en la concentración de ácido úrico en aproximadamente 20%.
En un estudio de una sola dosis en pacientes con diabetes tipo 2, el tratamiento con 300 mg antes de una comida mixta retrasó la absorción de glucosa intestinal y redujo la glucosa postprandial a través de mecanismos, tanto renales como no renales.
Electrofisiología cardiaca: En un estudio aleatorizado, doble ciego, placebo controlado con comparador activo con 4 formas de cruzamiento, 60 sujetos sanos recibieron una sola dosis oral de Canagliflozina de 300 mg, 1200 mg de Canagliflozina (4 veces la máxima dosis recomendada), moxifloxacina y placebo. No se observaron cambios significativos en el intervalo QTc con cualquiera de las dosis recomendadas de 300 mg o la de 1200 mg. Con la dosis de 1200 mg, las concentraciones plasmáticas máximas de Canagliflozina fueron casi 1.4 veces la máxima de estado estable después de una dosis de 300 mg diarios.
Eficacia clínica: Se estudió INVOKANA® como monoterapia, como tratamiento adyuvante con metformina, sulfonilurea, metformina y sulfonilurea, metformina y una tiazolidindiona (pioglitazona), y como tratamiento adyuvante con insulina (con o sin otros agentes antihiperglucémicos). Se comparó la eficacia de INVOKANA® con inhibidor de DPP-4 (sitagliptina) y una sulfonilurea (glimepirida). INVOKANA® también se valoró en pacientes de edad avanzada, aquellos con alteración renal moderada y enfermedad cardiovascular, o en alto riesgo de ésta última.
Un total de 10,285 pacientes con diabetes tipo 2 que recibieron fármaco, participaron en nueve estudios doble ciego, controlados de eficacia clínica y seguridad, realizados para valorar los efectos de INVOKANA® sobre el control de la glucemia. La distribución racial fue de 72% de raza blanca, 16% asiáticos, 4% afroamericanos y 8% de otros grupos. Aproximadamente 16% de los pacientes eran de origen hispanoamericano. Aproximadamente 58% fue de sexo masculino, todos tenían una media de edad total de 59.6 años (rango de 21 a 96), con 3082 pacientes de 65 años y mayores, y 510 ≥ 75 años. Se hizo un estudio en pacientes con alteración renal moderada con un TFGe de 30 a < 50 mL/min/1.73 m2 (N = 269) y otros tres estudios incluyeron pacientes con alteración renal moderada (eGFR de 30 a < 60 mL/min/1.73 m2) (N = 816).
En pacientes con diabetes tipo 2, el tratamiento con INVOKANA® produjo mejoras clínica y estadísticamente significativas en HbA1C, glucosa plasmática en ayuno (FPG), y glucosa 2 horas postprandial (PPG), en comparación con el placebo. INVOKANA® fue eficaz de disminuir la HbA1C en una amplia gama de pacientes, independientemente de la duración de la enfermedad y el uso concomitante de agentes antihiperglucémicos para tratar la diabetes tipo 2. Se observaron mejoras estadísticamente significativas en el control de la glucemia con relación al placebo con INVOKANA® cuando fue administrada como monoterapia, como tratamiento adyuvante inicial con metformina o una sulfonilurea, tratamiento adyuvante con metformina y una sulfonilurea, metformina y pioglitazona, o tratamiento adyuvante con insulina (con o sin otros agentes antihiperglucémicos). Además, se observó mejoría significativa de HbA1C con INVOKANA® en sujetos con alteración renal moderada (TFGe 30 a < 60 mL/min/1.73 m2) y en pacientes de edad avanzada. Se observaron disminuciones de HbA1C en todos los subgrupos, incluidos los de edad avanzada, género, raza, índice de masa corporal basal (IMC) y función basal de las células beta. Se observaron disminuciones mayores en HbA1C con relación al placebo en pacientes con HbA1C y TFGe basales mayores (ver Farmacodinamia).
Monoterapia: Un total de 584 pacientes con control inadecuado de la glucemia (HbA1C ≥ 7% a ≤10%) con dieta y ejercicio participó en un estudio clínico doble ciego, aleatorio controlado con placebo de tres grupos paralelos, multicéntrico para valorar la eficacia de INVOKANA® durante 26 semanas. La edad promedio fue 55 años. 44% de los pacientes eran hombres y la TFGe basal promedio fue 87 mL/min/1.73 m2. Los pacientes que tomaban otros agentes antihiperglucémicos (N = 281), discontinuaron el agente y se sometieron a dieta, ejercicio y un periodo de eliminación del medicamento de casi 8 semanas, seguido de inmediato por un periodo doble ciego de inclusión, de un solo ciego, con placebo, de 2 semanas. Los pacientes que no tomaban un agente antihiperglucémico oral (fuera de tratamiento por al menos 8 semanas) (N = 303) con control inadecuado de la glucemia, entraron a un periodo de inclusión con placebo uni cegado de 2 semanas. Los pacientes se distribuyeron en forma aleatoria para tomar 100 mg de INVOKANA®, 300 mg de INVOKANA® o placebo, administrados una vez al día. Como se muestra en la Tabla 1, se observaron mejoras significativas (p < 0.001) en HbA1C, FPG, PPG, peso corporal, y presión arterial sistólica, con relación al placebo. Además, un mayor porcentaje de pacientes alcanzó un HbA1C < 7.0% en comparación con quienes recibieron placebo.
Los pacientes que no fueron elegibles para inclusión en el principal estudio controlado con placebo por una hiperglucemia más grave (HbA1C > 10 y ≤ 12%), participaron en un subestudio de tratamiento activo separado (N = 91) y se trataron con 100 mg o 300 mg de INVOKANA® (ver Tabla 1).
Tabla 1. Resultados del estudio clínico de 26 semanas de INVOKANA® como monoterapia controlado con placebo1
|
Parámetro de Eficacia |
INVOKANA® 100 mg (N = 195) |
INVOKANA® 300 mg (N = 197) |
Placebo (N = 192) |
|
HbA1C (%) |
|||
|
Basal (media) |
8.06 |
8.01 |
7.97 |
|
Cambio respecto a basal (media ajustada) |
-0.77 |
-1.03 |
0.14 |
|
Diferencia respecto del placebo (media ajustada) (IC al 95%) |
-0.912 (-1.09; -0.73) |
-1.162 (-1.34; -0.99) |
N/A3 |
|
• Porcentaje de pacientes que alcanzaron una HbA1C < 7% |
44.52 |
62.42 |
371.00 |
|
• Glucosa plasmática en ayuno (mg/dL) |
|||
|
Basal (media) |
172.35 |
172.35 |
165.69 |
|
Cambio respecto al basal (media ajustada) |
-27.19 |
-34.93 |
8.38 |
|
Diferencia respecto del placebo (media ajustada) (IC al 95%) |
-1.972 (-2.34; -1.60) |
-2.412 (-2.78; -2.03) |
N/A3 |
|
• Glucosa 2 horas posprandial (mg/dL) |
|||
|
Basal (media) |
249.79 |
253.94 |
229.44 |
|
Cambio respecto al basal (media ajustada) |
-42.86 |
-58.89 |
5.22 |
|
Diferencia respecto de placebo (media ajustada) (IC al 95%) |
-2.672 (-3.28; -2.05) |
-3.552 (-4.17; -2.94) |
N/A3 |
|
• Peso corporal |
|||
|
Basal (media) en kg |
85.9 |
86.9 |
87.5 |
|
% del cambio respecto al basal (media ajustada) |
-2.8 |
-3.9 |
-0.6 |
|
Diferencia respecto del placebo (media ajustada) (IC al 95%) |
-2.22 (-2.9; -1.6) |
-3.32 (-4.0; -2.6) |
N/A3 |
|
• Presión sistólica (mm Hg) |
|||
|
Línea de base (media) |
126.7 |
128.5 |
127.7 |
|
Cambio respecto al basal (media ajustada) |
-3.3 |
-5.0 |
0.4 |
|
Diferencia respecto del placebo (media ajustada) (IC al 95%) |
-3.72 (-5.9; -1.6) |
-5.42 (-7.6; -3.3) |
N/A3 |
|
Subestudio separado del tratamiento activo de pacientes con cifras basales altas de HbA1C (> 10 a ≤ 12%) |
|||
|
• Parámetro de Eficacia |
INVOKANA® 100 mg (N = 47) |
INVOKANA® 300 mg (N = 44) |
|
|
• HbA1C (%) |
|||
|
Basal (media) |
10.59 |
10.62 |
|
|
Cambio respecto al basal (media ajustada) |
-2.13 |
-2.56 |
|
|
• Porcentaje de pacientes que alcanzan HbA1C < 7% |
17.4 |
11.6 |
|
|
• Glucosa plasmática en ayuno (mg/dL) |
|||
|
Basal (media) |
237.37 |
243.13 |
|
|
Cambio respecto al basal (media ajustada) |
-81.72 |
-86.22 |
|
|
• Glucosa 2 horas posprandial (mg/dL) |
|||
|
Basal (media) |
330.30 |
354.43 |
|
|
Cambio respecto al basal (media ajustada) |
-118.50 |
-125.70 |
|
|
• Peso corporal |
|||
|
Basal (media) en kg |
83.2 |
81.6 |
|
|
% de cambio respecto al basal (media ajustada) |
-3.0 |
-3.8 |
|
|
• Parámetro de eficacia |
INVOKANA® 100 mg (N = 47) |
INVOKANA® 300 mg (N = 44) |
|
|
• Presión arterial sistólica (mm Hg) |
|||
|
Basal (media) |
125.0 |
126.6 |
|
|
Cambio respecto al basal (media ajustada) |
-4.5 |
-5.0 |
|
•1 Población de intento de tratamiento utilizando la última observación en el estudio antes del tratamiento de rescate de la glucemia.
2 p < 0.001 en comparación con placebo.
3 N/A = No aplicable.
Tratamiento adyuvante:
Tratamiento adyuvante con metformina: Un total de 1284 pacientes con control inadecuado de glucemia (HbA1C de ≥ 7% a ≤ 10.5%) bajo monoterapia con metformina (2000 mg/día o al menos 1500 mg/día, si no se toleraba una dosis mayor), participaron en un estudio doble ciego aleatorio con placebo y producto activo, controlado de 4 grupos paralelos, multicéntrico para valorar la eficacia de INVOKANA® como tratamiento adyuvante de metformina durante 26 semanas. La edad promedio fue de 55 años; 47% de los pacientes eran hombres y la TFGe basal promedio fue 89 mL/min/1.73 m2. Los pacientes tratados con metformina (N = 1009) en el momento de la detección con un control inadecuado de la glucemia concluyeron un periodo de inclusión de 2 semanas doble ciego, con placebo. Otros pacientes con metformina y otro agente oral o una dosis menor que la requerida de metformina (N = 275), se cambiaron a un esquema de monoterapia con metformina. Después de al menos 8 semanas con una dosis doble de monoterapia con metformina, los pacientes ingresaron a un periodo de inclusión de 2 semanas, de un solo ciego, con placebo. Los pacientes se distribuyeron en forma aleatoria para la adición de 100 mg de INVOKANA®, 300 mg de INVOKANA®, sitagliptina 100 mg o placebo, administrado una vez al día.
Como se demuestra en la Tabla 2, se observaron mejoras estadísticamente significativas (p < 0.001) en HbA1C, FPG, PPG, peso corporal, y presión arterial sistólica con relación a placebo. Además, un mayor porcentaje de pacientes alcanzó un HbA1C < 7.0% en comparación con placebo. Menos pacientes con INVOKANA® requirieron tratamiento de rescate de la glucemia: 1.6% de quienes recibían 100 mg de INVOKANA®, 0.3% de quienes recibían 300 mg de INVOKANA® y 14.8% de los pacientes que recibían placebo.
Tabla 2. Resultados de un estudio clínico controlado con placebo de INVOKANA® como tratamiento adyuvante con metformina1
|
Parámetro de eficacia |
INVOKANA® + metformina 26 semanas |
Placebo + metformina (N = 183) |
|
|
100 mg (N = 368) |
300 mg (N = 367) |
||
|
HbA1C (%) |
|||
|
Basal (media) |
7.94 |
7.95 |
7.96 |
|
Cambio respecto al basal (media ajustada) |
-0.79 |
-0.94 |
-0.17 |
|
Diferencia respecto del placebo (media ajustada) (IC al 95%) |
-0.622 (-0.76; -0.48) |
-0.772 (-0.91; -0.64) |
N/A3 |
|
• Porcentaje de pacientes que alcanzaron una A1C < 7% |
45.52 |
57.82 |
29.8 |
|
• Glucosa plasmática en ayuno (mg/dL) |
|||
|
Basal (media) |
168.57 |
172.71 |
164.25 |
|
Cambio respecto al basal (media ajustada) |
-27.37 |
-37.82 |
2.54 |
|
Diferencia respecto del placebo (media ajustada) (IC al 95%) |
-1.652 (-1.99; -1.32) |
-2.232 (-2.57; -1.90) |
N/A3 |
|
• Glucosa 2 horas posprandial (mg/dL) |
|||
|
Basal (media) |
257.54 |
261.18 |
248.71 |
|
Cambio respecto al basal (media ajustada) |
-47.90 |
-57.09 |
-9.90 |
|
Diferencia respecto del placebo (media ajustada) (IC al 95%) |
-2.122 (-2.73; -1.51) |
-2.622 (-3.24; -2.01) |
N/A3 |
|
• Peso corporal |
|||
|
Basal (media) en kg |
88.7 |
85.4 |
86.7 |
|
Cambio % respecto al basal (media ajustada) |
-3.7 |
-4.2 |
-1.2 |
|
Diferencia respecto de placebo (media ajustada) (IC al 95%) |
-2.52 (-3.1; -1.9) |
-2.92 (-3.5; -2.3) |
N/A3 |
|
• Presión arterial sistólica (mm Hg) |
|||
|
Basal (media) |
128.0 |
128.7 |
128.0 |
|
Cambio respecto al basal (media ajustada) |
-3.8 |
-5.1 |
1.5 |
|
Diferencia respecto del placebo (media ajustada) (IC al 95%) |
-5.42 (-7.3; -3.4) |
-6.62 (-8.5; -4.6) |
N/A3 |
1 Población de intento de tratamiento con uso de la última observación en el estudio antes del tratamiento de rescate de la glucemia.
2 p < 0.001 en comparación con placebo.
3 N/A = No aplicable.
Estudio controlado de producto activo contra glimepirida como tratamiento adyuvante con metformina: Un total de 1450 pacientes con control inadecuado de la glucemia (concentración de HbA1C ≥ 7% a ≤ 9.5%) bajo tratamiento con monoterapia con metformina (≥ 2000 mg/día o al menos 1500 mg/día si no se toleraba la dosis mayor) participó en un estudio clínico aleatorio doble ciego controlado y con producto activo, de grupos paralelos, de 3 grupos, multicéntrico, para valorar la eficacia de INVOKANA® como tratamiento adyuvante con metformina durante 52 semanas. La edad promedio fue de 56 años; 52% de los pacientes eran hombres y la TFGe basal promedio fue 90 mL/min/1.73 m2. Los pacientes con metformina (N = 928) a una dosis especificada por protocolo estable, ingresaron a un periodo de inclusión con placebo de 2 semanas uni cegado . Otros pacientes (N = 522) entraron a un periodo de titulación de la dosis de metformina y estabilización de la dosis/eliminación del agente antihiperglucémico, seguido de inmediato por el periodo de inclusión de dos semanas. Después del periodo de inclusión, los pacientes con control inadecuado de la glucemia se distribuyeron en forma aleatoria para la adición de 100 mg de INVOKANA®, 300 mg de INVOKANA® o glimepirida (la titulación permitida durante el estudio de 52 semanas a 6 a 8 mg), administrada una vez al día.
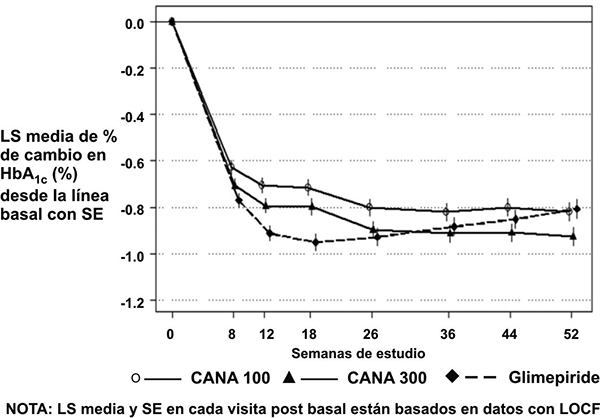
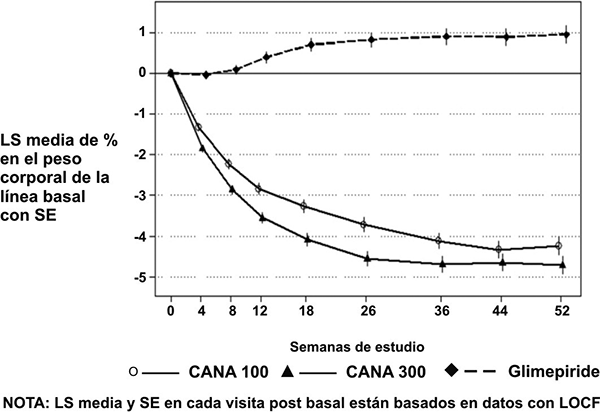
Como se muestra en la Tabla 3 y la Figura 1, después de 52 semanas, el tratamiento con 100 mg de INVOKANA® aportó disminuciones similares en la HbA1C respecto de la línea de base en comparación con glimepirida (con el límite superior del intervalo de confianza al 95% cercano a la diferencia entre grupos menor que el margen de no inferioridad pre especificado de 0.3%); 300 mg de INVOKANA® brindaron una mayor reducción (p < 0.05) respecto de la línea de base en HbA1C en comparación con glimepirida (con el límite superior del intervalo de confianza al 95% menor de 0). Se observaron mejoras estadísticamente significativas (p < 0.001) en el peso corporal con INVOKANA® en comparación con glimepirida. La incidencia de hipoglucemia con INVOKANA® fue significativamente menor (p < 0.001) en comparación con glimepirida. Menos pacientes con INVOKANA® requirieron tratamiento de rescate de la glucemia: 6.6% de los pacientes que recibían 100 mg de INVOKANA®, 4.9% de los que recibían 300 mg de INVOKANA® y 10.6% de los que recibían glimepirida.
En un subgrupo de pacientes (N = 208) que se sometió a DXA y TC abdominal para la valoración de la composición corporal, se mostró que casi 66% de la disminución de peso con canagliflozina se debía a pérdida de masa grasa, con cantidades similares de grasa subcutánea visceral y abdominal perdidas.
Tabla 3. Resultados de un estudio clínico de 52 semanas de comparación de INVOKANA® con la glimepirida como tratamiento adyuvante con metformina1
|
Parámetro de eficacia |
INVOKANA® + metformina 52 Semanas |
Glimepirida (titulada) + metformina (N = 482) |
|
|
100 mg (N = 483) |
300 mg (N = 485) |
||
|
HbA1C (%) |
|||
|
Basal (media) |
7.78 |
7.79 |
7.83 |
|
Cambio respecto al basal (media ajustada) |
-0.82 |
-0.93 |
-0.81 |
|
Diferencia con glimepirida (media ajustada) (IC al 95%) |
-0.012 (-0.11; 0.09) |
-0.122 (-0.22; -0.02) |
N/A3 |
|
• Porcentaje de pacientes que alcanzaron una HbA1C < 7% |
53.6 |
60.1 |
55.8 |
|
• Glucosa plasmática en ayuno (mg/dL) |
|||
|
Basal (media) |
165.33 |
163.71 |
165.69 |
|
Cambio respecto al basal (media ajustada) |
-24.31 |
-27.37 |
-1.02 |
|
Diferencia respecto de la glimepirida (media ajustada) (IC al 95%) |
-0.33 (-0.56; -0.11) |
-0.51 (-0.73; -0.28) |
N/A3 |
|
• Peso Corporal |
|||
|
Basal (media) en kg |
86.8 |
86.6 |
86.6 |
|
Cambio % respecto al basal (media ajustada) |
-4.2 |
-4.7 |
1.0 |
|
Diferencia respecto de glimepirida (media ajustada) (IC al 95%) |
-5.24 (-5.7; -4.7) |
-5.74 (-6.2; -5.1) |
N/A3 |
|
• Presión Arterial Sistólica (mm Hg)5 |
|||
|
Basal (media) |
130.0 |
130.0 |
129.5 |
|
Cambio respecto al basal (media ajustada) |
-3.3 |
-4.6 |
-0.2 |
|
Diferencia respecto de glimepirida (media ajustada) (IC al 95%) |
-3.5 (-4.9; -2.1) |
-4.8 (-6.2; -3.4) |
N/A3 |
•1 Población de intención de tratamiento utilizando la última observación en el estudio antes del tratamiento de rescate de la glucemia.
2 Cumplieron los criterios pre especificados de no inferioridad para la glimepirida (con el límite superior de IC al 95% cercano a la diferencia entre grupos menores que el margen de no inferioridad pre especificado de < 0.3%). En una valoración pre especificada, el límite superior del IC al 95% para INVOKANA® 300 mg, no así para 100 mg de INVOKANA® que fue < 0, lo que indica una disminución mayor (p < 0.05) en la A1C con 300 mg de INVOKANA® con relación a la glimepirida.
3 N/A = No aplicable.
4 p < 0.001.
5 Incluye sólo pacientes que tenían ambas cifras, basales y posbasales.
Figura 1. Medias del cambio respecto al basal para HbA1C (%) y peso corporal durante 52 Semanas en un estudio de comparación de INVOKANA® con glimepirida como tratamiento adyuvante con metformina
Tratamiento adyuvante con una sulfonilurea: Un total de 127 pacientes con control inadecuado de la glucemia (HbA1C de ≥ 7% a ≤ 10.5%) con la monoterapia con sulfonilurea, participaron en un subestudio aleatorio doble ciego, controlado con placebo, de tres grupos paralelos, multicéntrico, de un estudio cardiovascular para valorar la eficacia de la INVOKANA® como tratamiento adyuvante con una sulfonilurea durante 18 semanas. La edad promedio fue de 65 años; 57% de los pacientes eran hombres y la TFGe basal promedio fue 69 mL/min/1.73 m2. Los pacientes con monoterapia con sulfonilurea a una dosis preespecificada por protocolo estable (≥ 50% de la dosis máxima) durante al menos 10 semanas, concluyeron un periodo de inclusión con placebo uni cegado de dos semanas. Después del periodo de inclusión, los pacientes con control inadecuado de la glucemia se distribuyeron en forma aleatoria para la adición de 100 mg de INVOKANA®, 300 mg de la INVOKANA® o placebo, administrados una vez al día.
Como se muestra en la Tabla 4, se observaron mejoras estadísticamente significativas (p < 0.001) en HbA1C y FPG con relación al placebo en la semana 26. Además, un mayor porcentaje de pacientes alcanzó un HbA1C < 7.0% en comparación con placebo. Menos pacientes con INVOKANA® requirieron tratamiento de rescate de la glucemia: 4.8% de los que recibieron 100 mg de INVOKANA®, 0.0% de quienes recibieron 300 mg de INVOKANA® y 17.8% de quienes recibieron placebo. Los pacientes tratados con 300 mg de INVOKANA® mostraron disminuciones en el peso corporal, en comparación con los de placebo. Se observó una mayor incidencia de hipoglucemia en este estudio, compatible con el aumento esperado de la hipoglucemia cuando se agregaba a la sulfonilurea un agente no relacionado con la hipoglucemia (ver Precauciones generales y Reacciones secundarias y adversas).
Tabla 4. Resultados de un estudio clínico controlado con placebo de INVOKANA® como tratamiento adyuvante con sulfonilurea1
|
Parámetro de eficacia |
INVOKANA® + sulfonilurea 18 semanas |
Placebo + sulfonilurea (N = 45) |
|
|
100 mg (N = 42) |
300 mg (N = 40) |
||
|
HbA1C (%) |
|||
|
Basal (media) |
8.29 |
8.28 |
8.49 |
|
Cambio respecto de basal (media ajustada) |
-0.70 |
-0.79 |
0.04 |
|
Diferencia respecto del placebo (media ajustada) (IC al 95%) |
-0.742 (-1.15; -0.33) |
-0.832 (-1.24; -0.41) |
N/A4 |
|
• Porcentaje de pacientes que alcanzaron una HbA1C < 7% |
25.03 |
33.33 |
5.0 |
|
• Glucosa plasmática en ayuno (mg/dL) |
|||
|
Basal (media) |
185.32 |
178.10 |
184.96 |
|
• Cambio respecto de basal (media ajustada) |
-25.39 |
-36.02 |
12.06 |
|
• Diferencia respecto de placebo (media ajustada) (IC al 95%) |
-2.072 (-2.99; -1.15) |
-2.662 (-3.59; -1.74) |
N/A4 |
|
• Peso corporal |
|||
|
Línea de base (media) en kg |
85.1 |
80.4 |
85.5 |
|
Cambio % respecto de basal (media ajustada) |
-0.6 |
-2.0 |
-0.2 |
|
Diferencia respecto de placebo (media ajustada) (IC al 95%) |
-0.4 (-1.8; 1.0) |
-1.8 (-3.2; -0.4) |
N/A4 |
|
• Presión arterial sistólica (mm Hg) |
|||
|
Basal (media) |
138 |
133.5 |
137.3 |
|
Cambio respecto de basal (media ajustada) |
-3.5 |
-5.1 |
-3.4 |
|
Diferencia respecto de placebo (media ajustada) (IC al 95%) |
-0.1 (-6.5; 6.2) |
-1.8 (-8.2; 4.7) |
N/A4 |
•1 Población de intención de tratamiento con uso de la última observación en estudio antes del tratamiento de rescate de la glucemia.
2 p < 0.001 en comparación con placebo.
3 p < 0.01.
4 N/A = No aplicable.
Tratamiento adyuvante con metformina y sulfonilurea: Un total de 469 pacientes con control inadecuado de la glucemia (cifra de HbA1C ≥ 7% a ≤ 10.5%) con la combinación de metformina (2000 mg/día o al menos 1500 mg/día si no se toleraba una dosis mayor) y sulfonilurea (dosis máxima o cercana a la máxima eficaz) participó en un estudio clínico doble ciego, controlado con placebo, de 3 grupos paralelos, multicéntrico para valorar la eficacia de INVOKANA® como tratamiento adyuvante de metformina y sulfonilurea durante 26 semanas. La edad promedio fue 57 años, 51% de los pacientes eran hombres y la TFGe basal promedio fue 89 mL/min/1.73 m2. Los pacientes con dosis eficaces cercanas a la máxima y máxima eficaz de metformina y sulfonilurea (N = 372), ingresaron a un periodo de inclusión de 2 semanas, uni cegado con placebo. Otros pacientes (N = 97) ingresaron a un periodo de titulación de dosis de metformina y sulfonilurea y estabilización de la dosis/eliminación del agente antihiperglucémico de 12 semanas, seguido de inmediato por un periodo de inclusión de 2 semanas. Después del periodo de inclusión, los pacientes con control inadecuado de la glucemia, se distribuyeron en forma aleatoria para la adición de 100 mg de INVOKANA®, 300 mg de INVOKANA® o placebo, administrados una vez al día. Como se muestra en la Tabla 5, se observaron mejoras estadísticamente significativas (p < 0.001) en HbA1C, FPG y peso corporal con relación al placebo. Además, un mayor porcentaje de pacientes alcanzó una HbA1C < 7.0% en comparación con placebo. Menos pacientes con INVOKANA® requirieron tratamiento de rescate de la glucemia: 1.3% de los que recibían 100 mg de INVOKANA®, 1.9% de quienes recibían 300 mg de INVOKANA®, y 12.8% de quienes recibían placebo. Se observó una mayor incidencia de hipoglucemia en este estudio, consistente con el aumento esperado de la hipoglucemia cuando se agregaba a la sulfonilurea un agente no vinculado con la hipoglucemia (ver Precauciones generales y Reacciones secundarias y adversas).
Tabla 5. Resultados de un estudio clínico controlado con placebo de 26 Semanas de INVOKANA® como tratamiento adyuvante de metformina y sulfonilurea1
|
Parámetro de eficacia |
INVOKANA® + metformina y sulfonilurea 26 semanas |
Placebo + metformina y sulfonilurea (N = 156) |
|
|
100 mg (N = 157) |
300 mg (N = 156) |
||
|
HbA1C (%) |
|||
|
Basal (media) |
8.13 |
8.13 |
8.12 |
|
Cambio respecto de basal (media ajustada) |
-0.85 |
-1.06 |
-0.13 |
|
Diferencia respecto del placebo (media ajustada) (IC al 95%) |
-0.712 (-0.90; -0.52) |
-0.922 (-1.11; -0.73) |
N/A3 |
|
• Porcentaje de pacientes que alcanzaron una HbA1C < 7% |
43.22 |
56.62 |
18.0 |
|
• Glucosa plasmática en ayuno (mg/dL) |
|||
|
Basal (media) |
172.89 |
168.21 |
169.65 |
|
Cambio respecto de basal (media ajustada) |
-18.19 |
-30.43 |
4.14 |
|
Diferencia respecto de placebo (media ajustada) (IC al 95%) |
-1.242 (-1.75; -0.73) |
-1.922 (-2.43; -1.41) |
N/A3 |
|
• Peso corporal |
|||
|
Basal (media) en kg |
93.5 |
93.5 |
90.8 |
|
Cambio % respecto de basal (media ajustada) |
-2.1 |
-2.6 |
-0.7 |
|
Diferencia respecto del placebo (media ajustada) (IC al 95%) |
-1.42 (-2.1; -0.7) |
-2.02 (-2.7; -1.3) |
N/A3 |
|
• Presión arterial sistólica (mm Hg) |
|||
|
Basal (media) |
130.4 |
130.8 |
130.1 |
|
Cambio respecto de basal (media ajustada) |
-4.9 |
-4.3 |
-2.6 |
|
Diferencia respecto del placebo (media ajustada) (IC al 95%) |
-2.2 (-4.7; 0.2) |
-1.6 (-4.1; 0.9) |
N/A3 |
•1 Población de intento de tratamiento utilizando la última observación en el estudio antes del tratamiento de rescate de la glucemia.
2 p < 0.001 en comparación con placebo.
3 N/A = No aplicable o no medida en este estudio.
Estudio controlado con el producto activo contra sitagliptina como tratamiento adyuvante con metformina y sulfonilurea: Un total de 755 pacientes con control inadecuado de la glucemia (cifra de HbA1C ≥ 7.0% a ≤ 10.5%) con la combinación de metformina (2000 mg/día o al menos 1500 mg/día si no se toleraba una dosis mayor) y sulfonilurea (dosis casi máxima o máxima eficaz) participó en un estudio clínico de 2 grupos paralelos, doble ciego, con producto activo controlado, multicéntrico para valorar la eficacia de 300 mg de INVOKANA® como tratamiento adyuvante con metformina y sulfonilurea contra sitagliptina 100 mg como tratamiento adyuvante con metformina y sulfonilurea durante 52 semanas. La edad promedio fue 57 años; 56% de los pacientes eran hombres y la TFGe basal promedio fue 88 mL/min/1.73 m2. Aquellos con dosis casi máximas o máximas eficaces de metformina y sulfonilurea (N = 716), ingresaron a un periodo de inclusión de 2 semanas uni cegado con placebo. Otros (N = 39) ingresaron a un periodo de titulación de dosis de metformina y sulfonilurea y estabilización de la dosis de hasta 12 semanas, seguido de inmediato por el periodo de inclusión de 2 semanas. Después del periodo de inclusión, los pacientes con control inadecuado de la glucemia se distribuyeron en forma aleatoria para la adición de 300 mg de INVOKANA® o 100 mg de sitagliptina.
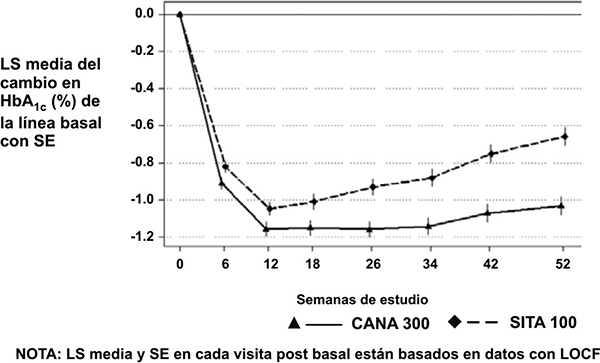
Como se muestra en la Tabla 6 y la Figura 2, después de 52 semanas, 300 mg de INVOKANA® aportaron una mayor reducción (p < 0.05) en la HbA1C en comparación con 100 mg de sitagliptina (con el límite superior del intervalo de confianza al 95% cerca de la diferencia entre grupos menor de 0). Además, un mayor porcentaje de pacientes alcanzó una HbA1C de < 7.0% con 300 mg de INVOKANA® con relación a sitagliptina: 47.6% de los pacientes que recibían 300 mg de INVOKANA® y 35.3% de los que recibían sitagliptina. Aquellos tratados con 300 mg de INVOKANA® mostraron un decremento medio significativo en el cambio porcentual respecto al basal del peso corporal, en comparación con quienes recibieron 100 mg de sitagliptina. Se observó una incidencia similarmente aumentada de hipoglucemia con 300 mg de INVOKANA® y sitagliptina en este estudio, lo que es compatible con el aumento esperado de la hipoglucemia cuando agentes no vinculados con la hipoglucemia se agregan a una sulfonilurea (ver Precauciones generales y Reacciones secundarias y adversas). El porcentaje de los pacientes que cumplió con los criterios de retiro de la glucemia (con base en FPG hasta la Semana 26 y HbA1C después) fue menor con 300 mg de INVOKANA® 10.6%) en comparación con 100 mg de sitagliptina (22.5%).
Tabla 6. Resultados de un estudio clínico de 52 Semanas de comparación de INVOKANA® con sitagliptina como tratamiento adyuvante con metformina y sulfonilurea1
|
Parámetro de eficacia |
300 mg de INVOKANA® + metformina y sulfonilurea (N = 377) |
100 mg de sitagliptina + metformina y sulfonilurea (N = 378) |
|
HbA1C (%) |
||
|
Basal (media) |
8.12 |
8.13 |
|
Cambio respecto al basal (media ajustada) |
-1.03 |
-0.66 |
|
Diferencia respecto de sitagliptina (media ajustada) (IC al 95%) |
-0.372 (-0.50; -0.25) |
N/A4 |
|
• Porcentaje de pacientes que alcanzaron la HBA1C < 7% |
47.6 |
35.3 |
|
• Glucosa plasmática en ayuno (mg/dL) |
||
|
Basal (media) |
169.65 |
163.71 |
|
Cambio respecto al basal (media ajustada) |
-29.89 |
-5.76 |
|
Diferencia respecto de sitagliptina (media ajustada) (IC al 95%) |
-1.34 (-1.66; -1.01) |
N/A4 |
|
• Peso corporal |
||
|
Basal (media) en kg |
87.6 |
89.6 |
|
Cambio % respecto al basal (media ajustada) |
-2.5 |
-0.3 |
|
Diferencia respecto de sitagliptina (media ajustada) (IC al 95%) |
-2.83 (-3.3; -2.2) |
N/A4 |
|
• Presión arterial sistólica (mm Hg) |
||
|
Basal (media) |
131.2 |
130.1 |
|
Cambio respecto al basal (media ajustada) |
-5.1 |
0.9 |
|
Diferencia respecto de sitagliptina (media ajustada) (IC al 95%) |
-5.93 (-7.6; -4.2) |
N/A4 |
•1 Población de intento de tratamiento con uso de la última observación en el estudio antes del tratamiento de rescate de la glucemia.
2 Cumplieron criterios preespecificados de no inferioridad de la sitagliptina (con el límite superior del IC al 95% cerca de la diferencia entre grupos menor que el margen de no inferioridad preespecificado de < 0.3%); en una valoración preespecificada, el límite superior del IC % para 300 mgg de INVOKANA® fue < 0, lo que indica una mayor disminución (p < 0.05) en A1C con 300 mg de INVOKANA® en relación con sitagliptina.
3 p < 0.001.
4 N/A = No aplicable.
Figura 2. Media de cambio respecto al basal de HbA1C (%) durante 52 semanas en un estudio de comparación de INVOKANA® con sitagliptina como tratamiento adyuvante de metformina y sulfonilurea
Tratamiento adyuvante con metformina y pioglitazona: Un total de 342 pacientes con control inadecuado de la glucemia (cifra de HbA1C ≥ 7.0% a ≤ 10.5%) con la combinación de metformina (2000 mg/día o al menos 1500 mg/día si no se toleraba una dosis mayor) y pioglitazona (30 o 45 mg/día) participó en un estudio clínico aleatorio, doble ciego, controlado con placebo de 3 grupos paralelos, multicéntrico para valorar la eficacia de INVOKANA® como tratamiento adyuvante de metformina y pioglitazona durante 26 semanas. La edad promedio fue 57 años; 63% de los pacientes eran hombres y la TFGe basal promedio fue 86 mL/min/1.73 m2. Los pacientes ya con dosis especificadas por protocolo de metformina y pioglitazona (N = 163), ingresaron a un periodo de inclusión uni cegado de 2 semanas con placebo. Otros pacientes (N = 181) entraron a un periodo de titulación de dosis con metformina y pioglitazona y estabilización de dosis durante hasta 12 semanas con al menos 8 con dosis estables de metformina y pioglitazona, seguidas de inmediato por un periodo de inclusión de 2 semanas. A continuación del periodo de inclusión, los pacientes con control inadecuado de la glucemia se distribuyeron en forma aleatoria (N = 344) para la adición de 100 mg de INVOKANA®, 300 mg de INVOKANA® o placebo, administrados una vez al día. Como se muestra en la Tabla 7, se observaron mejoras estadísticamente significativas (p < 0.001) en A1C, FPG, y peso corporal con relación a placebo para INVOKANA® en la semana 26. Además, un mayor porcentaje de pacientes alcanzó un HbA1C de < 7.0% en comparación con placebo. Menos pacientes con INVOKANA® requirieron tratamiento de rescate de la glucemia: 0.9% de los que recibían 100 mg de INVOKANA®, 0.0% de los que recibían 300 mg de INVOKANA® y 12.2% de quienes recibían placebo.
Tabla 7. Resultados del estudio clínico controlado con placebo de 26 semanas de INVOKANA® como tratamiento adyuvante de metformina y pioglitazona1
|
Parámetro de eficacia |
INVOKANA® + metformina y pioglitazona 26 semanas |
Placebo + metformina y pioglitazona (N = 115) |
|
|
100 mg (N = 113) |
300 mg (N = 114) |
||
|
HbA1C (%) |
|||
|
Basal (media) |
7.99 |
7.84 |
8.00 |
|
Cambio respecto al basal (media ajustada) |
-0.89 |
-1.03 |
-0.26 |
|
Diferencia respecto del placebo (media ajustada) (IC al 95%) |
-0.622 (-0.81; -0.44) |
-0.762 (-0.95; -0.58) |
N/A3 |
|
• Porcentaje de pacientes que alcanzaron una HbA1C < 7% |
46.92 |
64.32 |
32.5 |
|
• Glucosa plasmática en ayuno (mg/dL) |
|||
|
Basal (media) |
168.93 |
164.07 |
164.43 |
|
Cambio respecto al basal (media ajustada) |
-26.83 |
-33.13 |
2.52 |
|
• Diferencia respecto de placebo (media ajustada) (IC al 95%) |
-1.632 (-2.05; -1.21) |
-1.982 (-2.41; -1.56) |
N/A4 |
|
• Peso corporal |
|||
|
Basal (media) en kg |
94.2 |
94.4 |
94 |
|
Cambio % respecto al basal (media ajustada) |
-2.8 |
-3.8 |
-0.1 |
|
Diferencia respecto del placebo (media ajustada) (IC al 95%) |
-2.72 (-3.6; -1.8) |
-3.72 (-4.6; -2.8) |
N/A3 |
|
• Presión arterial sistólica (mm Hg) |
|||
|
Basal (media) |
126.4 |
126.7 |
128.2 |
|
Cambio respecto al basal (media ajustada) |
-5.3 |
-4.7 |
-1.2 |
|
Diferencia respecto de placebo (media ajustada) (IC al 95%) |
-4.1 (-6.9; -1.3) |
-3.5 (-6.3; -0.6) |
N/A3 |
•1 Población de intento de tratamiento con uso de la última observación en el estudio antes del tratamiento de rescate de la glucemia.
2 p < 0.001 en comparación con placebo.
3 N/A = No aplicable o no determinado en este estudio.
Tratamiento adyuvante con insulina (con o sin otros agentes antihiperglucémicos): Un total de 1718 pacientes con control inadecuado de la glucemia (cifra de HbA1C ≥ 7.0 a ≤ 10.5%) con insulina ≥ 30 unidades/día o tratamiento adyuvante con otros agentes antihiperglucemiantes, participó en un estudio aleatorio doble ciego, controlado con placebo de 3 grupos paralelos, de un subestudio multicéntrico de un estudio cardiovascular; en el cual se valoró la eficacia de la INVOKANA® como tratamiento adyuvante con insulina (con o sin otros agentes antihiperglucémicos) durante 18 semanas. La edad promedio fue 63 años, 66% de los pacientes eran hombres y la TFGe basal promedio fue 75 mL/min/1.73 m2. Los pacientes con insulina basal, en dosis en bolo, o basal/dosis en bolo, con la mayoría en un esquema basal/de dosis en bolo de insulina, durante al menos 10 semanas, entraron a un periodo de inclusión de 2 semanas uni cegado con placebo. Después de éste, los pacientes con control inadecuado de la glucemia se distribuyeron en forma aleatoria para la adición de 100 mg de la INVOKANA®, 300 mg de la INVOKANA® o placebo, una vez al día. La dosis media de insulina diaria línea de base fue de 83 unidades, similar entre los grupos de tratamiento.
Como se muestra en la Tabla 8, se observaron mejoras estadísticamente significativas (p < 0.001) en HbA1C, FPG y peso corporal con relación a placebo para INVOKANA® en la semana 18. Además, un mayor porcentaje de pacientes alcanzó una HbA1C < 7.0% en comparación cono el placebo. Menos pacientes con INVOKANA® requirieron tratamiento de rescate de la glucemia: 4.1% de los que recibieron 100 mg de INVOKANA®, 3.1% de los que recibieron 300 mg de INVOKANA® y 8.7% de los que recibieron placebo. Se observó una mayor incidencia de hipoglucemia en este estudio, que es compatible con el aumento esperado de la hipoglucemia cuando se agrega a la insulina un agente no relacionado con la hipoglucemia (ver Precauciones generales y Reacciones secundarias y adversas).
Tabla 8. Resultados de un estudio clínico controlado con placebo de 18 semanas de INVOKANA® como adyuvante de la Insulina ≥ 30 unidades/día (con o sin otros agentes antihiperglucémicos)1
|
Parámetro de eficacia |
INVOKANA® + Insulina 18 Semanas |
Placebo + Insulina (N = 565) |
|
|
100 mg (N = 566) |
300 mg (N = 587) |
||
|
HbA1C (%) |
|||
|
Basal (media) |
8.33 |
8.27 |
8.20 |
|
Cambio respecto al basal (media ajustada) |
-0.63 |
-0.72 |
0.01 |
|
Diferencia respecto del placebo (media ajustada) (IC 95%) |
-0.652 (-0.73; -0.56) |
-0.732 (-0.82; -0.65) |
N/A3 |
|
• Porcentaje de pacientes que alcanzaron una HbA1C < 7% |
19.8 |
24.72 |
7.72 |
|
• Glucosa plasmática en ayuno (mg/dL) |
|||
|
Basal |
169.83 |
168.31 |
168.93 |
|
Cambio respecto al basal (media ajustada) |
-18.55 |
-30.43 |
3.96 |
|
• Diferencia respecto de placebo (media ajustada) (IC 97.5%) |
-1.252 (-1.55; -0.96) |
-1.612 (-1.90; -1.31) |
N/A3 |
|
Peso corporal |
|||
|
Basal (media) en kg |
96.9 |
96.7 |
97.7 |
|
Cambio % respecto al basal (media ajustada) |
-1.8 |
-2.3 |
0.1 |
|
Diferencia respecto del placebo (media ajustada) (IC 97.5%) |
-1.92 (-2.2; -1.5) |
-2.42 (-2.8; -2.0) |
N/A3 |
|
Presión arterial sistólica (mm Hg) |
|||
|
Basal (media) |
137.0 |
138.2 |
138.2 |
|
Cambio respecto al basal (media ajustada) |
-5.1 |
-6.9 |
-2.5 |
|
Diferencia respecto del placebo (media ajustada) (IC 97.5%) |
-2.62 (-4.1; -1.1) |
-4.42 (-5.8; -2.9) |
N/A3 |
•1 Población de intento de tratamiento utilizando la última observación en el estudio antes del tratamiento de rescate de la glucemia.
2 p < 0.001 en comparación con placebo.
3 N/A = No aplicable.
Estudios en poblaciones especiales:
Estudio en pacientes de edad avanzada:
Un total de 714 pacientes de edad avanzada (≥ 55 a ≤ 80 años de edad) con control inadecuado de la glucemia (HbA1C línea de base ≥ 7.0 a ≤ 10.0%) bajo tratamiento actual de la diabetes (con dieta y ejercicio solos, o en combinación con agentes orales o parenterales), participó en un estudio doble ciego, aleatorio, controlado con placebo para valorar la eficacia de INVOKANA® como adyuvante del tratamiento actual de diabetes durante 26 semanas. La edad promedio fue 64 años, 55% de los pacientes eran hombres y la TFGe basal promedio fue 77 mL/min/1.73 m2. Los pacientes con control inadecuado de la glucemia con su tratamiento actual de la diabetes se distribuyeron en forma aleatoria para agregar 100 mg de INVOKANA®, 300 mg de INVOKANA® o placebo, administrados una vez al día. Como se muestra en la Tabla 9, se observaron cambios estadísticamente significativos (p < 0.001) respecto a la basal en HbA1C, FPG y peso corporal con INVOKANA® en la semana 26. Además, un mayor porcentaje de pacientes alcanzó un HbA1C < 7.0% en comparación con placebo. Menos pacientes con INVOKANA® requirieron tratamiento de rescate de la glucemia: 2.1% de los que recibían 100 mg de INVOKANA®, 0.4% de los que recibían 300 mg de INVOKANA® y 11% de los que recibían placebo (ver Propiedades Farmacocinéticas-Poblaciones especiales).
Un subgrupo de pacientes (n = 211) participó en el subestudio de composición corporal con uso de análisis de la composición corporal por DXA. Esto demostró que casi 66% de la disminución de peso con INVOKANA® se debía a la pérdida de masa grasa con relación a placebo. No hubo cambios significativos de las porciones trabecular y cortical en la densidad ósea.
Tabla 9. Resultados de un estudio clínico controlado con placebo de 26 semanas de INVOKANA® como tratamiento adyuvante de agentes antihiperglucémicos en pacientes de edad avanzada controlados inadecuadamente con agentes antihiperglucémicos (AHA)1
|
Parámetro de eficacia |
INVOKANA® + AHA actual 26 Semanas |
Placebo + AHA actual N = 237 |
|
|
100 mg N = 241 |
300 mg N = 236 |
||
|
HbA1C (%) |
|||
|
Basal (media) |
7.77 |
7.69 |
7.76 |
|
Cambio respecto al basal (media ajustada) |
-0.60 |
-0.73 |
-0.03 |
|
Diferencia respecto de placebo (media ajustada) (IC al 95%) |
-0.572 (-0.71; -0.44) |
-0.702 (-0.84; -0.57) |
N/A3 |
|
• Porcentaje de pacientes que alcanzaron HbA1C < 7% |
47.72 |
58.52 |
28.0 |
|
• Glucosa plasmática en ayuno (mg/dL) |
|||
|
Línea de base (media) |
160.82 |
152.90 |
156.32 |
|
Cambio respecto al basal (media ajustada) |
-18.01 |
-20.35 |
7.38 |
|
Diferencia respecto del placebo (media ajustada) (IC al 95%) |
-25.392 (-49.29; -19.27) |
-23.112 (-33.85; -21.43) |
N/A3 |
|
• Peso corporal |
|||
|
Basal (media) en kg |
88.4 |
88.8 |
91.3 |
|
Cambio % respecto al basal (media ajustada) |
-2.4 |
-3.1 |
-0.1 |
|
Diferencia respecto del placebo (media ajustada) (IC al 95%) |
-1.412 (-1.76; -1.07) |
-1.542 (-1.88; -1.19) |
N/A3 |
|
• Presión arterial sistólica (mm Hg) |
|||
|
Basal (media) |
130.6 |
131.1 |
131.4 |
|
Cambio respecto al basal (media ajustada)2 |
-3.5 |
-6.8 |
1.1 |
|
Diferencia respecto del placebo (media ajustada)2 (IC al 95%) |
-4.62 (-6.9; -2.4) |
-7.92 (-10.1; -5.6) |
N/A3 |
•1 Poblaciones de intención de tratamiento con uso de la última observación en el estudio antes del tratamiento de rescate de la glucemia.
2 p < 0.001 en comparación con placebo.
3 N/A = No aplicable.
Pacientes con alteración renal: Un total de 269 pacientes con alteración renal moderada y TFGe de 30 a < 50 mL/min/1.73 m2 inadecuadamente controlados con el tratamiento actual de diabetes (cifras basales de HbA1C ≥ 7.0 a ≤ 10.5%) participaron en un estudio clínico aleatorio doble ciego, controlado con placebo para valorar la eficacia de INVOKANA® como tratamiento adyuvante del actual de la diabetes (dieta o agente antihiperglucémico, con la mayoría de los pacientes que recibía insulina, una sulfonilurea, o ambas) durante 26 semanas. La edad promedio fue 68 años, 61% de los pacientes eran hombres y la TFGe basal promedio fue 39 mL/min/1.73 m2. Los pacientes con control inadecuado de la glucemia con su tratamiento actual de diabetes se distribuyeron en forma aleatoria para agregar 100 mg de INVOKANA®, 300 mg de INVOKANA® o placebo, administrados a diario. La TFGe media en este estudio clínico fue de 39.4 mL/min/1.73 m2, que fue similar entre todos los grupos de tratamiento.
Como se muestra en la Tabla 10, se observaron mejorías significativas en HbA1C con relación a placebo para 100 mg y 300 mg de INVOKANA®, respectivamente, en la semana 26. Además, un mayor porcentaje de pacientes alcanzó una HbA1C < 7.0% en comparación con placebo. Menos pacientes que recibían INVOKANA® requirieron tratamiento de rescate de la glucemia: 4.4% de los que recibían 100 mg de INVOKANA®, 3.4% de los que recibían 300 mg de INVOKANA® y 14.4% de los que recibían placebo. Los pacientes tratados con INVOKANA® mostraron decrementos medios en el cambio porcentual respecto del peso corporal basal en comparación con placebo. Se observó una mayor incidencia de hipoglucemia en este estudio, compatible con el aumento esperado de hipoglucemia cuando se agregó a la insulina, sulfonilurea o ambas un agente no vinculado con hipoglucemia (ver Precauciones generales, Reacciones secundarias y adversas y Propiedades de farmacocinética-Poblaciones especiales).
Tabla 10. Resultados del estudio clínico controlado con placebo de 26 semanas de INVOKANA® como tratamiento adyuvante de Agentes antihiperglucémicos (AHA) en pacientes con alteración renal moderada1
|
Parámetro de eficacia |
INVOKANA® + AHA (si acaso) 26 semanas |
Placebo + AHA (si acaso) N = 90 |
|
|
100 mg N = 90 |
300 mg N = 89 |
||
|
HbA1C (%) |
|||
|
Basal (media) |
7.89 |
7.97 |
8.02 |
|
Cambio respecto al basal (media ajustada) |
-0.33 |
-0.44 |
-0.03 |
|
Diferencia respecto del placebo (media ajustada) (IC al 95%) |
-0.30 (0.53; -0.07) |
-0.402 (-0.63; -0.17) |
N/A3 |
|
• Porcentaje de pacientes que alcanzaron una HbA1C < 7% |
27.3 |
32.6 |
17.2 |
|
• Glucosa plasmática en ayuno (mg/dL) |
|||
|
Basal (media) |
170.41 |
158.48 |
160.82 |
|
Cambio respecto al basal (media ajustada) |
-14.94 |
-11.70 |
0.54 |
|
Diferencia respecto de placebo (media ajustada) (IC al 95%) |
-0.85 (-1.58; -0.13) |
-0.67 (-1.41; 0.06) |
N/A3 |
|
• Peso corporal |
|||
|
Basal (media) en kg |
90.5 |
90.2 |
92.7 |
|
Cambio % respecto al basal (media ajustada) |
-1.2 |
-1.5 |
0.3 |
|
Diferencia respecto de placebo (media ajustada) (IC al 95%) |
-1.62 (-2.3; -0.8) |
-1.82 (-2.6; -1.0) |
N/A3 |
|
• Presión arterial sistólica (mm Hg) |
|||
|
Basal (media) |
135.9 |
136.7 |
132.1 |
|
Cambio respecto al basal (media ajustada) |
-6.0 |
-6.4 |
-0.3 |
|
Diferencia respecto de placebo (media ajustada) (IC al 95%) |
-5.7 (-9.5; -1.9) |
-6.1 (-10.0; -2.3) |
N/A3 |
•1 Población de intento de tratamiento con uso de la última observación en el estudio antes del tratamiento de rescate de la glucemia.
2 p < 0.001 en comparación con placebo.
3 N/A = No aplicable.
Análisis integrado de los pacientes con alteración renal moderada: Se hizo un análisis de la población acumulada de pacientes (N = 1085) con alteración renal moderada (TFGe línea de base de 30 a < 60 mL/min/1.73 m2) de cuatro estudios controlados y con placebo para valorar el cambio en la basal en HbA1C y el cambio porcentual con respecto a la basal en el peso corporal de los pacientes. La TFGe media en este análisis fue de 48.2 mL/min/1.73 m2, similar en todos los grupos de tratamiento. La mayoría estaba recibiendo insulina, sulfonilurea o ambas.
Este análisis demostró que INVOKANA® aportó mejoras estadísticamente significativas (p < 0.001) en HbA1C y peso corporal en comparación con el placebo (ver Tabla 11). Se observó una mayor incidencia de hipoglucemia en este análisis integrado, concordante con el incremento esperado de la hipoglucemia cuando se agregaba a la insulina, la sulfonilurea o ambas un agente no vinculado con hipoglucemia (ver Precauciones generales y Reacciones secundarias y adversas).
Tabla 11. Análisis integrado de cuatro estudios clínicos de Fase 3 en pacientes con alteración renal moderada1
|
Parámetro de eficacia |
INVOKANA® + AHA (si acaso) |
Placebo + AHA (si acaso) N = 382 |
|
|
100 mg N = 338 |
300 mg N = 365 |
||
|
HbA1C (%) |
|||
|
Basal (media) |
8.10 |
8.10 |
8.01 |
|
Cambio respecto al basal (media ajustada) |
-0.52 |
-0.62 |
-0.14 |
|
Diferencia respecto de placebo (media ajustada) (IC al 95%) |
-0.382 (-0.50; -0.26) |
-0.472 (-0.59; -0.35) |
N/A3 |
|
• Peso corporal |
|||
|
Basal (media) en kg |
90.3 |
90.1 |
92.4 |
|
Cambio % respecto al basal (media ajustada) |
-2.0 |
-2.4 |
-0.5 |
|
Diferencia respecto de placebo (media ajustada) (IC al 95%) |
-1.62 (-2.0; -1.1) |
-1.92 (-2.3; -1.5) |
N/A3 |
•1 Población de intento de tratamiento con uso de la última observación en el estudio antes del tratamiento de rescate de la glucemia.
2 p < 0.001.
3 N/A = No aplicable.
Trastornos comórbidos de la Diabetes:
Presión arterial: En un análisis de cuatro estudios de 26 semanas, controlados y con placebo (N = 2313), la disminución media en la presión arterial sistólica con respecto a placebo, se observó con 100 mg de INVOKANA® (- 3.9 mm Hg), 300 mg de INVOKANA® (- 5.3 mm Hg), y placebo (-0.1 mm Hg), independientemente del uso de medicamentos antihipertensivos en la basal. En esta misma población hubo un efecto más pequeño sobre la presión arterial diastólica con medias de cambios de -2.1 mm Hg con 100 mg de INVOKANA®, -2.5 mm Hg con INVOKANA® 300 mg, y -0.3 mm Hg con placebo, independientemente del uso de medicamentos antihipertensivos en la basal. No hubo cambio discernible en la frecuencia cardiaca.
Efectos en los lípidos: En un análisis integrado de cuatro estudios controlados y con placebo de 26 semanas, los pacientes con diabetes tipo 2 tratados con ambas dosis de INVOKANA® tuvieron incremento de sus cifras séricas de colesterol total, LDL-C y HDL-C (colesterol de lipoproteínas de alta densidad) en comparación con cambios menores con placebo, en tanto que las cifras séricas de triglicéridos disminuyeron en comparación con placebo (ver Tabla 12). En la semana 26, el cociente LDL-C/HDL-C cambió en forma mínima en comparación con la basal en los tres grupos de tratamiento. A semejanza de los cambios en el número de partículas no-HDL-C, apolipoproteína B y LDL-C (medidas en el estudio de monoterapia y el de tratamiento adyuvante de metformina de 26 semanas) aumentó en un grado menor en comparación con los cambios de LDL-C (ver Reacciones secundarias y adversas).
Tabla 12. Efecto de INVOKANA® sobre parámetros de lípidos en cuatro estudios controlados y con placebo de 26 semanas1
|
INVOKANA® 100 mg (N = 833) |
INVOKANA® 300 mg (N = 834) |
Placebo (N = 646) |
||
|
Colesterol total |
||||
|
Basal media (mediana) en mg/dL |
189.09 (186.00) |
186.00 (182.90) |
191.80 (188.32) |
|
|
Cambio de la media de mínimos cuadrados (mediana) en mg/dL |
3.86 (3.86) |
6.96 (8.12) |
-0.77 (-1.54) |
|
|
Cambio % en la media de mínimos cuadrados (mediana) del colesterol total |
3.4 (2.0) |
5.2 (4.7) |
0.9 (-0.8) |
|
|
• LDL-C |
||||
|
Basal media (mediana) en mg/dL |
106.72 (105.95) |
104.40 (102.08) |
2108.27 (105.95) |
|
|
Cambio de la media de mínimos cuadrados (mediana) en mg/dL |
2.32 (1.93) |
5.80 (5.80) |
-2.32 (-1.93) |
|
|
Cambio % de la media de mínimos cuadrados (mediana) de LDL-C |
5.7 (2.0) |
9.3 (6.0) |
1.3 (-2.3) |
|
|
• HDL-C |
||||
|
Basal media (mediana) en mg/dL |
46.01 (44.08) |
46.40 (44.85) |
45.24 (44.08) |
|
|
Cambio de la media de mínimos cuadrados (mediana) en mg/dL |
3.48 (3.09) |
4.25 (4.25) |
1.16 (1.93) |
|
|
Cambio % de la media de mínimos cuadrados (mediana) en HDL-C |
9.4 (7.8) |
10.3 (9.6) |
4.0 (3.5) |
|
|
• No-HDL-C |
||||
|
Basal media (mediana) en mg/dL |
143.07 (139.21) |
131.86 (136.11) |
146.55 (143.07) |
|
|
Cambio de la media de mínimos cuadrados (mediana) en mg/dL |
-0.00 (-0.38) |
2.70 (3.09) |
-2.32 (-3.09) |
|
|
Cambio % de la media de mínimos cuadrados (mediana) en no-HDL-C |
2.2 (-0.3) |
4.3 (2.0) |
0.7 (-2.4) |
|
|
• Cociente LDL-C/HDL-C |
||||
|
Basal media (mediana) |
2.5 (2.4) |
2.4 (2.3) |
2.5 (2.4) |
|
|
Cambio en la media de mínimos cuadrados (mediana) |
-0.1 (-0.1) |
-0.1 (-0.1) |
-0.2 (-0.1) |
|
|
Cambio en el cociente de la media de mínimos cuadrados (mediana) % |
1.4 (-5.2) |
0.8 (-2.1) |
0.8 (-6.5) |
|
|
• Triglicéridos |
||||
|
Basal media (mediana) en mg/dL |
182.45 (153.22) |
180.68 (150.56) |
185.99 (163.85) |
|
|
Cambio de la media de mínimos cuadrados (mediana) en mg/dL |
-9.74 (-8.85) |
-19.48 (-11.51) |
-0.00 (-2.65) |
|
|
Cambio % de la media de mínimos cuadrados (mediana) en triglicéridos |
2.4 (-6.0) |
0.0 (-9.2) |
7.6 (-2.2) |
|
1 Como monoterapia o tratamiento adyuvante con metformina, metformina y sulfonilurea, y metformina y pioglitazona.
Pacientes con HbA1c > 10% a < 12%: Un subestudio de pacientes con HbA1c basal > 10 a < 12% con canagliflozina como monoterapia, mostró resultados en la disminución de HbA1c desde la basal, de -2.13% y -2.56% con canagliflozina 100 mg y 300 mg respectivamente.
Glucosa plasmática en ayuno: En cuatro estudios controlados con placebo, el tratamiento con INVOKANA® como monoterapia o tratamiento adyuvante con uno o dos fármacos antihiperglucémicos orales, produjo cambios medios respecto a la basal con relación al placebo en FPG, de -21.6 mg/dL a -34.2 mg/dL para INVOKANA® 100 mg, y -34.2 mg/dL a -43.2 mg/dL para 300 mg de INVOKANA®, respectivamente. Estas disminuciones se sostuvieron durante el periodo de tratamiento y fueron casi máximas después del primer día de la terapéutica.
Glucosa posprandial: Con el uso de una prueba de tolerancia de una comida mixta estandarizada, se determinó la glucosa posprandial en tres estudios clínicos controlados y con placebo, como monoterapia o tratamiento adyuvante con uno o dos fármacos antihiperglucémicos. INVOKANA® produjo cambio de disminución media respecto al basal con relación al placebo en la glucosa posprandial de -27. 1 mg/dL a -48.62 mg/dL para 100 mg y -37.82 mg/dL a -63.03 mg/dL para 300 mg, respectivamente, debido a decrementos en la concentración de glucosa preprandial y menores variaciones en la glucosa posprandial.
Función de las células Beta: Los estudios clínicos en un subgrupo de pacientes con diabetes tipo 2 (N = 297) con INVOKANA® durante 26 semanas indican mejor función de las células Beta con base en parámetros como la valoración del modelo de homeostasia para la función de las células Beta (HOMA2-B) y la tasa de secreción de insulina mejorada con la prueba de tolerancia de una comida mixta.
Farmacocinética: La farmacocinética de la canagliflozina es esencialmente similar en sujetos sanos y pacientes con diabetes tipo 2. Después de una administración oral de 100 mg y 300 mg en sujetos sanos, la canagliflozina se absorbió rápidamente, con concentraciones plasmáticas máximas (Tmáx media) que se presentaron de 1 a 2 horas después de la dosis. La Cmáx y el ABC de la canagliflozina aumentaron en una forma proporcional a la dosis de 50 a 300 miligramos. La vida media aparente (t1/2) (expresada como ± desviación estándar) para las dosis de 100 y 300 mg fue de 10.6 ± 2.13 horas, y 13.1 ± 3.28 horas, respectivamente. Se alcanzó el estado estable después de 4 a 5 días de dosificación una vez al día de 100 mg a 300 mg de canagliflozina, la cual no muestra farmacocinética dependiente de la hora y se acumuló en plasma hasta 36% después de dosis múltiples de 100 y 300 mg.
Absorción: La biodisponibilidad oral media absoluta de la canagliflozina es de casi 65%. La coadministración de canagliflozina con una comida rica en grasa no tuvo efecto sobre su farmacocinética; por lo tanto, se puede tomar INVOKANA® con o sin alimentos. Sin embargo, con base en el potencial de disminuir las variaciones de la glucosa plasmática posprandial, debido a una absorción tardía de la glucosa intestinal, se recomienda que INVOKANA® se tome preferentemente antes de la primera comida del día (ver Dosis y vía de administración).
Distribución: El volumen medio de estado estable de distribución de la canagliflozina después de una sola administración intravenosa en solución en sujetos sanos fue de 119 L, lo que sugiere una extensa distribución en los tejidos. Canagliflozina se une extensamente a las proteínas plasmáticas (99%), sobre todo a la albúmina. La unión es independiente de la concentración plasmática de canagliflozina y no se altera de manera significativa en los pacientes con alteración renal o hepática.
Metabolismo: La principal vía de eliminación metabólica de la canagliflozina es la O-glucoronización, principalmente por UGT1A9 y UGT2B4, hasta dos metabolitos, O-glucorónidos inactivos. Se observaron aumentos en el ABC de canagliflozina (26% y 18%) en sujetos portadores del alelo UGT1A9*3 y UGT2B4*2 respectivamente . Estos aumentos en la exposición a canagliflozina no se espera que sean clínicamente relevantes. El metabolismo de canagliflozina (oxidativo) mediado por la CYP3A4 es mínimo (aproximadamente 7%) en los seres humanos.
Eliminación: Después de la administración de una sola dosis oral de canagliflozina [14C] a sujetos saludables, se recuperó 41.5, 7.0 y 3.2% de la dosis radiactiva administrada en las heces, como canagliflozina, un metabolito hidroxilado y un metabolito O-glucorónido, respectivamente. La circulación enterohepática de la canagliflozina fue mínima.
Casi 33% de la dosis radiactiva administrada se excretó en la orina, principalmente como metabolito glucorónido (30.5%). Se eliminó menos de 1% de la dosis sin cambios, como canagliflozina, en la orina. La depuración renal de las dosis de 100 y 300 mg varió de 1.30 a 1.55 mL/minuto.
Canagliflozina es un fármaco de lenta depuración, con una eliminación sistémica media de casi 192 mL/min en sujetos sanos después de su administración intravenosa.
Poblaciones especiales:
Alteración renal: En un estudio abierto de una sola dosis, se valoró la farmacocinética de 200 mg de canagliflozina en sujetos con grados variables de alteración renal (clasificados utilizando la fórmula de Modificación de Dieta en la Enfermedad Renal (MDRD)-TFGe) en comparación con sujetos sanos. El estudio incluyó a 3 sujetos con función renal normal (TFGe ≥ 90 mL/min/1.73 m2), 10 sujetos con alteración renal leve (TFGe 60 a < 90 mL/min/1.73 m2), 9 sujetos con alteración renal moderada (TFGe 30 a < 60 mL/min/1.73 m2) y 10 con alteración renal grave (TFGe 15 a < 30 mL/min/1.73 m2) así como 8 sujetos con nefropatía terminal (IRCT), en proceso de hemodiálisis.
La Cmáx de canagliflozina aumento moderadamente 13%, 29% y 29% en sujetos con insuficiencia renal leve, moderada y severa, respectivamente, pero no en sujetos en hemodiálisis. En comparación con sujetos sanos, el ABC plasmática de canagliflozina aumentó aproximadamente 17%, 63% y 50% en sujetos con insuficiencia renal leve, moderada y grave, respectivamente, pero fue similar para los sujetos con IRCT y los sanos. (ver Dosis y vía de administración, Precauciones generales y Reacciones secundarias y adversas).
Canagliflozina se eliminó mínimamente por hemodiálisis.
Alteración hepática: Con relación a sujetos con función hepática normal, los cocientes medios geométricos de la Cmáx y el ABC8 de canagliflozina fueron 107% y 110% respectivamente, en sujetos con Child-Pugh A (alteración hepática leve), y 96% y 111% respectivamente, en sujetos con Child-Pugh B (alteración hepática moderada) después de la administración de una sola dosis de 300 mg de Canagliflozina.
Estas diferencias no se consideran clínicamente significativas. No es necesario un ajuste de dosis en los pacientes con alteración hepática leve o moderada. No hay experiencia clínica en pacientes con alteración hepática Child-Pugh C (grave) y, por lo tanto, no se recomienda INVOKANA® para usarse en esta población.
Ancianos (≥ 65 años de edad): La edad no tuvo efecto clínicamente significativo sobre la farmacocinética de canagliflozina, con base en un análisis de farmacocinética de población (ver Dosis y vía de administración, Precauciones generales y Reacciones secundarias y adversas).
Pacientes pediátricos (< 18 años de edad): No se han hecho estudios de caracterización de la farmacocinética de canagliflozina en pacientes pediátricos.
Otras poblaciones: No se necesita ajuste de la dosis con base en género, raza/grupo étnico o índice de masa corporal. Estas características no tuvieron efecto clínico significativo sobre la farmacocinética de canagliflozina, con base en análisis de farmacocinética de la población.
CONTRAINDICACIONES: Hipersensibilidad a la sustancia activa o cualquiera de sus excipientes (ver Reacciones secundarias y adversas).
INVOKANA® no se ha estudiado en pacientes con diabetes tipo 1, por lo tanto, no se recomienda su uso en estos pacientes.
INVOKANA® no debe usarse para el tratamiento de cetosis diabética o en pacientes con tasa de filtración glomerular estimada < 45 mL/min/1.73 m2 (depuración de creatinina < 45 mL/min), ya que no será eficaz en esos pacientes.
RESTRICCIONES DE USO DURANTE EL EMBARAZO Y LA LACTANCIA:
Embarazo: No hay estudios adecuados y bien controlados en mujeres embarazadas. En animales no se señalan efectos lesivos directos o indirectos con respecto a la toxicidad reproductiva (ver Precauciones en relación con efectos de carcinogénesis mutagénesis, teratogénesis y sobre la fertilidad). Se deben considerar terapias alternativas apropiadas durante el embarazo, especialmente durante el segundo y tercer trimestre INVOKANA® sólo debe ser usada durante el embarazo si el beneficio potencial justifica el riesgo potencial al feto.
Lactancia: Los datos farmacodinámicos/toxicológicos disponibles en animales han mostrado excreción de canagliflozina en la leche. No se sabe si canagliflozina se excreta en la leche humana. No podría descartarse el riesgo para el lactante. Se debe tomar la decisión si se continua la lactancia o el tratamiento con INVOKANA®, tomando en cuenta el beneficio de la lactancia materna para el lactante y el beneficio del tratamiento para la mujer (ver Precauciones en relación con efectos de carcinogénesis, mutagénesis, teratogénesis y sobre la fertilidad).
REACCIONES SECUNDARIAS Y ADVERSAS: Se trata de eventos que se consideraron razonablemente vinculados con el uso de canagliflozina con base en una valoración amplia de la información disponible de eventos adversos. No se puede establecer una relación causal con INVOKANA® de manera confiable en casos individuales. Debido a que los estudios clínicos se hacen bajo condiciones ampliamente variables, la tasa de reacciones adversas observadas en los estudios clínicos de un fármaco; no pueden compararse directamente con las de estudios clínicos de otro y pueden no reflejar las observadas en la práctica clínica.
Se valoró la seguridad de INVOKANA® (Canagliflozina) en 10,285 pacientes con diabetes tipo 2, incluyendo 3,139 tratados con 100 mg de INVOKANA® y 3,506 tratados con 300 mg de INVOKANA® que recibieron fármaco en nueve estudios de Fase 3, doble ciego, controlados.
Se hicieron análisis de seguridad en pacientes que recibieron INVOKANA® como monoterapia o como tratamiento adyuvante con otros agentes antihiperglucémicos. Se estudió INVOKANA® como monoterapia en un ensayo clínico controlado con placebo de 26 semanas de duración que incluyó un subestudio de tratamiento activo en pacientes con hiperglucemia más intensa (HbA1c [A1C] > 10 y ≤ 12%). En cinco estudios controlados con placebo o fármaco activo se investigó INVOKANA® como tratamiento adyuvante con otros agentes antihiperglucémicos: dos como metformina (26 y 52 semanas); dos con metformina y sulfonilurea (26 y 52 semanas), y uno como metformina y pioglitazona (26 semanas). En dos estudios controlados y con placebo se investigó el uso de INVOKANA® agregado al esquema actual de tratamiento de la diabetes, uno en pacientes de edad avanzada y otro con pacientes con alteración renal moderada. En un estudio cardiovascular particular, actualmente en curso en pacientes con diabetes tipo 2; se hicieron análisis de seguridad donde se investigó INVOKANA® como tratamiento adyuvante con una sulfonilurea e insulina.
Se hizo una valoración primaria de la seguridad y tolerabilidad en un análisis acumulado (N = 2,313) de cuatro estudios clínicos de 26 semanas controlado con placebo (monoterapia y tratamiento adyuvante con metformina, metformina y sulfonilurea, y metformina y pioglitazona). En este análisis acumulado, las reacciones adversas de más frecuente informe durante el tratamiento (≥ 5%) fueron candidiasis vulvovaginal, infección de vías urinarias, y poliuria o polaquiuria. Las reacciones adversas que llevaron a la discontinuación de ≥5% de todos los pacientes tratados con INVOKANA® en estos estudios fueron candidiasis vulvovaginal (0.7% de las mujeres) y balanitis o balanopostitis (0.5% de los hombres). Se hicieron análisis de seguridad adicionales (incluyendo aquéllos a largo plazo) de datos a través de todo el programa de canagliflozina (estudios controlados con placebo y sustancia activa) para valorar los eventos adversos comunicados a fin de identificar las reacciones adversas.
En la Tabla 13 se listan las diversas reacciones comunicadas en ≥ 2% de los pacientes tratados con INVOKANA® en los cuatro estudios clínicos acumulados de 26 semanas, controlados y con placebo (N = 2,313).
Tabla 13. Reacciones adversas de cuatro estudios acumulados de 26 semanas, controlados y con placebo1 informados en ≥ 2% de los pacientes tratados con INVOKANA®
|
Órgano, aparato o sistema Reacciones adversas |
INVOKANA® 100 mg (N = 833) % |
INVOKANA® 300 mg (N = 834) % |
Placebo (N = 646) % |
|
Trastornos gastrointestinales |
|||
|
Estreñimiento |
15 (1.8) |
19 (2.3) |
6 (0.9) |
|
Náusea |
18 (2.2) |
19 (2.3) |
10 (1.5) |
|
Sed2 |
23 (2.8) |
19 (2.3) |
1 (0.2) |
|
Trastornos renales y urinarios |
|||
|
Poliuria o polaquiuria3 |
44 (5.3) |
38 (4.6) |
5 (0.8) |
|
Infección de vías urinarias4 |
49 (5.9) |
36 (4.3) |
26 (4.0) |
|
Trastornos del aparato reproductor y de mama |
|||
|
Balanitis o balanopostitis5 |
17 (4.2) |
15 (3.7) |
2 (0.6) |
|
Candidiasis vulvovaginal6 |
44 (10.4) |
49 (11.4) |
10 (3.2) |
1 Incluye monoterapia y tratamiento agregado con metformina, metformina y sulfonilurea, y metformina y pioglitazona.
2 La sed incluye al término sed con incidencias (1.3%, 1.9%, 0.2%) para 100 mg de INVOKANA®, 300 mg de INVOKANA® y placebo, respectivamente y también las denominaciones boca seca y polidipsia, con incidencia < 1% en cualquier grupo de tratamiento.
3 Poliuria y polaquiuria incluyen los términos poliuria con incidencias (0.7%, 1.4%, 0.0%) y polaquiuria (4.2%, 3.1%, 0.6%) para 100 mg de INVOKANA®, 300 mg de INVOKANA® y placebo, respectivamente, y también incluye las denominaciones aumento del gasto urinario, urgencia Miccional y Nocturia nocturna con incidencias < 1% para cualquier grupo de tratamiento.
4 La infección de vías urinarias incluye la denominación infección de vías Urinarias con incidencias (5.5%, 4.1%, 4.0%) para 100 mg de INVOKANA®, 300 mg de INVOKANA® y placebo, respectivamente, y también incluye las denominaciones cistitis, infección renal e infección urinaria, con incidencias < 1% en cualquier grupo de tratamiento. No hubo desequilibrio entre 100 mg de INVOKANA®, 300 mg de INVOKANA® y placebo para las infecciones renales o infección urinaria.
5 Balanitis o balanopostitis incluyen los términos balanitis (2.2%, 1.7%, 0.0%) y balanopostitis (1.0%, 0.7%, 0.3%) en las incidencias para 100 mg de INVOKANA®, 300 mg de INVOKANA® y placebo, respectivamente, y también incluye las denominaciones balanitis por especies de cándida e infección genital micótica con incidencia < 1% en cualquier grupo de tratamiento.
6 La candidiasis vulvovaginal incluye las denominaciones candidiadis vulvovaginal (1.6%, 2.8%, 1.0%), infección micótica vulvovaginal (5.9%, 5.3%, 1.3%), vulvovaginitis (1.9%, 1.6%, 0.0%), e infección vaginal (1.2%, 1.6%, 0.6%) en las incidencias para 100 mg de INVOKANA®, 300 mg de INVOKANA® y placebo, respectivamente, y también incluye las denominaciones vulvitis e infección micótica genital, con incidencia < 1% en cualquier grupo de tratamiento.
Otras reacciones adversas en estudios clínicos de INVOKANA® que ocurrieron con una tasa < 2% en estudios controlados y con placebo fueron reacciones adversas relacionadas con disminución del volumen intravascular (mareo postural, hipotensión ortostática, hipotensión, deshidratación y síncope), exantema, y urticaria.
Las características de seguridad en el estudio clínico de pacientes en edad avanzada, el estudio clínico en pacientes con disfunción renal moderada y los dos estudios controlados con producto activo fueron en general consistentes con las reacciones adversas identificadas en la Tabla 13. En el estudio cardiovascular extenso, las características de seguridad también fueron, en general, consistentes con otras reacciones adversas relacionadas con la disminución del volumen intravascular (ver Dosis y vía de administración, Reacciones secundarias y adversas y Precauciones generales).
Descripción de reacciones adversas selectas:
Disminución del volumen intravascular: En el análisis acumulado de cuatro estudios controlados con placebo de 26 semanas, y dos estudios controlados y con activo, la incidencia de reacciones adversas relacionadas con un menor volumen intravascular (por ejemplo, vértigo postural, hipotensión ortostática, hipotensión, deshidratación y síncope) fue 1.2% para INVOKANA® 100 mg, 1.3% para INVOKANA® 300 mg y 1.1% para placebo. La incidencia con el tratamiento con INVOKANA® en los 2 estudios controlados con activo fue similar a los componentes.
En el estudio cardiovascular dedicado, donde los pacientes generalmente eran mayores, con una mayor incidencia de comorbilidades, las incidencias de reacciones adversas relacionadas con un menor volumen intravascular fueron 2.8% con 100 mg de INVOKANA®, 4.6% con 300 mg y 1.9% placebo.
Para valorar los factores de riesgo de éstas, se hizo un análisis acumulado más grande (N = 9,439) de los pacientes de ocho estudios de Fase 3 controlados, incluyendo ambas dosis de INVOKANA®, en el cual, los pacientes con diuréticos de asa, aquellos con alteración renal moderada (TFGe 30 a < 60 mL/min/1.73 m2, depuración de creatinina 30 a < 60 mL/min/1.73 m2), y los pacientes ≥ 75 años, tuvieron mayores incidencias de esas reacciones adversas. Para los pacientes con tratamiento con diuréticos de asa, las incidencias fueron de 3.2% para 100 mg de INVOKANA® y 8.8% para 300 mg de INVOKANA®, en comparación con 4.7% en el grupo de control. Para los pacientes con TFGe basal < 60 mL/min/1.73 m2 (depuración de creatinina < 60 mL/min/1.73 m2), las incidencias fueron de 4.8% con 100 mg de INVOKANA® y 8.1% con 300 mg de INVOKANA® en comparación con 2.6% en el grupo de control. En los pacientes > 75 años de edad, las indicaciones fueron de 4.9% con 100 mg de INVOKANA® y 8.7% con 300 mg de INVOKANA® en comparación con 2.6% en el grupo de control (ver Dosis y vía de administración, Precauciones generales y Farmacocinética-poblaciones especiales).
En el estudio cardiovascular dedicado y el análisis acumulado más grande, la descontinuación por reacciones adversas relacionadas a la reducción del volumen intravascular y las reacciones adversas graves relacionadas con disminución del volumen intravascular no aumentaron con INVOKANA®.
Hipoglucemia en el tratamiento adyuvante con insulina o secretagogos de insulina: La frecuencia de hipoglucemia fue baja (< 6%) entre los grupos de tratamiento cuando se usó como monoterapia y como terapia adjunta a antihiperglucémicos no asociados con hipoglucemia. Cuando se usó INVOKANA® como tratamiento adyuvante con insulina o sulfonilurea, se observó hipoglucemia con mayor frecuencia, lo que es compatible con el incremento esperado de la hipoglucemia cuando se agrega a la insulina o uno de sus componentes (por ejemplo, sulfonilurea) un agente no relacionado con la hipoglucemia. En el subestudio de 18 semanas, cuando INVOKANA® se agregó al tratamiento con insulina, se observó hipoglucemia en 49.3%, 48.2% y 36.8% de los pacientes tratados con 100 mg de INVOKANA®, 300 mg de INVOKANA® y placebo, respectivamente. Cuando INVOKANA® se adicionó al tratamiento con sulfonilureas, se observó hipoglucemia en 4.1%, 12.5% y 5.8% de los pacientes tratados con 100 mg de INVOKANA®, 300 mg de INVOKANA® y placebo, respectivamente, sin informes de hipoglucemia grave en grupos de tratamiento alguno (ver Dosis y vías de administración y Precauciones generales).
Infecciones micóticas genitales: Se reporto candidiasis vulvovaginal (incluyendo vulvovaginitis e infección micótica vulvovaginal) en 10.4% y 11.4% de las mujeres tratadas con INVOKANA® 100 mg y 300 mg respectivamente, en comparación con 3.2% en las mujeres tratadas con placebo. Muchos reportes de candidiasis vulvovaginal se presentaron durante los primeros 4 meses de tratamiento con canagliflozina. Entre las mujeres bajo tratamiento con INVOKANA®, 2.3% experimentaron más de una infección. En general, 0.7% de todas las mujeres descontinuaron el tratamiento con INVOKANA® debido a candidiasis vulvovaginal (ver Advertencias).
Se reportó balanitis o balanopostitis en 4.2% y 3.7% de los hombres tratados con INVOKANA® 100 mg y 300 mg respectivamente, en comparación con 0.6% de los hombres tratados con placebo. Entre los hombres bajo tratamiento con INVOKANA®, 0.9% experimentaron más de una infección. En general, 0.5% de los hombres descontinuaron el tratamiento con INVOKANA® debido a balanitis candidiásica o balanopostitis. En el análisis acumulado de 8 estudios clínicos controlados, se reportó fimosis en 0.3% de los hombres no circuncidados, en este análisis también se reportó en circuncisión en 0.2% de los hombres tratados con canagliflozina (ver Precauciones generales).
Infecciones de vías urinarias: Las infecciones de tracto urinario fueron más frecuentemente reportadas en los pacientes bajo tratamiento con INVOKANA® 100 mg y 300 mg (5.9% vs. 4.3% respectivamente) en comparación con placebo (4.0%). La mayoría fueron de leves a moderadas, sin aumento en la frecuencia de eventos adversos serios. Los sujetos respondieron al tratamiento estándar mientras continuaban con canagliflozina. La incidencia de infecciones recurrentes no aumentó con canagliflozina.
Fractura ósea: En un estudio cardiovascular de 4.327 pacientes con alto riesgo de enfermedad cardiovascular conocido, las tasas de incidencia de fractura ósea fueron 16.3, 16.4 y 10.8 por 1000 pacientes-año de exposición a INVOKANA® 100 mg, INVOKANA® 300 mg y placebo, respectivamente, el desequilibrio de la fractura se produce inicialmente dentro de las primeras 26 semanas de terapia. En otros estudios de diabetes tipo 2 con INVOKANA®, que incluyó una población general de la diabetes de aproximadamente 5800 pacientes, no se observó diferencia en el riesgo de fractura en relación al control. Después de 104 semanas de tratamiento, la canagliflozina no afectó negativamente a la densidad mineral ósea.
Reacciones adversas en poblaciones específicas:
Ancianos: El perfil de seguridad en pacientes ancianos fue, generalmente, consistente con el de pacientes jóvenes. Pacientes ≥ 75 años de edad presentaban mayor incidencia de reacciones adversas relacionadas con disminución del volumen intravascular (vértigo postural, hipotensión ortostática, hipotensión) con incidencias de 4 9%, 8.7% y 2 6% con INVOKANA® 100 mg, 300 mg y el grupo control respectivamente. Se reportó disminución en la TFGe (-3.6% y -5.2%) con INVOKANA® 100 mg y 300 mg respectivamente, en comparación con el grupo control (-3.0%) (ver Dosis y vía de administración y Precauciones generales).
Pacientes con TFGe de 45 a < 60 mL/min/1.73 m2 (depuración de creatinina de 45 a < 60 mL/min): En un análisis de pacientes con TFGe de 45 a < 60 mL/min/1.73 m2 (depuración de creatinina de 45 a < 60 mL/min), la incidencia de reacciones adversas relacionadas con la reducción del volumen intravascular fue 4.6% con INVOKANA® 100 mg y 7.1% con INVOKANA® 300 mg en relación a 3.4% con placebo (ver Dosis y vía de administración y Precauciones generales). Los niveles de creatinina sérica aumentaron 4.9% y 7.3% con INVOKANA® 100 mg y 300 mg, respectivamente, en relación a 0.2% con placebo. Los niveles de BUN aumentaron 13.2% y 13.6% con INVOKANA® 100 mg y 300 mg, respectivamente, en relación a 0.7% con placebo. La proporción de pacientes con grandes disminuciones en la TFGe (> 30%) en cualquier momento durante el tratamiento fue 6.1%, 10.4% y 4.3% con INVOKANA® 100 mg, 300 mg y placebo respectivamente. Cuando se alcanzó el objetivo primario, 2.3% de los pacientes tratados con INVOKANA® 100 mg, 4.3% de los pacientes tratados con INVOKANA® 300 mg y 3.5% con placebo, presentaban esas disminuciones (ver Precauciones generales).
La incidencia de potasio sérico elevado (> 5.4 mEq/L y 15% mayor a la basal) fue 5.2% con INVOKANA® 100 mg, 9.1% con INVOKANA® 300 mg y 5.5% con placebo (ver Precauciones generales). En raras ocasiones, las elevaciones fueron observadas en pacientes con insuficiencia renal moderada que tenían elevaciones previas de potasio y/o estaban bajo tratamiento con múltiples medicamentos para reducir la excreción de potasio, como diuréticos ahorradores de potasio e inhibidores de la enzima convertidora de angiotensina (IECA). En general, la elevación fue transitoria y no requirió tratamiento específico.
Los niveles de fosfato sérico aumentaron 3.3% y 4.2% con INVOKANA® 100 mg y 300 mg respectivamente, en comparación con 1.1% con placebo. La incidencia de elevación de fosfato sérico (> 1.65 mmol/L y 25% arriba de la basal) fue 1.4% con INVOKANA® 100 mg, 1.3% con INVOKANA® 300 mg y 0.4% con placebo. En general, la elevación fue transitoria y no requirió tratamiento específico.
PRECAUCIONES EN RELACIÓN CON EFECTOS DE CARCINOGÉNESIS, MUTAGÉNESIS, TERATOGÉNESIS Y SOBRE LA FERTILIDAD: Ningún dato clínico reveló riesgo particular para los seres humanos con base en estudios convencionales de farmacología de seguridad, toxicidad de dosis repetidas, genotoxicidad y toxicidad para la reproducción y el desarrollo. Un estudio en ratas juveniles en el que se administró canagliflozina desde el día posnatal 21 hasta el día 90, no mostró aumento en la sensibilidad en comparación con los efectos observados en ratas adultas.
Carcinogenicidad y mutagenicidad: Canagliflozina no aumentó la incidencia de tumores en ratones machos y hembras, en un estudio de 2 años a dosis de 10, 30 y 100 mg/kg. La dosis más alta, de 100 mg/kg aportó hasta 14 veces la dosis clínica de 300 mg con base en el ABC de la exposición. Canagliflozina aumentó la incidencia de tumores testiculares de células de Leydig en ratas macho con todas las dosis probadas (10, 30 y 100 mg/kg) la dosis más baja, de 10 mg/kg, es aproximadamente 1.5 veces la dosis clínica de 300 mg con base en el ABC de la exposición. Las dosis mayores de canagliflozina (100 mg/kg) en ratas macho y hembra, aumentaron la incidencia de feocromocitomas y tumores de células tubulares renales. Con base en el ABC de la exposición, el nivel del efecto no observable (NOEL) de 30 mg/kg/día para feocromocitomas y tumores de los tubulos renales es casi 4.5 veces la correspondiente para la dosis clínica diaria de 300 mg. Con base en estudios mecánicos preclínicos y clínicos, los tumores de células de Leydig y tubulares renales y feocromocitomas se deben a mecanismos considerados como específicos para ratas. Los tumores de células de túbulos renales inducidos por canagliflozina y los feocromocitomas en ratas parecen causados por absorción deficiente de los carbohidratos como consecuencia del efecto inhibitorio de canagliflozina en SGLT-1 del intestino de las ratas. Los estudios mecánicos de clínica no han mostrado absorción deficiente de carbohidratos en seres humanos a dosis de canagliflozina de hasta 2 veces la dosis clínica máxima recomendada. Los tumores de células de Leydig se vinculan con un aumento en la hormona luteinizante (LH), que es un mecanismo conocido. En un estudio clínico de 12 semanas, la LH no estimulada no aumentó en las ratas macho tratadas con canagliflozina.
La canagliflozina no fue mutagénica con y sin activación metabólica en el ensayo de Ames. La canagliflozina fue mutagénica en el ensayo in vitro de linfoma en ratón con, pero no sin activación metabólica. La canagliflozina no fue mutagénica o clastogénica en un ensayo de micronúcleos oral in vivo en ratas y un ensayo Cometa oral in vivo en ratas.
Toxicología reproductiva: En un estudio de desarrollo pre y posnatal, canagliflozina administrada en ratas hembra desde el día de gestación 6 al día 20 de lactancia, dio como resultado disminución del peso corporal en crías machos y hembras a dosis tóxicas maternas > 30 mg/kg/día (exposición ≥ 5.9 veces la exposición humana a canagliflozina con las dosis máximas recomendadas en humanos). La toxicidad materna se limitó a disminución de peso.
En estudios de fertilidad en ratas macho y hembra, canagliflozina no tuvo efectos adversos sobre el desarrollo embrionario temprano, el apareamiento y la fertilidad hasta la dosis más alta de 100 mg/kg (hasta 19 veces la dosis clínica de 300 mg, con base en el ABC de la exposición).
Fertilidad: No se ha estudiado el efecto de canagliflozina sobre la fertilidad en humanos. No se observaron efectos sobre la fertilidad en estudios en animales (ver Precauciones en relación con efectos de carcinogénesis, mutagénesis, teratogénesis y sobre la fertilidad).
INTERACCIONES MEDICAMENTOSAS Y DE OTRO GÉNERO:
Valoración de interacciones in vitro: Canagliflozina se metaboliza principalmente por conjugación con glucurónido, mediado por la enzima UDP glucoronil transferasa 1A9 (UGT1A9) y 2B4 (UGT2B4).
En estudios in vitro, canagliflozina no causo inhibición de las enzimas del citocromo P450: CYP1A2, CYP2A6, CYP2C19, CYP2D6, CYP2E1, CYP2B6, CYP2C8 y CYP2C9. A concentraciones más altas que las terapéuticas, tampoco indujo la expresión de las enzimas: CYP1A2, CYP2C19, CYP2B6, CYP3A4. Canagliflozina inhibió débilmente la enzima CYP3A4 in vitro; sin embargo, basado en un estudio clínico, no se observó interacción clínicamente relevante. Por lo tanto, no se espera que canagliflozina altere la depuración metabólica de los medicamentos coadministrados que son metabolizados por estas enzimas.
Canagliflozina es un sustrato de la glicoproteína P (gpP), e inhibe con baja potencia el transporte de digoxina mediado por glucoproteína P.
Valoración de las interacciones in vivo: Se llevaron a cabo estudios de interacción farmacológica específicos para investigar los efectos de los inhibidores o inductores de las enzimas del metabolismo farmacológico UGT1A9 y UGT2B4 y los transportadores gpP y MRP2 sobre la farmacocinética de canagliflozina, así como para valorar los efectos de canagliflozina sobre la farmacocinética del sustrato de gpP digoxina.
Efectos de canagliflozina sobre otros fármacos: Se evaluaron los efectos de otros medicamentos sobre canagliflozina en estudios clínicos. Se observó que ciclosporina, hidroclorotiazida, anticonceptivos orales (etinilestradiol y levonorgestrel), metformina y probenecid no tuvieron efectos clínicamente relevantes en la farmacocinética de canagliflozina.
Rifampicina: La coadministración con rifampicina, un inductor no selectivo de varias enzimas UGT y transportadores farmacológicos incluyendo UGT1A9, UGT2B4, gpP y MRP2 disminuyeron la exposición a canagliflozina. Si es necesario coadministrar la combinación con un inductor de estos UGT y sistemas de transporte farmacológico (por ejemplo, rifampicina, fenitoína, barbitúricos, fenobarbital, ritonavir carbamazepina, efavirenz y hierba de San Juan [Hypericum perforatum]) con INVOKANA®, vigile la HbA1c en pacientes que reciben 100 mg de INVOKANA® una vez al día, considerando el aumento de dosis a 300 mg una vez al día si se requiere control glucémico adicional. En pacientes con TFGe de 45 a < 60 mL/min/1.73 m2 (depuración de creatinina 45 a < 60 mL/min) bajo tratamiento con INVOKANA® 100 mg que están tomando de forma concomitante inductores de UGT y que requieren control glucémico adicional, se debe considerar el uso de otros tratamientos antihiperglucémicos.
Tabla 14. Efecto de los medicamentos coadministrados sobre la exposición sistémica a canagliflozina
|
Medicamento coadministrado |
Dosis del medicamento coadministrado1 |
Dosis de canagliflozina1 |
Cociente medio geométrico (cociente con/sin fármaco coadministrado) ningún efecto = 1 |
|
|
ABC2 (IC al 90%) |
Cmáx (IC al 90%) |
|||
|
No se requiere ajuste de dosis de INVOKANA® para los siguientes |
||||
|
Ciclosporina |
400 mg |
300 mg una vez al día por 8 días |
1.23 (1.19 ; 1.27) |
1.01 (0.91 ; 1.11) |
|
Etinilestradiol y levonorgestrel |
0.03 mg etinilestradiol y 0.15 mg de levonorgestrel |
200 mg una vez al día o por 6 días |
0.91 (0.88 ; 0.94) |
0.92 (0.84 ; 0.99) |
|
Hidroclorotiazida |
25 mg una vez al día durante 35 días |
300 mg una vez al día por 7 días |
1.12 (1.08 ; 1.17) |
1.15 (1.06 ; 1.25) |
|
Metformina |
2000 mg |
300 mg una vez al día por 8 días |
1.10 (1.05 ; 1.15) |
1.05 (1.96 ; 1.16) |
|
Probenecid |
500 mg dos veces al día por 3 días |
300 mg una vez al día por 17 días |
1.21 (1.16 ; 1.25) |
1.13 (1.00 ; 1.28) |
|
Rifampicina |
600 mg una vez al día por 8 días |
300 mg |
0.49 (0.44 ; 0.54) |
0.72 (0.61 ; 0.84) |
1 Dosis única a menos que se indique lo contrario.
2 ABCinf para los medicamentos administrados como dosis única y ABC 24 h para medicamentos administrados como dosis múltiples.
En estudios de interacción farmacológica llevados a cabo en sujetos sanos, canagliflozina en estado estacionario no tuvo efecto clínicamente importante sobre la farmacocinética de metformina, anticonceptivos orales (etinilestradiol y levonorgestrel), gliburida, simvastatina, paracetamol o warfarina.
Digoxina: La combinación de canagliflozina 300 mg 1 vez al día por 7 días con una sola dosis de 0.5 mg de digoxina, seguida de 0.25 mg/día por 6 días, dio como resultado 20% de aumento en el ABC y 36% de aumento en la Cmax de digoxina posiblemente debido a la interacción a nivel de gpP. Por lo tanto, los pacientes bajo tratamiento con digoxina u otros glucósidos cardiacos (digoxina, por ejemplo) deberían vigilarse apropiadamente.
Tabla 15. Efecto de canagliflozina sobre la exposición sistémica de medicamentos coadministrados
|
Medicamento coadministrado |
Dosis del medicamento coadministrado1 |
Dosis de Canagliflozina1 |
Cociente medio geométrico (cociente con/sin fármaco coadministrado) ningún efecto = 1 |
||
|
ABC2 (IC al 90%) |
Cmax (IC al 90%) |
||||
|
No se requiere ajuste de la dosis del fármaco coadministrado para los siguientes |
|||||
|
Digoxina |
0.5 mg una vez el primer día, seguido por 0.25 mg una vez al día por 6 |
300 mg una vez al día por 7 días |
digoxina |
1.20 (1.12; 1.28) |
1.36 (1.21; 1.53) |
|
Etinilestradiol y levonorgestrel |
0.03 mg de etinilestradiol y 0.15 mg de levonogestrel |
200 mg una vez al día durante 6 días |
etinilestradiol |
1.07 (0.99; 1.15) |
1.22 (1.21; 1.53) |
|
levonorgestrel |
1.06 (1.00; 1.13) |
1.22 (1.11; 1.35) |
|||
|
Gliburida |
1.25 mg |
200 mg una vez al día durante 6 días |
gliburida |
1.02 (0.98; 1.07) |
0.93 (0.85; 1.01) |
|
3-cis-hidroxigliburida |
1.01 (0.96; 1.07) |
0.99 (0.91; 1.08) |
|||
|
4-trans-hidroxigliburida |
1.03 (0.97; 1.09) |
0.96 (0.88; 1.04) |
|||
|
Hidroclorotiaxida |
25 mg una vez al día durante 35 días |
300 mg una vez al día por 7 días |
Hidroclorotiazida |
0.99 (0.95; 1.04) |
0.94 (0.87; 1.01) |
|
Metformina |
2000 mg |
300 mg una vez al día por 8 días |
metformina |
1.20 (1.08; 1.34) |
1.06 (0.93; 1.20) |
|
Paracetamol |
1000 mg |
300 mg dos veces al día por 25 días |
paracetamol |
1.063 (0.98; 1.14) |
1.00 (0.92; 1.09) |
|
Simvastatina |
40 mg |
300 mg una vez al día por 7 días |
simvastatina |
1.12 (0.94; 1.33) |
1.09 (0.91; 1.31) |
|
simvastatina ácida |
1.18 (1.03; 1.35) |
1.26 (1.10; 1.45) |
|||
|
Warfarina |
300 mg |
300 mg una vez al día por 12 días |
(R)-warfarina |
1.01 (0.96; 1.06) |
1.03 (0.94; 1.13) |
|
(S)-warfarina |
1.06 (1.00; 1.12) |
1.01 (0.90; 1.13) |
|||
|
INR |
1.00 (0.98; 1.03) |
1.05 (0.99; 1.12) |
|||
1 Dosis única, a menos que se indique lo contrario.
2 ABCinf para medicamentos administrados como dosis única y ABC 24 h para medicamentos administrados en dosis múltiples.
3 ABC0-12h.
ALTERACIONES EN LOS RESULTADOS DE PRUEBAS DE LABORATORIO:
Prueba de 1, 5 anhidroglucitol: El aumento en la excreción urinaria de glucosa con INVOKANA® puede falsamente disminuir los niveles de 1, 5 anhidroglucitol y hacer que este parámetro no sea confiable para la evaluación del control glucémico. Por lo tanto, la prueba de 1, 5 anhidroglucitol no se debe usar para la valoración del control glucémico en pacientes bajo tratamiento con canagliflozina Para mayores detalles, se recomienda contactar al fabricante de la prueba.
Prueba urinaria de glucosa: Debido a su mecanismo de acción, los pacientes bajo tratamiento con INVOKANA® mostrarán resultados positivos en esta prueba.
Las tasas de incidencias de resultados de laboratorio anormales señaladas a continuación, se derivan del análisis de estudios clínicos controlados con placebo durante 26 semanas, a menos que se especifique lo contrario.
Incremento en el potasio sérico: Se observaron cambios promedio en el potasio sérico, desde la basal en 0.5% y 1.0% con INVOKANA® 100 mg y 300 mg, respectivamente, en comparación con 0.6% con placebo. Se observaron episodios de potasio sérico elevado (> 5.4 mEq/L y 15% por arriba del basal) en 4.4% de los pacientes tratados con 100 mg de INVOKANA®, 7.0% de los tratados con 300 mg y 4.8% de los tratados con placebo. En general, las elevaciones fueron medias (6.5 mEq/L), transitorias y no requirieron de tratamiento específico.
Aumentos en la creatinina sérica y el nitrógeno de urea en sangre (BUN): Se observaron cambios promedio desde la basal en la creatinina sérica con decrementos conmensurables en la TFGe de 2.8% y 4 0% con INVOKANA® 100 mg y 300 mg respectivamente en comparación con 1.5% con placebo. Los cambios promedio desde la basal en el BUN fueron 17.1% y 18.0% con INVOKANA® 100 mg y 300 mg, respectivamente, en comparación con 2.7% con placebo. Estos fueron cambios generalmente observados durante las primeras 6 semanas desde el inicio del tratamiento, subsecuentemente, la concentración de creatinina sérica tendió gradualmente hacia el basal y los niveles de BUN se mantuvieron estables.
El porcentaje de pacientes con decrementos mayores en la eGFR (> 30%) respecto a la basal que ocurrieron en cualquier momento durante el tratamiento, fue de 2.0% con 100 mg de INVOKANA® y 4.1% con 300 mg de INVOKANA® con relación a 2.1% con placebo. Estos decrementos en la TFGe, a menudo fueron transitorios, con menos pacientes que presentaron este grado de decremento en el punto terminal del estudio y menos al llegar al objetivo primario; 0.7% de los pacientes con 100 mg de INVOKANA®, 1.4% con 300 mg de INVOKANA® y 0.5% de los tratados con placebo (ver Precauciones generales).
Después de la discontinuación del tratamiento con INVOKANA®, estos cambios en las cifras de laboratorio mejoraron o regresaron a la basal.
Cambios en los lípídos: El promedio porcentual de cambio desde la basal con relación a placebo en las lipoproceínas de colesterol de baja densidad (LDL-C) fue 4.4 mg/dL (4.5%) y 8.2 mg/dL (8.2%) INVOKANA® 100 mg y 300 mg, respectivamente. Se observaron pequeños aumentos en el colesterol total de 2.5% y 4.3% en relación con placebo para INVOKANA® 100 mg y 300 mg, respectivamente . Asimismo, hubo aumento en las lipoproteínas de colesterol de alta densidad (HDL-C) con relación a placebo de 5.4% y 6.3% con INVOKANA® 100 mg y 300 mg, respectivamente. Los aumentos en el colesterol no HDL en relación a placebo fueron 0.05 mmol/L (1.5%) y 0.13 mmol/L (3.6%) con INVOKANA® 100 mg y 300 mg, respectivamente. La relación de colesterol LDL/HDL no mostró cambios con ninguna dosis de INVOKANA® en relación con placebo. Las concentraciones de ApoB y LDL-C (medidas en 2 estudios) y colesterol no HDL aumentaron en una menor extensión, en comparación con los cambios de LDL-C.
Aumentos en hemoglobina: Los cambios promedio (cambios porcentuales) en la concentración de hemoglobina desde la basal fueron 4.7 g/L (3.5%) con INVOKANA® 100 mg, 5.1 g/L (3.8%) con INVOKANA® 300 mg y -1.8 g/L (-1.1%) con placebo. De manera conmensurable, se observaron pequeños incrementos en el cambio porcentual medio desde la basal en eritrocitos séricos y hematócrito. Al final del tratamiento, 4.0%, 2.7% y 0.8% de los pacientes tratados con INVOKANA® 100 mg, 300 mh y placebo, respectivamente, presentaban niveles de hemoglobina mayores al límite superior normal.
Aumentos en el fósforo sérico: Se observaron aumentos relacionados con la dosis en los niveles de fósforo sérico con INVOKANA®. En el acumulado de 4 estudios clínicos controlados con placebo, el cambio promedio porcentual en los niveles de fósforo sérico fue 3.6% y 5.1% con INVOKANA® 100 mg y 300 mg, respectivamente, en comparación con 1.5% con placebo. Los episodios de elevación en el fósforo sérico (> 1.65 mmol/L 25% arriba de la basal) fueron observados en 0.6% y 1.6% de los pacientes tratados con INVOKANA® 100 mg y 300 mg respectivamente, en comparación con 1.3% de los pacientes tratados con placebo.
Decrementos en el urato sérico: Se observaron decrementos moderados en el porcentaje de cambio medio desde la basal en el urato sérico en los grupos de 100 mg y 300 mg de INVOKANA® (-10.1% y -10.6%, respectivamente) en comparación con placebo, donde hubo un ligero incremento respecto a la línea basal (1.9%). Las disminuciones en el urato sérico en los grupos de INVOKANA® fueron máximas o casi máximas para la semana 6 y se mantuvieron con las dosis. Se observó un incremento transitorio en la excreción de ácido úrico, que no fue persistente. En un análisis acumulado (N = 9,439) de pacientes de 8 estudios Fase 3 controlados incluidas ambas dosis de INVOKANA®, no aumentaron los eventos de nefrolitiasis.
Seguridad cardiovascular: Se realizó un metaanálisis prospectivo y preespecífico de eventos cardiovasculares adjudicados de manera independiente. Los datos fueron tomados de los estudios Fases 2 y 3 en 9,632 pacientes con diabetes tipo 2, incluyendo 4,327 que están participando en un estudio cardiovascular en curso (pacientes con enfermedad cardiovascular o en alto riesgo de enfermedad cardiovascular). El cociente de riesgo para el objetivo primario (tiempo al evento en muerte cardiovascular compuesta, EVC no fatal, infarto al miocardio no fatal, angina inestable que requiere hospitalización) para INVOKANA® (ambas dosis) vs. activo combinado y placebo, fue 0.91 (IC 95%: 0.68, 1.22). La razón de riesgo para 100 mg y 300 mg fue similar. Por lo tanto, no hay evidencia de aumento en el riesgo cardiovascular tanto con INVOKANA® 100 mg como INVOKANA® 300 mg en relación con los comparadores.
PRECAUCIONES GENERALES:
General: INVOKANA® no ha sido estudiada en pacientes con Diabetes tipo 1 y por lo tanto no se recomienda.
INVOKANA® no debe ser usada en pacientes con TFGe < 45 mL/min/1.73 m2 (depuración de creatinina < 45 mL/min) ya que no sería efectiva para estas afecciones.
Disminución del volumen intravascular: Debido a su mecanismo de acción, INVOKANA® aumenta la excreción urinaria de glucosa (EUG) e induce diuresis osmótica. Los pacientes que pueden ser más susceptibles a las reacciones adversas relacionadas con la reducción de volumen intravascular (por ejemplo, vértigo postural, hipotensión ortostática o hipotensión) son aquellos bajo tratamiento con diuréticos de asa, que cursan con alteración renal moderada o ≥ 75 años de edad (ver Dosis y vías de administración, y Reacciones secundarias y adversas).
En estudios clínicos controlados con placebo de INVOKANA®, se observó aumento de las reacciones adversas relacionadas con la disminución de volumen intravascular más a menudo con la dosis de 300 mg y se presentaron con mayor frecuencia en los primeros tres meses (ver Reacciones secundarias y adversas). Debido a la reducción del volumen intravascular se observaron generalmente durante las primeras 6 semanas de tratamiento con INVOKANA®, aumentos dosis-dependiente en la creatinina plasmática y disminución concomitante en la TFGe. En pacientes susceptibles a mayor disminución del volumen intravascular, como se describió antes, a veces se observaron decrementos mayores en la TFGe (> 30%) que posteriormente mejoraron y rara vez requirieron interrupción del tratamiento con INVOKANA® (ver Reacciones secundarias y adversas).
Debe recomendarse al paciente comunicar los síntomas de disminución de volumen intravascular. Estas reacciones adversas con poca frecuencia llevan a suspender el tratamiento con INVOKANA® y a menudo se trataron con la modificación del esquema de tratamiento antihipertensivo (incluyendo diuréticos) mientras se continuaba el tratamiento con INVOKANA®. En pacientes con depleción de volumen debe considerarse la corrección de este trastorno antes de iniciar INVOKANA®.
La función renal debe ser valorada previo al inicio del tratamiento con INVOKANA®. Se recomienda monitoreo frecuente de la función renal en pacientes con TFGe < 60 mL/min/1.73 m2 (depuración de creatinina < 60 mL/min). INVOKANA® no debe ser usada en pacientes con TFGe < 45 mL/min/1.73 m2 (depuración de creatinina < 45 mL/min).
Hipoglucemia por el tratamiento adyuvante con otros agentes antihiperglucémicos: Cuando se utilizó solo o como tratamiento adyuvante con agentes antihiperglucémicos no relacionados con hipoglucemia, INVOKANA® mostró baja incidencia de hipoglucemia. Se sabe que la insulina y sus secretagogos (por ejemplo, sulfonilurea) causan hipoglucemia. Cuando se usó INVOKANA® como tratamiento adyuvante con insulina o secretagogos de insulina (por ejemplo, sulfonilurea), la incidencia de hipoglucemia aumentó con respecto a placebo.
Por lo tanto, para disminuir el riesgo de hipoglucemia puede considerarse disminución de la dosis de insulina o uno de sus secretagogos (véase Dosis y vías de administración, y Reacciones secundarias y adversas).
Infecciones micóticas y genitales: Consistente con el mecanismo de acción; inhibición de SGLT2 y aumento de la excreción urinaria de glucosa (EUG). Estudios clínicos reportaron candidiasis vulvovaginal, balanitis y balanopostitis (ver Reacciones secundarias y adversas). Tanto hombres como mujeres con antecedentes de infecciones micóticas genitales fueron más susceptibles a desarrollar infección. Tanto balanitis como balanopostitis se presentaron principalmente en hombres no circuncidados, así como fimosis. En un análisis de 8 estudios clínicos, 0.2% de los pacientes se sometieron a circunsición. La mayoría de las infecciones micóticas genitales fueron tratadas con antimicóticos tópicos prescritos por un profesional de la salud o por auto tratamiento mientras se continuaba el tratamiento con INVOKANA®.
Efectos sobre la capacidad para manejar y usar máquinas: Canagliflozina no tiene influencia conocida sobre la capacidad de conducir y usar máquinas. Sin embargo, debería alertarse a los pacientes del riesgo de hipoglucemia cuando se usa INVOKANA® como tratamiento adjunto a insulina o un secretagogo de insulina, y el riesgo elevado de reacciones adversas con relación a la disminución del volumen intravascular, como vértigo postural (véase Dosis y vía de administración, y Reacciones secundarias y adversas).
DOSIS Y VÍA DE ADMINISTRACIÓN:
Administración: INVOKANA® debe tomarse vía oral una vez al día, preferentemente antes del primer alimento del día (ver Farmacocinética). Las tabletas se deben tragarse completas.
Adultos de 18 años de edad y mayores: La dosis recomendada de INVOKANA® es 100 o 300 mg una vez al día. La tableta de 300 mg debe ser considerada en pacientes con tasa de filtración glomerular estimada (TFGe) ≥ 60 mL/min/1.73 m2 (depuración de creatinina ≥ 60 mL/min), que necesitan control glucémico más estrecho y aquellos con bajo riesgo de reacciones adversas asociadas con reducción del volumen intravascular por el tratamiento con INVOKANA® (ver Precauciones generales)
La dosis inicial de 100 mg una vez al día debe ser usada en pacientes bajo tratamiento con diuréticos de asa y pacientes ≥75 años de edad. En pacientes con evidencia de disminución en el volumen intravascular, se recomienda corregir esta condición antes de iniciar el tratamiento con INVOKANA®. En aquellos pacientes que toleran INVOKANA® 100 mg y que necesitan control glucémico más estrecho, la dosis se puede aumentar a 300 mg (ver Precauciones generales).
Cuando se usa INVOKANA® como tratamiento adyuvante con insulina o un secretagogo de la insulina (por ejemplo, sulfonilurea), se puede considerar disminución de la dosis de insulina o el secretagogo de la insulina para reducir el riesgo de hipoglucemia (ver Precauciones generales y Reacciones secundarias y adversas).
Dosis olvidada: Si se olvida una dosis, deberá tomarse tan pronto como el paciente lo recuerde; sin embargo, no deberá tomarse dosis doble en el mismo día.
Poblaciones especiales:
Pacientes pediátricos (< 18 años de edad): No se ha establecido la seguridad y eficacia de INVOKANA® en pacientes pediátricos.
Ancianos: En pacientes ≥ 75 años la dosis inicial es de 100 mg una vez al día. Se deben tomar en cuenta la función renal y el riesgo de depleción de volumen (ver Precauciones generales y Reacciones secundarias y adversas).
Alteración Renal: En pacientes con tasa de filtración glomerular estimada de TFGe 45 a < 60 mL/min/1.73 m2 (depuración de creatinina 45 a < 60 ml/min) la dosis se limita a 100 mg una vez al día.
No se debe iniciar el tratamiento con INVOKANA® en pacientes TFGe < 45 mL/min/1.73 m2 (depuración de creatinina < 45 mL/min).
Se debe descontinuar el tratamiento con INVOKANA® cuando la TFGe sea persistentemente < 45 mL/min/1.73 m2 (depuración de creatinina < 45 mL/min) (ver Precauciones generales y Reacciones secundarias y adversas).
MANIFESTACIONES Y MANEJO DE LA SOBREDOSIFICACIÓN O INGESTA ACCIDENTAL:
Signos y síntomas: Las dosis únicas de hasta 1,600 mg de INVOKANA® en sujetos sanos y 300 mg de INVOKANA® dos veces al día durante 12 semanas en pacientes con diabetes tipo 2, fueron generalmente bien toleradas.
Tratamiento: En caso de sobredosis es razonable usar las medidas de sostén usuales, por ejemplo, retirar el material no absorbido del tracto digestivo, vigilancia clínica e iniciar tratamiento de sostén, según esté indicado por el estado clínico del paciente. Durante una sesión de hemodiálisis de 4 horas, hubo mínima eliminación de canagliflozina. No se espera que canagliflozina sea dializable mediante diálisis peritoneal.
NOMBRE Y DOMICILIO DEL LABORATORIO:
JANSSEN-CILAG, S.A. de C.V.
Carret. Federal México-Puebla Km. 81.5
San Mateo Capultitlán, C.P. 74160
Huejotzingo, Puebla, México
PRESENTACIONES:
INVOKANA® Tabletas: Caja con 30 tabletas de 100 mg o 300 mg.
RECOMENDACIONES SOBRE ALMACENAMIENTO: INVOKANA® Tabletas: Consérvese a temperatura ambiente a menos de 30°C y en lugar seco.
LEYENDAS DE PROTECCIÓN:
No se administre a menores de 18 años. Su venta requiere receta médica. No se deje al alcance de los niños. No se use durante el embarazo y lactancia. Literatura exclusiva para médicos.

