
ENTRESTO
SACUBITRIL, VALSARTÁN
Comprimidos
1 Caja, 30 Comprimidos, 50 mg
1 Caja, 30 Comprimidos, 100 mg
1 Caja, 60 Comprimidos, 100 mg
1 Caja, 60 Comprimidos, 200 mg
FORMA FARMACÉUTICA Y FORMULACIÓN:
Cada COMPRIMIDO contiene:
Sacubitrilo valsartán sódico hidratado equivalen a 50 mg, 100 mg, 200 mg de Sacubitrilo valsartán
Excipiente cbp 1 comprimido
ENTRESTO® contiene un complejo salino integrado por las formas aniónicas de sacubitrilo y valsartán, por cationes de sodio y por moléculas de agua en proporción molar de 1:1:3:2.5, respectivamente. Tras la administración oral, el complejo se disocia en sacubitrilo (que es metabolizado a LBQ657 [sacubitrilato]) y valsartán
INDICACIONES TERAPÉUTICAS:
ENTRESTO® está indicado para reducir el riesgo de muerte cardiovascular y hospitalización por insuficiencia cardiaca en pacientes adultos con insuficiencia cardiaca crónica. Los beneficios son más evidentes en pacientes con fracción de eyección ventricular izquierda (FEVI) por debajo de lo normal.
FARMACOCINÉTICA Y FARMACODINAMIA:
Grupo farmacoterapéutico, ATC:
Grupo farmacoterapéutico: Agentes que actúan sobre el sistema renina-angiotensina; antagonistas del receptor de la angiotensina II, otras combinaciones.
Código ATC: C09DX04.
Mecanismo de acción:
ENTRESTO® presenta el mecanismo de acción novedoso de un inhibidor de la neprilisina y del receptor de la angiotensina (INRA) al inhibir la neprilisina (endopeptidasa neutra; NEP) por medio del sacubitrilato, que es el metabolito activo del profármaco sacubitrilo y antagonizar simultáneamente el receptor de la angiotensina II de tipo 1 (AT1) por medio de valsartán. Los efectos renales y beneficios cardiovasculares complementarios de ENTRESTO® en pacientes con insuficiencia cardiaca se atribuyen al aumento de las concentraciones de los péptidos que son degradados por la neprilisina, como los péptidos natriuréticos [PN]), resultado de la acción del sacubitrilato y a la inhibición simultánea de los efectos nocivos de la angiotensina II por parte del valsartán. Los PN ejercen sus efectos mediante la activación de los receptores presentes en las membranas celulares que están acoplados a una guanilil-ciclasa, dando por resultado un aumento de las concentraciones del segundo mensajero, el monofosfato de guanosina cíclico (cGMP), lo cual promueve la vasodilatación, la natriuresis y la diuresis, el aumento de la filtración glomerular y el flujo sanguíneo renal, la inhibición de la liberación de renina y aldosterona, la reducción de la actividad simpática, así como efectos antihipertróficos y antifibróticos.
La activación sostenida del sistema renina-angiotensina-aldosterona (SRAA) causa vasoconstricción, retención renal de sodio y de líquidos, activación del desarrollo y la proliferación celular y, como consecuencia de ello, un remodelado cardiovascular inadaptado. El valsartán inhibe los efectos perjudiciales de la angiotensina II sobre los sistemas cardiovascular y renal porque antagoniza selectivamente el receptor AT1, además de inhibir la liberación de aldosterona dependiente de la angiotensina II.
Farmacocinética:
Absorción: Tras la administración oral, ENTRESTO® se disocia en sacubitrilo, que es metabolizado a sacubitrilato y valsartán, los cuales alcanzan concentraciones plasmáticas máximas al cabo de 0.5 horas, 2 horas y 1.5 horas, respectivamente. Se estima que la biodisponibilidad absoluta por vía oral es mayor o igual al 60% en el caso del sacubitrilo y del 23% en el caso del valsartán. El valsartán en ENTRESTO® está más biodisponible que el valsartán en otras formulaciones de tabletas en el mercado.
Con la administración de ENTRESTO® dos veces al día se alcanzan concentraciones estables de sacubitrilo, sacubitrilato y valsartán al cabo de 3 días. Al tener concentraciones estables no se observa acumulación significativa de sacubitrilo o valsartán, mientras que la concentración de sacubitrilato se acumula por 1.6 veces más. La administración de ENTRESTO® con alimentos no tiene efectos clínicamente significativos sobre la exposición sistémica al sacubitrilo, sacubitrilato y valsartán. Aunque la exposición a valsartán disminuye cuando se administra ENTRESTO® con alimentos, el descenso no se acompaña de una reducción clínicamente significativa del efecto terapéutico. Por consiguiente, se puede administrar ENTRESTO® con o sin alimentos.
Distribución: ENTRESTO® muestra un alto grado de unión a proteínas plasmáticas (94-97%). Según la comparación entre las concentraciones plasmáticas y las del LCR, el sacubitrilato atraviesa la barrera hematoencefálica en grado limitado (0.28%). ENTRESTO® tiene un volumen de distribución aparente comprendido entre 75 L y 103 L.
Biotransformación/metabolismo: Por la acción de esterasas, el sacubitrilo se biotransforma rápidamente en sacubitrilato, que más no se metaboliza en grado significativo. El valsartán se metaboliza en grado mínimo, ya que apenas el 20% de la dosis, aproximadamente, se recupera en forma de metabolitos. Se ha identificado un hidroximetabolito en el plasma en concentraciones bajas (<10%). Dado que las isoformas de CYP450 desempeñan un papel mínimo en el metabolismo del sacubitrilo y el valsartán, no se prevé que la coadministración con fármacos que afecten a dichas isoformas repercuta en la farmacocinética.
Eliminación: Tras la administración oral, entre el 52% al 68% del sacubitrilo (principalmente en forma de sacubitrilato) y aproximadamente el 13% del valsartán y sus metabolitos se excretan en la orina; entre el 37% y el 48% del sacubitrilo (principalmente en forma de sacubitrilato) y el 86% del valsartán y sus metabolitos se excretan en las heces.
El sacubitrilo, el sacubitrilato y el valsartán se eliminan del plasma con una vida media de eliminación (T1/2) promedio que ronda 1.43 horas, 11.48 horas y 9.90 horas, respectivamente.
Linealidad/no linealidad: La farmacocinética del sacubitrilo, el sacubitrilato y el valsartán es lineal en el intervalo posológico estudiado (50-400 mg de ENTRESTO®).
Poblaciones especiales:
Pacientes pediátricos (menores de 18 años): No se ha estudiado ENTRESTO® en pacientes pediátricos.
Pacientes ancianos (mayores de 65 años): En los sujetos de edad avanzada, la exposición a sacubitrilato y a valsartán aumenta un 42% y un 30%, respectivamente, en comparación con los sujetos más jóvenes. Sin embargo, este aumento no se acompaña de efectos clínicamente significativos, por lo que no es necesario ajustar la dosis.
Género: La farmacocinética de ENTRESTO® (sacubitrilo, sacubitrilato y valsartán) es similar en uno y otro género.
Raza/etnia: La farmacocinética de ENTRESTO® (sacubitrilo, sacubitrilato y valsartán) es comparable en las distintas razas y grupos étnicos (caucásicos, negros, asiáticos, japoneses y otros).
Insuficiencia renal: Se observó una correlación entre la función renal y la exposición sistémica al sacubitrilato, pero no al valsartán. En pacientes con insuficiencia renal entre leve (60 mL/min/1.73 m2 ≤TFGe <90 mL/min/1.73 m2) y moderada (30 mL/min/1.73 m2 ≤TFGe <60 mL/min/1.73 m2), el ABC del sacubitrilato era hasta 2 veces mayor. No es preciso ajustar la dosis en pacientes con insuficiencia renal leve o moderada. En los pacientes con insuficiencia renal grave (TFGe <30 mL/min/1.73 m2), el ABC del sacubitrilato era 2.7 veces mayor. Se recomienda una dosis inicial de 50 mg dos veces al día en los pacientes con insuficiencia renal grave. Se recomienda proceder con precaución cuando se administre ENTRESTO® a esta población porque la información al respecto es escasa.
No se han llevado a cabo estudios en pacientes sometidos a diálisis, pero dado que el sacubitrilato y el valsartán circulan muy unidos a proteínas plasmáticas, es improbable que se eliminen eficazmente mediante diálisis.
Insuficiencia hepática: En comparación con los sujetos sanos, en los pacientes con insuficiencia hepática leve o moderada, la exposición al sacubitrilo fue 1.5 y 3.4 veces mayor, la exposición al sacubitrilato fue 1.5 y 1.9 veces mayor, y la exposición al valsartán fue 1.2 y 2.1 veces mayor, respectivamente. No se recomienda ajustar la dosis cuando se administre ENTRESTO® a pacientes con insuficiencia hepática leve (grado A de la clasificación de Child-Pugh), incluidos los aquejados de trastornos biliares obstructivos. Se recomienda una dosis inicial de 50 mg dos veces al día en los pacientes con insuficiencia hepática moderada (grado B de la clasificación de Child-Pugh). No se ha estudiado la administración de ENTRESTO® a pacientes con insuficiencia hepática grave, por lo que no se recomienda su uso en esta población.
Farmacodinamia: Se evaluaron los efectos farmacodinámicos de ENTRESTO® tras la administración de dosis únicas y repetidas a sujetos sanos y a pacientes con insuficiencia cardiaca, y son congruentes con una inhibición de la neprilisina y del SRAA ejercidos simultáneamente. En un estudio controlado de 7 días de duración con valsartán, llevado a cabo en pacientes con fracción de eyección del ventrículo izquierdo reducida (FEVI-r), la administración de ENTRESTO® aumentó de forma significativa y no sostenida la natriuresis, elevó la concentración de cGMP en la orina y redujo las concentraciones plasmáticas del fragmento de la región media del propéptido natriurético auricular (MR-proANP) y el fragmento aminoterminal del propéptido natriurético auricular de tipo B (NT-proBNP) en comparación con valsartán. En un estudio de 21 días de duración llevado a cabo en pacientes con FEVI-r, ENTRESTO® elevó significativamente las concentraciones urinarias de péptido natriurético auricular (PNA) y cGMP y las concentraciones plasmáticas de cGMP, y redujo las concentraciones plasmáticas de NT-proBNP, aldosterona y endotelina 1 en comparación con las basales. Asimismo, ENTRESTO® bloqueó el receptor AT1, como se puso de manifiesto por el aumento de la actividad de la renina plasmática y las concentraciones plasmáticas de renina.
En el estudio PARADIGM-HF, ENTRESTO® redujo la concentración plasmática de NT-proBNP, troponina y el ST2 soluble (sST2) y aumento la concentración de cGMP en la orina en comparación con valsartán. Mientras que el BNP es un sustrato de la neprilisina, el NT-proBNP no lo es. Por lo que, el NT-proBNP (a diferencia del BNP) es un biomarcador adecuado para el seguimiento de los pacientes con insuficiencia cardiaca tratados con ENTRESTO®.
En un estudio clínico minucioso del intervalo QTc llevado a cabo en varones sanos, dosis únicas de 400 mg y 1200 mg de ENTRESTO® no ejercieron efectos sobre la repolarización cardiaca.
La neprilisina es una de las muchas enzimas que intervienen en la depuración del amiloide β (Aβ) del cerebro y el líquido cefalorraquídeo (LCR). La administración a sujetos sanos de 400 mg de ENTRESTO® una vez al día durante dos semanas se asoció con un aumento de Aβ1-38 en el LCR en comparación con el placebo; no se observaron variaciones de las concentraciones de Aβ 1-40 y Aβ 1-42 en el LCR. Se desconoce la relevancia clínica de este hallazgo (ver Precauciones en relación con efectos de carcinogénesis, teratogénesis, mutagénesis y sobre la fertilidad).
Estudios clínicos: La administración en los estudios clínicos se basó en la cantidad total de ambos componentes de ENTRESTO®; es decir, 24/26 mg, 49/51 mg y 97/103 mg correspondientes a 50 mg, 100 mg y 200 mg, respectivamente.
Paradigm-hf: El ensayo PARADIGM-HF fue un estudio multinacional, aleatorizado y con doble enmascaramiento, llevado a cabo en 8,442 pacientes en los que se comparó ENTRESTO® con enalapril, ambos administrados a pacientes adultos con insuficiencia cardiaca crónica de clase II-IV de la NYHA y disfunción sistólica (fracción de eyección del ventrículo izquierdo [FEVI] ≤40%), además de otro tratamiento de la insuficiencia cardiaca. La variable principal de valoración se componía de la muerte por causas cardiovasculares (CV) y la hospitalización por insuficiencia cardiaca (IC).
Antes de participar en el estudio, los pacientes estaban correctamente tratados con un tratamiento de referencia que comprendía un inhibidor de la enzima convertidora de la angiotensina (IECA) o un antagonista del receptor de la angiotensina II (ARA) (>99%), betabloqueadores (94%), antagonistas de los mineralocorticoides (58%) y diuréticos (83%). La mediana de la duración del seguimiento fue de 27 meses, y se trató a los pacientes durante un máximo de 4.3 años.
Los pacientes debían suspender definitivamente el tratamiento con el IECA o el ARA e ingresar en un periodo secuencial de preinclusión con enmascaramiento simple durante el cual recibieron un tratamiento con 10 mg de enalapril dos veces al día, seguido de un tratamiento con 100 mg de ENTRESTO® dos veces al día, dosis que luego se aumentó a 200 mg dos veces al día. Los pacientes pasaron seguidamente al periodo de doble enmascaramiento, en el que fueron asignados aleatoriamente a recibir 200 mg de ENTRESTO® o 10 mg de enalapril dos veces al día [ENTRESTO® (n=4,209); enalapril (n=4,233)].
La mediana de edad de la población estudiada era de 64 años, y el 19% de los pacientes tenían 75 años o más. En el momento de la aleatorización, el 70% de los pacientes tenían insuficiencia cardiaca de clase II de la NYHA y el 25% era de la clase III-IV.
Al final del estudio, el 76% de los pacientes del grupo de ENTRESTO® seguían recibiendo la dosis prevista de 200 mg dos veces al día (dosis diaria media de 375 mg). En el grupo de enalapril, el 75% de los pacientes seguían recibiendo la dosis prevista de 10 mg dos veces al día al final del estudio (dosis diaria media de 18.9 mg).
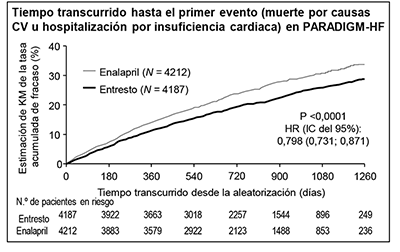
ENTRESTO® demostró una superioridad clínica y estadísticamente significativa respecto a enalapril, ya que, comparado con este, redujo el riesgo de muerte por causas CV o de hospitalizaciones por IC en un 20% (cociente de riesgos instantáneos [hazard ratio; HR]: 0.80; IC del 95% [0.73; 0.87], p = 0.0000002 en la prueba unilateral). Este efecto se observó pronto y se mantuvo durante todo el ensayo. La reducción del riesgo absoluto fue del 4.69%. Se observó una reducción estadísticamente significativa de las muertes por causas CV y de las primeras hospitalizaciones por IC (muerte por causas CV, reducción del riesgo relativo [RRR]: 20%, HR: 0.80; IC del 95% [0.71; 0.89], p = 0.00004 en la prueba unilateral, y hospitalización por IC, RRR: 21%; HR: 0,79; IC del 95% [0.71; 0.89], p = 0.00004 en la prueba unilateral); véanse la Tabla 1 y la Figura 1. La muerte súbita representó el 45% de las muertes por causas CV y se redujo un 20% en los pacientes tratados con ENTRESTO® en comparación con los que recibieron enalapril (HR: 0.80, p = 0.0082). El fallo de la bomba representó el 26% de las muertes por causas CV y se redujo un 21% en los pacientes tratados con ENTRESTO® en comparación con los que recibieron enalapril (HR: 0.79, p = 0.0338).
Esta reducción del riesgo se observó sistemáticamente en subgrupos tales como los establecidos por edad, sexo, raza, ubicación geográfica, clase de la NYHA, fracción de eyección, función renal, antecedentes de diabetes o de hipertensión arterial, fracaso de un tratamiento anterior de la IC, y fibrilación auricular.
ENTRESTO® también redujo la mortalidad por cualquier causa de forma significativa, en un 16%, en comparación con enalapril (RRR: 16%, HR: 0.84; IC del 95% [0.76; 0.93], p = 0.0005 en la prueba unilateral) (Tabla 1). La reducción del riesgo absoluto fue del 2.84%.
Tabla 1. Efecto del tratamiento sobre la variable principal (compuesta) de valoración, sus componentes y la mortalidad por cualquier causa – PARADIGM-HF
|
ENTRESTO® N = 4,187# n (%) |
Enalapril N = 4,212# n (%) |
Cociente de riesgos instantáneos (HR) (IC del 95%) |
Reducción del riesgo relativo |
Valor de p*** |
|
|
Variable principal (compuesta) de valoración: muerte por causas CV y hospitalización por IC* |
914 (21.83) |
1117 (26.52) |
0,80 (0.73; 0.87) |
20% |
0.0000002 |
|
Componentes individuales de la variable principal (compuesta) de valoración |
|||||
|
Muerte por causas CV** |
558 (13.33) |
693 (16.45) |
0,80 (0.71; 0.89) |
20% |
0.00004 |
|
Primera hospitalización por IC |
537 (12.83) |
658 (15.62) |
0,79 (0.71; 0.89) |
21% |
0.00004 |
|
Variable de valoración secundaria |
|||||
|
Mortalidad por cualquier causa |
711 (16.98) |
835 (19.82) |
0,84 (0.76; 0.93) |
16% |
0.0005 |
* La variable de valoración principal se definió como el tiempo transcurrido hasta el primer evento.
** La muerte por causas CV comprende todos los pacientes que fallecieron hasta la fecha de cierre de la base de datos, con independencia de que hubiera hospitalizaciones anteriores.
*** Valor de p en la prueba unilateral.
# Población completa de análisis.
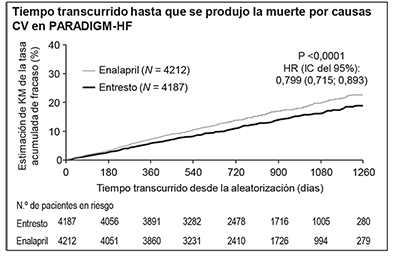
La curva de Kaplan-Meier de la figura que aparece a continuación (izquierda) muestra el tiempo transcurrido hasta que se produjo el primer evento de los considerados en la variable principal (compuesta) de valoración; esto es, la muerte por causas CV o la hospitalización por IC. El efecto terapéutico de ENTRESTO® se manifestó pronto y se mantuvo a lo largo de todo el estudio. La curva de Kaplan-Meier de la figura que aparece a continuación (derecha) muestra el tiempo transcurrido hasta que se produjo la muerte por causas CV.
Figura 1. Curvas de Kaplan-Meier correspondientes a la variable principal (compuesta) de valoración y a su componente muertes por causas CV – PARADIGM-HF
En conjunto, se produjeron menos hospitalizaciones por cualquier causa entre los pacientes tratados con ENTRESTO® que entre los que recibieron enalapril, lo que incluye una reducción relativa del riesgo del 12% en el caso de la primera hospitalización (HR: 0,88 [IC del 95%: 0.82; 0.94], p <0.001), y una reducción relativa de la tasa del 16% en el caso del número total de hospitalizaciones (HR: 0.84 [IC del 95%: 0.78; 0.91], p <0.001).
Según la evaluación basada en el cuestionario de Kansas City para la autoevaluación de la calidad de vida en la miocardiopatía (Kansas City Cardiomyopathy Questionnaire, KCCQ), con ENTRESTO® se obtuvo una puntuación clínica global significativamente mejor en los ámbitos relacionados con los síntomas de IC y las limitaciones físicas. La proporción de pacientes en los que la clase funcional de la NYHA mejoró entre el inicio y el mes 8 fue mayor en el grupo de ENTRESTO® (16%) que en el de enalapril (14%), y la proporción de pacientes en los que la clase funcional de la NYHA empeoró fue menor (10% y 13%, respectivamente).
Paragon-hf: PARAGON-HF fue un ensayo multicéntrico, aleatorizado, doble ciego que comparó ENTRESTO® y valsartán en 4,796 pacientes adultos con insuficiencia cardiaca sintomática con fracción de eyección preservada (fracción de eyección del ventrículo izquierdo ≥45%) y cardiopatía estructural [agrandamiento de la aurícula izquierda (LAE) o hipertrofia ventricular izquierda (HVI)]. Se excluyeron los pacientes con una presión arterial sistólica de <110 mmHg y los pacientes con cualquier FEVI ecocardiográfica previa <40% en el momento del cribado.
El criterio de valoración principal de PARAGON-HF fue la combinación de hospitalizaciones por insuficiencia cardiaca (IC) total (primera y recurrente) y muerte cardiovascular (CV). Después de suspender el tratamiento con IECA o ARA existentes, los pacientes ingresaron en periodos de preinclusión con enmascaramiento simple durante los cuales recibieron valsartán 80 mg dos veces al día, seguido de ENTRESTO® 100 mg dos veces al día. Los pacientes con dosis bajas previas de un IECA o ARA II comenzaron el periodo de preinclusión recibiendo 40 mg de valsartán dos veces al día durante 1-2 semanas. Los pacientes que completaron con éxito los periodos de preinclusión secuenciales fueron aleatorizados para recibir ENTRESTO® 200 mg (N = 2,419) dos veces al día o valsartán 160 mg (N = 2,403) dos veces al día. La duración media del seguimiento fue de 35 meses y los pacientes fueron tratados durante un máximo de 4.7 años.
La edad media de la población estudiada fue de 73 años y el 52% eran mujeres. En la aleatorización, el 77% de los pacientes eran de clase II de la NYHA, el 19% eran de clase III de la NYHA y el 0.4% eran de clase IV de la NYHA. La mediana de la fracción de eyección del ventrículo izquierdo fue del 57%. La causa subyacente de la insuficiencia cardiaca fue de etiología isquémica en el 36% de los pacientes. Además, el 96% tenía antecedentes de hipertensión, el 23% tenía antecedentes de infarto de miocardio, el 46% tenía una TFGe <60 mL/min/1.73 m2 y el 43% tenía diabetes mellitus. La mayoría de los pacientes tomaban betabloqueadorres (80%) y diuréticos (95%).
En PARAGON-HF, ENTRESTO® redujo la tasa del criterio de valoración combinado de hospitalizaciones por IC totales (primera y recurrente) y muerte CV, según un análisis que utiliza un modelo de tasas proporcionales, en un 13% en comparación con valsartán (razón de tasas [RR]; 0.87; IC del 95% [0.75; 1.01], p = 0.059). El efecto del tratamiento se debió principalmente a la reducción del 15% del total de hospitalizaciones por IC en los pacientes asignados al azar a ENTRESTO® (RR 0.85; IC del 95% [0.72, 1.00]).
ENTRESTO® redujo en un 14% la tasa del criterio de valoración combinado de empeoramiento total de la insuficiencia cardiaca (hospitalizaciones por IC y visitas urgentes por IC) y muerte CV (RR 0.86; IC del 95% [0.75, 0.99]).
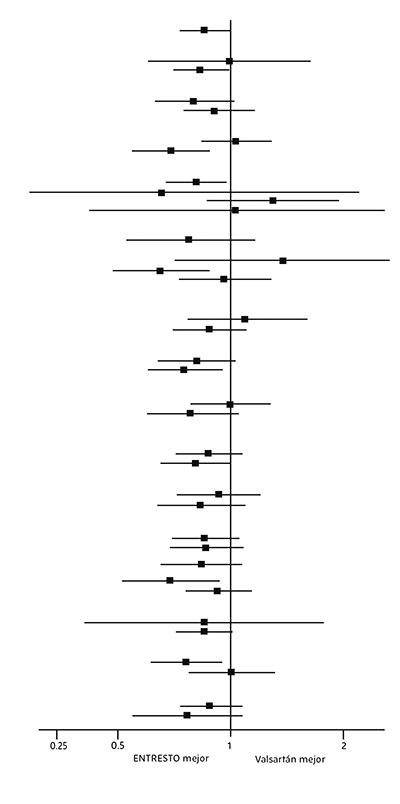
Se examinó una amplia gama de características demográficas, características basales de la enfermedad y medicamentos concomitantes basales para determinar su influencia en los resultados (Figura 2).
Figura 2. Variable principal (compuesta) de valoración: muerte por causas CV y hospitalización por IC; Análisis de subgrupos – PARAGON-HF
|
Subgrupo |
ENTERESTO |
Valsartán |
Razón de tasas |
|
|
n/N (EAR) |
n/N (EAR) |
Estimación (IC del 95%) |
||
|
Global |
894/2407 (12.8) |
1009/2389 (14.6) |
|
0.8 (0.75. 1.01) |
|
Edad |
||||
|
<65 años ≥65 años |
138/412 (11.4) 756/1995 (13.1) |
138/413 (11.4) 871/1976 (15.3) |
0.99 (0.64, 1.53) 0.85 (0.73, 0.99) |
|
|
Edad |
||||
|
<75 años ≥75 años |
425/1307 (11.1) 469/1100 (15.0) |
513/1290 (13.5) 496/1099 (16.0) |
0.82 (0.66, 1.02) 0.92 (0.76, 1.11) |
|
|
Sexo biológico |
||||
|
Varón Mujer |
503/1166 (15.1) 391/1241 (10.8) |
477/1151 (14.6) 532/1238 (14.7) |
1.03 (0.85, 1.25) 0.73 (0.59, 0.90) |
|
|
Raza |
||||
|
Caucásica Negra Asiática Otra |
709/1963 (12.3) 37/52 (23.6) 128/297 (16.3) 20/95 (7.9) |
833/1944 (14.6) 52/50 (35.8) 109/310 (13.0) 15/85 (7.1) |
0.83 (0.71, 0.97) 0.69 (0.24, 1.99) 1.25 (0.87, 1.79) 1.03 (0.47, 2.28) |
|
|
Región |
||||
|
Norteamérica Latinoamérica Europa occidental Europa central Asia/Pacífico y otras |
223/288 (25.1) 49/191 (10.3) 225/699 (10.7) 228/856 (9.1) 169/373 (16.9) |
255/271 (31.0) 34/179 (7.8) 319/691 (15.6) 233/859 (9.4) 163/389 (15.4) |
0.80 (0.57, 1.14) 1.33 (0.75, 2.36) 0.69 (0.53. 0.89) 0.97 (0.76, 1.24) 1.10 (0.79, 1.52) |
|
|
Diabetes al inicio (aleat.) |
||||
|
Sí No |
500/1049 (16.8) 349/1358 (9.9) |
541/1020 (18.4) 468/1369 (11.8) |
0.89 (0.74, 1.09) 0.84 (0.68, 1.03) |
|
|
FEVI |
||||
|
≤Mediana (57%) >Mediana (57%) |
457/1239 (12.8) 437/1168 (12.9) |
591/1256 (16.4) 418/1133 (12.7) |
0.78 (0.64, 0.95) 1.00 (0.81, 1.23) |
|
|
FA según ECG al inicio (aleat.) |
||||
|
Sí No |
279/717 (13.3) 607/1672 (12.6) |
314/679 (15.9) 694/1698 (14.2) |
0.81 (0.63, 1.04) 0.89 (0.75, 1.06) |
|
|
FA según antecedentes al inicio (aleat.) |
||||
|
Sí No |
520/1246 (14.3) 374/1161 (11.2) |
620 1275 (16.7) 389/1114 (12.2) |
0.83 (0.69, 1.00) 0.94 (0.75, 1.18) |
|
|
Nt-proNBP en el cribado |
||||
|
≤Mediana (911 pg/mL) >Mediana (911 pg/mL) |
329/1199 (9.2) 558/1189 (16.7) |
379/1180 (10.9) 625/1189 (18.6) |
0.85 (0.67, 1.08) 0.87 (0.73, 1.05) |
|
|
TAS en el cribado |
||||
|
≤Mediana (137 mmHg) >Mediana (137 mmHg) |
461/1220 (13.2) 433/1158 (12.5) |
523/1230 (14.9) 486/1159 (14.4) |
0.88 (0.72, 1.07) 0.86 (0.69, 1.06) |
|
|
Uso de ARM al inicio (aleat.) |
||||
|
Sí No |
218/592 (12.9) 676/1815 (12.8) |
327/647 (17.6) 682/1742 (13.5) |
0.73 (0.56, 0.95) 0.94 (0.79, 1.12) |
|
|
Intolerancia a IECA |
||||
|
Sí No |
40/123 (10.7) 854/2284 (13.0) |
46/139 (11.5) 963/2250 (14.8) |
0.87 (0.46, 1.65) 0.87 (0.75, 7.01) |
|
|
FGe al inicio (aleat.) |
||||
|
≤60mL/min/1.73 m2 ≤50mL/min/1.73 m2 |
493/1164 (14.9) 401/1243 (11.0) |
622/1177 (18.8) 386/1211 (10.8) |
0.79 (0.66, 0.95) 1.01 (0.80, 1.27) |
|
|
Clase NYHA al inicio (aleat.) |
||||
|
MI III/IV |
670/1939 (11.9) 222/466 (16.4) |
732/1904 (13.3) 277/485 (20.0) |
0.90 (0.76, 1.06) 0.79 (0.59, 1.06) |
|
Nota: La figura anterior presenta los efectos en varios subgrupos, todos los cuales son características de referencia. Los límites de confianza del 95% que se muestran no tienen en cuenta el número de comparaciones realizadas y pueden no reflejar el efecto de un factor en particular después del ajuste para todos los demás factores.
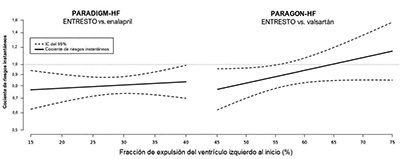
En un análisis de la relación entre la FEVI y el resultado en PARADIGM-HF y PARAGON-HF, los pacientes con FEVI por debajo de lo normal (hasta aproximadamente 60%) tratados con ENTRESTO® experimentaron una mayor reducción del riesgo (Tabla 2, Figura 3, y Figura 4). La FEVI es una medida variable que puede cambiar con el tiempo y el rango normal difiere según las características del paciente y el método de evaluación; los prescriptores deben usar el juicio clínico para decidir a quién tratar. En ambos estudios, el efecto del tratamiento con ENTRESTO® se demostró temprano y se mantuvo durante la duración de los ensayos (Figura 1 y 4).
Tabla 2. Efecto del tratamiento sobre la variable de valoración compuesta (primaria y expandida) y los
componentes para FEVI ≤60% – PARAGON-HF
|
ENTRESTO® N = 1,688 |
Valsartán N = 1,683 |
Tamaño del efecto (95% CI) |
|||
|
Criterio de validación de eficacia |
n |
Tasa de eventosa |
n |
Tasa de eventosa |
|
|
Criterio de valoración compuesto del total de hospitalizaciones por IC (primera y recurrente) y muerte CV |
619 |
12.7 |
761 |
15.9 |
RR = 0.79 (0.67, 0.94) |
|
Criterio de valoración compuesto de empeoramiento total de la ICb y muerte CV |
653 |
13.3 |
798 |
16.7 |
RR = 0.80 (0.67, 0.94) |
|
Componentes Individuales de los Criterios de Valoración Compuestos |
|||||
|
Hospitalizaciones totales por IC |
469 |
9.6 |
594 |
12.4 |
RR = 0.76 (0.62, 0.92) |
|
Muerte CV |
150 |
3.1 |
167 |
3.5 |
HR = 0.88 (0.71, 1.10) |
|
Empeoramiento total de la ICb |
503 |
10.3 |
631 |
13.2 |
RR = 0.75 (0.62, 0.91) |
|
Criterios de Valoración Secundarios |
n/N |
Cambio desde el inicio (SE) |
n/N |
Cambio desde el inicio (SE) |
Diferencia de Tratamiento (IC del 95% |
|
Cambio en la puntuación de resumen clínico (CSS) del KCCQ a los 8 meses |
1578/1677 |
-1.67 (0.42) |
1571/1671 |
-2.71 (0.42) |
LSM = 1.03 (-0.13, 2.20) |
|
n/N |
Tasa de eventos |
n/N |
Tasa de eventos |
Diferencia de Tratamiento (IC del 95% |
|
|
Cambio favorable de la clasificación NYHA a los 8 meses |
1481/1625 |
N/A |
1452/1618 |
N/A |
OR = 1.42 (1.08, 1.88)c |
|
Criterio de valoración combinado renald |
22/1688 |
0.45 |
47/1683 |
0.99 |
HR = 0.45 (0.27, 0.75) |
|
Muerte por todas las causas |
256/1688 |
5.23 |
267/1683 |
5.57 |
HR = 0.94 (0.79, 1.11) |
Abreviaturas: RR = razón de tasas, HR = razón de riesgo, OR = razón de probabilidades, SE = error estándar.
a Tasa de eventos por 100 pacientes-año.
b La combinación de empeoramiento de la IC incluyó el total (primera y recurrente) de visitas urgentes por IC y hospitalizaciones por IC. Una visita urgente por IC se definió como una evaluación urgente y no planificada por un médico, por ejemplo, en un Departamento de Emergencias y requiriendo tratamiento intravenoso.
c La razón de probabilidades para el cambio de clase de la NYHA representa la razón de probabilidades común basada en el modelo de mejora y no empeoramiento, con OR>1 que refleja cambios favorables en el grupo de ENTRESTO.
Figura 3. Número de eventos promedio a lo largo del tiempo para el criterio principal de valoración compuesto
del total de hospitalizaciones por IC y muerte CV en pacientes con FEVI ≤60% – PARAGON-HF
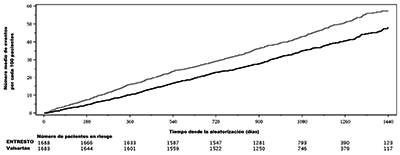
Figura 4. Efecto del tratamiento para el criterio de valoración compuesto del tiempo transcurrido hasta la primera hospitalización por IC o muerte CV por FEVI en PARADIGM-HF y PARAGON-HF
Titration: TITRATION fue un ensayo de 12 semanas de duración en el que se estudiaron la seguridad y la tolerabilidad en 538 pacientes con insuficiencia cardiaca crónica (clase II-IV de la NYHA) y disfunción sistólica (FEVI ≤35%) que no habían sido tratados anteriormente con un IECA ni con un ARA o estaban recibiendo estos fármacos en diversas dosis antes de ingresar en el estudio. Los pacientes empezaron tomando 50 mg de ENTRESTO® dos veces al día, pasaron luego a recibir 100 mg dos veces al día y por último recibieron la dosis prevista de 200 mg dos veces al día, todo ello según un régimen terapéutico de 3 o de 6 semanas.
En conjunto, el 76% de los pacientes llegaron a recibir la dosis prevista de 200 mg de ENTRESTO® dos veces al día y la mantuvieron durante 12 semanas sin ninguna interrupción del tratamiento ni reducción de la dosis. Entre los pacientes que nunca habían sido tratados con un IECA o un ARA o habían recibido dosis bajas de estos fármacos (equivalentes a <10 mg/día de enalapril), la proporción de ellos que llegaron a recibir y mantener la dosis de 200 mg de ENTRESTO® fue mayor en el grupo en el que se aumentó la dosis a lo largo de 6 semanas que en el grupo en el que dicho aumento se hizo en 3 semanas.
Paramount: PARAMOUNT fue un ensayo aleatorizado y con doble enmascaramiento, llevado a cabo en pacientes con FEVI ≥45%, en el que se comparó la administración de 200 mg de ENTRESTO® (n = 149) con la de 160 mg de valsartán (n=152) dos veces al día; se demostró una mayor reducción de las concentraciones de NT-proBNP entre el inicio y la semana 12, y la diferencia fue estadísticamente significativa (p = 0.0050). En los pacientes tratados con ENTRESTO®, la reducción de la concentración de NT-proBNP respecto a la inicial fue similar en la semana 12 y la 36, mientras que en los tratados con valsartán dicha concentración descendió entre la semana 12 y la 36. En la semana 36 se observaron reducciones significativas del tamaño de la aurícula izquierda, tanto del índice de volumen auricular izquierdo (p = 0.0069) como de la dimensión de la aurícula izquierda (p = 0.0337). En la semana 36 se observó una mejoría estadísticamente significativa de la IC expresada como clase de la NYHA (p = 0.0488).
CONTRAINDICACIONES:
• Hipersensibilidad a la sustancia activa, al sacubitrilo, al valsartán o a cualquiera de los excipientes.
• Coadministración con un inhibidor de la enzima convertidora de angiotensina (IECA) (ver Precauciones generales, Dosis y vía de administración e Interacciones medicamentosas y de otro género). No debe administrarse ENTRESTO® hasta 36 horas después de haber suspendido el tratamiento con el IECA.
• Antecedentes conocidos de angioedema relacionado con un tratamiento anterior a base de un IECA o un antagonista de los receptores de angiotensina (ARA).
• Angioedema hereditario.
• Coadministración con aliskiren en pacientes con diabetes de tipo 2 (ver Precauciones generales e interacciones medicamentosas y de otro género).
• Embarazo (ver Restricciones de uso durante el embarazo y la lactancia).
RESTRICCIONES DE USO DURANTE EL EMBARAZO Y LA LACTANCIA:
Resumen de riesgos: Como ocurre con otros fármacos que también actúan directamente sobre el SRAA, no se debe utilizar ENTRESTO® durante el embarazo (ver CONTRAINDICACIONES). ENTRESTO® actúa antagonizando los efectos de la angiotensina II, por lo que no se puede descartar que incluya riesgos para el feto. Se han comunicado casos de lesiones del feto en desarrollo (como aborto espontáneo, oligohidramnios y disfunción renal del neonato) en embarazadas que habían tomado valsartán. Se debe indicar a las pacientes que, si quedan embarazadas, deben suspender de inmediato la toma de ENTRESTO® e informar a su médico.
Datos en animales: El tratamiento con ENTRESTO® durante la organogénesis produjo un aumento de la letalidad embriofetal en ratas que recibieron dosis ≥100 mg/kg/día [≤0.72 veces la dosis máxima recomendada en el ser humano (DMRH) según el ABC] y en conejos que recibieron dosis ≥10 mg/kg/día [2 veces y 0.03 veces la DMRH según el ABC del valsartán y el sacubitrilato, respectivamente]. ENTRESTO® se considera teratógeno porque se observó una baja incidencia de hidrocefalia fetal, asociada con dosis tóxicas para la madre, en conejos que recibieron dosis ≥10 mg/kg/día. Los efectos adversos de ENTRESTO® sobre el embrión y el feto se atribuyen a su actividad antagonista del receptor de la angiotensina.
Los estudios de desarrollo prenatal y posnatal llevados a cabo en ratas que recibieron sacubitrilo en dosis de hasta 750 mg/kg/día (2.2 veces la DMRH según el ABC) y valsartán en dosis de hasta 600 mg/kg/día (0.86 veces la DMRH según el ABC) indican que el tratamiento con ENTRESTO® durante la organogénesis, la gestación y la lactancia puede afectar al desarrollo y la supervivencia de las crías.
Lactancia:
Resumen de riesgos: No se sabe si ENTRESTO® se excreta en la leche materna humana. Sus componentes, el sacubitrilo y el valsartán, se excretaron en la leche de ratas lactantes (ver Precauciones en relación con efectos de carcinogénesis, mutagénesis, teratogénesis y sobre la fertilidad). Dado que puede haber riesgo de reacciones adversas en el lactante, no se recomienda que la madre reciba ENTRESTO® durante la lactancia. Es preciso decidir si se deja de amamantar o bien se deja de recibir ENTRESTO® durante la lactancia, teniendo en cuenta la importancia de este medicamento para la madre.
Mujeres y varones en edad fértil: Se debe informar a las pacientes con posibilidad de quedar embarazadas de las consecuencias de la exposición a ENTRESTO® durante el embarazo y de la necesidad de que utilicen métodos anticonceptivos durante el tratamiento y hasta una semana después de la última dosis de ENTRESTO®.
Fertilidad: No hay datos relativos a los efectos de ENTRESTO® sobre la fertilidad humana. ENTRESTO® no mostró efectos sobre la fertilidad ni sobre el desarrollo embrionario temprano en ratas que recibieron dosis de hasta 150 mg/kg/día (≤1.0 veces y ≤0.18 veces la DMRH según el ABC del valsartán y el sacubitrilato, respectivamente).
REACCIONES SECUNDARIAS Y ADVERSAS:
Resumen del perfil de seguridad: Un total de 6,622 pacientes con insuficiencia cardiaca fueron tratados con ENTRESTO® en los estudios clínicos PARADIGM-HF (frente a enalapril) y PARAGON-HF (frente a valsartán). De estos, 5,085 estuvieron expuestos durante al menos 1 año.
PARADIGM-HF.
Se evaluó la seguridad de ENTRESTO® en pacientes con insuficiencia cardiaca crónica con FEVI ≤40% (fracción de eyección del ventrículo izquierdo reducida) en el estudio pivotal Fase 3 PARADIGM-HF, en el que se compararon pacientes que recibieron dos veces al día 200 mg de ENTRESTO® (n = 4203) o 10 mg de enalapril (n = 4229). Los pacientes asignados aleatoriamente al grupo de ENTRESTO® recibieron el tratamiento durante un máximo de 4.3 años, con una duración promedio de la exposición de 24 meses; en 3271 pacientes el tratamiento duró más de un año. En el estudio PARADIGM-HF, 450 (10.71%) de los pacientes tratados con ENTRESTO® y 516 (12.20%) de los tratados con enalapril tuvieron que interrumpir el tratamiento debido a un evento adverso (EA) durante el periodo de doble ciego. Los eventos que con mayor frecuencia obligaron a ajustar la dosis o a interrumpir el tratamiento fueron hipotensión arterial, hiperpotasemia e insuficiencia renal.
En pacientes con insuficiencia cardiaca, la incidencia general de reacciones adversas (RA) fue comparable con ENTRESTO® y con enalapril. La distribución de las RA concuerda con la farmacología de ENTRESTO® y los padecimientos de fondo de los pacientes.
La frecuencia general de las reacciones adversas no guardaba relación con el género, la edad ni la raza.
Las reacciones adversas se agrupan mediante la clasificación de Órganos y Sistemas, y dentro de cada clase se enumeran en orden de frecuencia decreciente según la convención siguiente: muy frecuente (≥1/10); frecuente (≥1/100 a <1/10); infrecuente (≥1/1,000 a <1/100); rara (≥1/10,000 a <1/1,000); muy rara (<1/10,000), incluidas las notificaciones aisladas. En cada categoría de frecuencia, las reacciones adversas se clasifican en orden de gravedad decreciente.
Tabla 3. Reacciones adversas en el estudio PARADIGM-HF; población de análisis de la seguridad
|
Reacciones adversas |
ENTRESTO® 200 mg dos veces al día (%)* |
Enalapril 10 mg dos veces al día (%)* |
Categoría de frecuencia |
|
Trastornos del metabolismo y la nutrición |
|||
|
Hiperpotasemia |
11.61 |
14.00 |
Muy frecuente |
|
Hipopotasemia |
3.31 |
2.53 |
Frecuente |
|
Trastornos del sistema nervioso |
|||
|
Mareo |
6.33 |
4.87 |
Frecuente |
|
Mareo postural |
0.57 |
0.28 |
Infrecuente |
|
Cefalea |
2.45 |
2.51 |
Frecuente |
|
Trastornos del oído y el laberinto |
|||
|
Vértigo |
1.45 |
1.40 |
Frecuente |
|
Trastornos vasculares |
|||
|
Hipotensión arterial |
17.61 |
11.97 |
Muy frecuente |
|
Síncope |
2.24 |
2.70 |
Frecuente |
|
Hipotensión ortostática |
1.52 |
0.80 |
Frecuente |
|
Trastornos respiratorios, torácicos y mediastínicos |
|||
|
Tos |
8.78 |
12.60 |
Frecuente |
|
Trastornos gastrointestinales |
|||
|
Diarrea |
4.62 |
4.47 |
Frecuente |
|
Náusea |
2.09 |
2.36 |
Frecuente |
|
Trastornos de la piel y del tejido subcutáneo |
|||
|
Angioedema |
0.45 |
0.24 |
Infrecuente |
|
Trastornos renales y urinarios |
|||
|
Disfunción renal |
10.14 |
11.52 |
Muy frecuente |
|
Insuficiencia renal (insuficiencia renal, insuficiencia renal aguda) |
4.76 |
5.30 |
Frecuente |
|
Trastornos generales |
|||
|
Fatiga |
2.97 |
3.05 |
Frecuente |
|
Astenia |
2.09 |
1.84 |
Frecuente |
* Población de análisis de la seguridad
Paragon-hf: La seguridad de ENTRESTO® en pacientes con insuficiencia cardiaca crónica y FEVI ≥45% (fracción de eyección del ventrículo izquierdo preservada) se evaluó en el estudio pivotal Fase 3 PARAGON-HF, que comparó pacientes tratados dos veces al día con ENTRESTO® 200 mg (n = 2,419) o valsartán 160 mg (n = 2,402). El perfil de seguridad de ENTRESTO® fue consistente con el perfil de seguridad en pacientes con insuficiencia cardiaca con fracción de eyección reducida.
Reacciones adversas provenientes de reportes espontáneos y casos en la literatura (frecuencia desconocida): Las siguientes reacciones adversas se han derivado de la experiencia postcomercialización con ENTRESTO® a través de informes de casos espontáneos y casos de literatura. Debido a que estas reacciones se informan voluntariamente de una población de tamaño incierto, no es posible estimar con fiabilidad su frecuencia, que por lo tanto se categoriza como desconocida. Las reacciones adversas a los medicamentos se enumeran de acuerdo con las clases de órganos del sistema en MedDRA.
Tabla 4. Reacciones adversas provenientes de reportes espontáneos y casos en la literatura (frecuencia desconocida)
|
Desórdenes del sistema inmune |
|
Hipersensibilidad (incluye erupción, prurito y anafilaxis) |
PRECAUCIONES EN RELACIÓN CON EFECTOS DE CARCINOGÉNESIS, MUTAGÉNESIS, TERATOGÉNESIS Y SOBRE LA FERTILIDAD:
Los estudios toxicológicos preclínicos llevados a cabo con ENTRESTO® comprendieron la evaluación de la seguridad farmacológica, la toxicidad de dosis repetidas, genotoxicidad, carcinogénesis y la toxicidad para la reproducción y el desarrollo. ENTRESTO® no tuvo efectos adversos sobre los sistemas de órganos vitales. La mayoría de los hallazgos observados en los estudios de toxicidad tras dosis repetidas fueron reversibles y atribuibles a la farmacología del bloqueo del receptor AT1.
Carcinogénesis, mutagénesis y genotoxicidad: En los estudios de carcinogénesis llevados a cabo en ratones y ratas que recibieron sacubitrilo y valsartán no se hallaron indicios de que ENTRESTO® tuviera potencial carcinógeno alguno. Las dosis de sacubitrilo estudiadas (dosis altas de 1200 y 400 mg/kg/día en ratones y ratas, respectivamente) eran unas 29 y 19 veces más altas, respectivamente, que la dosis máxima recomendada en el ser humano (DMRH) si se calculan en mg/m2. Las dosis de valsartán estudiadas (dosis altas de 160 y 200 mg/kg/día en ratones y ratas, respectivamente) eran unas 4 y 10 veces más altas, respectivamente, que la DMRH si se calculan en mg/m2.
Los estudios de mutagénesis y clastogenicidad llevados a cabo con ENTRESTO®, sacubitrilo y valsartán no pusieron de manifiesto efectos a escala génica ni cromosómica.
Fertilidad, reproducción y desarrollo: ENTRESTO® no mostró efectos sobre la fertilidad ni sobre el desarrollo embrionario temprano de ratas que recibieron dosis de hasta 150 mg/kg/día (≤1.0 veces y ≤0.18 veces la DMRH según el ABC del valsartán y sacubitrilato, respectivamente).
El tratamiento con ENTRESTO® durante la organogénesis produjo un aumento de la letalidad embriofetal en ratas que recibieron dosis ≥100 mg/kg/día [≤0.72 veces la DMRH según el ABC] y en conejos que recibieron dosis ≥10 mg/kg/día [2 veces y 0.03 veces la DMRH según el ABC del valsartán y el sacubitrilato, respectivamente]. ENTRESTO® se considera teratógeno porque se observó una baja incidencia de hidrocefalia fetal, asociada con dosis tóxicas para la madre, en conejos que recibieron dosis ≥10 mg/kg/día. Los efectos adversos de ENTRESTO® sobre el embrión y el feto se atribuyen a su actividad antagonista del receptor de la angiotensina (ver Restricciones de uso en el embarazo y la lactancia).
Los estudios de desarrollo pre y posnatal llevados a cabo en ratas que recibieron sacubitrilo en dosis de hasta 750 mg/kg/día [2.2 veces la DMRH según el ABC] y valsartán en dosis de hasta 600 mg/kg/día [0.86 veces la DMRH según el ABC] indican que el tratamiento con ENTRESTO® durante la organogénesis, la gestación y la lactancia puede afectar al desarrollo y la sobrevida de las crías.
Otros resultados preclínicos: Se evaluaron los efectos de ENTRESTO® sobre las concentraciones de amiloide β en el líquido cefalorraquídeo (LCR) y el tejido cerebral de macacos de Java jóvenes (2 a 4 años de edad) que recibieron el medicamento en dosis de 50 mg/kg/día durante 2 semanas. En este estudio se observó que ENTRESTO® ejerció un efecto farmacodinámico sobre la depuración del Aβ del LCR de tal modo que las concentraciones de Aβ 1-40, 1-42 y 1-38 en el LCR aumentaban; no se produjo un aumento correspondiente en las concentraciones cerebrales de Aβ En un estudio de dos semanas de duración en voluntarios sanos no se observaron aumentos de las concentraciones de Aβ1-40 y 1-42 en el LCR (ver FARMACOCINÉTICA Y FARMACODINAMIA). Además, en un estudio toxicológico llevado a cabo en macacos de Java que recibieron ENTRESTO® en dosis de 300 mg/kg/día durante 39 semanas no se produjo acumulación cerebral de amiloide β.
INTERACCIONES MEDICAMENTOSAS Y DE OTRO GÉNERO:
IECA: Está contraindicado la administración conjunta de ENTRESTO® y un IECA porque la concomitancia de la inhibición de la neprilisina (NEP) y la acción del IECA puede elevar el riesgo de angioedema. No se debe iniciar el tratamiento con ENTRESTO® hasta 36 horas después de haber tomado la última dosis del IECA. Y tampoco se debe empezar un tratamiento con un IECA hasta que hayan pasado 36 horas desde la última dosis de ENTRESTO® (ver Contraindicaciones y dosis y vía de administración).
Aliskiren: Está contraindicada la administración de ENTRESTO® junto con aliskiren en pacientes con diabetes tipo 2 (ver Contraindicaciones).
Reacciones previstas por las que no se recomienda la coadministración: Dado que ENTRESTO® actúa como antagonista de los receptores de la angiotensina II, no debe combinarse con un ARA (ver Precauciones generales).
Se debe evitar el uso concomitante de ENTRESTO® con aliskiren en pacientes con disfunción renal (TFGe <60 mL/min/1.73 m2) (ver Precauciones generales).
Interacciones observadas que deben tenerse en cuenta:
Estatinas: Datos obtenidos in vitro indican que el sacubitrilo inhibe los transportadores de aniones orgánicos OATP1B1 y OATP1B3. Por consiguiente, ENTRESTO® puede aumentar la exposición sistémica a sustratos de OATP1B1 y OATP1B3 al igual que las estatinas. La coadministración de ENTRESTO® llega a duplicar la Cmáx de la atorvastatina y sus metabolitos, y el ABC es hasta 1.3 veces mayor.
Se debe, pues, proceder con cautela cuando se coadministren ENTRESTO® y una estatina. No se observó interacción fármaco-fármaco clínicamente relevante cuando se administró conjuntamente simvastatina y ENTRESTO®.
Sildenafilo: En pacientes con hipertensión arterial, la adición de una dosis única de sildenafilo al tratamiento con ENTRESTO® se asoció con reducción de la tensión arterial mayor que la observada con ENTRESTO® solo. Por consiguiente, se debe proceder con precaución cuando se empiece a administrar sildenafilo u otro inhibidor de la fosfodiesterasa de tipo 5 (PDE-5) a pacientes en tratamiento con ENTRESTO®.
Interacciones previstas que deben tenerse en cuenta: Potasio: La administración junto con diuréticos ahorradores de potasio (por ejemplo: triamtereno, amilorida), antagonistas del receptor de mineralocorticoides (por ejemplo: espironolactona, eplerenona), suplementos de potasio o sustitutos de la sal común (de mesa) que contengan potasio puede aumentar las concentraciones de potasio y creatinina séricas. Si se administra ENTRESTO® junto con estos fármacos se recomienda vigilar la concentración sérica de potasio (ver Precauciones generales).
Antiinflamatorios no esteroideos (AINE), incluidos los inhibidores selectivos de la cicloxigenasa 2 (COX-2): En los pacientes ancianos, los pacientes hipovolémicos (incluidos los tratados con diuréticos) y los pacientes cuya función renal esté afectada, la administración de ENTRESTO® y un AINE puede elevar el riesgo de empeoramiento de la función renal. Por ello se recomienda vigilar dicha función cuando se inicie o modifique el tratamiento en pacientes que estén recibiendo al mismo tiempo ENTRESTO® y un AINE.
Litio: No se ha investigado la posibilidad de que se produzcan interacciones farmacológicas entre ENTRESTO® y el litio. Se han notificado elevaciones reversibles de las concentraciones séricas de litio y manifestaciones de toxicidad durante la administración de litio y un IECA o un ARA. Por consiguiente, se recomienda vigilar estrechamente las concentraciones séricas de litio durante el tratamiento conjunto con este y ENTRESTO®. Si también se está administrando un diurético, puede haber aún mayor riesgo de toxicidad del litio.
Transportadores: El metabolito farmacológicamente activo del sacubitrilo (sacubitrilato) y valsartán son sustratos de OATP1B1, OATP1B3 y OAT3; el valsartán es también sustrato de la proteína de resistencia a múltiples fármacos 2 (MRP2). Por consiguiente, la coadministración de ENTRESTO® y un inhibidor de OATP1B1, OATP1B3, OAT3 (por ejemplo: rifampicina, ciclosporina) o MRP2 (por ejemplo: ritonavir) puede aumentar la exposición sistémica a sacubitrilato o a valsartán, respectivamente. Se debe proceder con el cuidado adecuado cuando se comience o finalice un tratamiento concomitante con alguno de estos fármacos.
Interacciones no significativas: No se observaron interacciones farmacológicas clínicamente significativas con la coadministración de ENTRESTO® y furosemida, digoxina, warfarina, hidroclorotiazida, amlodipino, metformina, omeprazol, carvedilol, nitroglicerina por vía intravenosa o combinación de levonorgestrel/etinilestradiol. No se prevé ninguna interacción con atenolol, indometacina, glibenclamida ni cimetidina.
Interacciones con el CYP450: Los estudios de metabolismo in vitro indican que el riesgo de interacciones farmacológicas relacionadas con el CYP450 es bajo porque las isoformas de este citocromo desempeñan un papel limitado en el metabolismo de ENTRESTO®. ENTRESTO® no induce ni inhibe las isoformas de CYP450.
ALTERACIONES EN LOS RESULTADOS DE PRUEBAS DE LABORATORIO:
Hipocalcemia/hipercalcemia: Dentro de los estudios clínicos se reportaron las reacciones adversas hipocalcemia (frecuente) e hipercalcemia (muy frecuente), por lo que se debe monitorear adecuadamente la concentración sérica de calcio durante el tratamiento con ENTRESTO®.
Hemoglobina y hematocrito: Se observaron disminuciones de la hemoglobina/hematocrito menores al 20% en aproximadamente el 5%, tanto en el grupo tratado con ENTRESTO® como en el grupo tratado con enalapril, en el periodo a doble ciego del estudio PARADIGM-HF.
Creatinina sérica: Dentro del estudio clínico, se observaron incrementos de creatinina sérica mayores del 50% en el 1.4% de los pacientes en el grupo tratado con enalapril y en el 2.2% del grupo de pacientes tratados con ENTRESTO®. Durante el periodo a doble ciego, aproximadamente el 16% de los pacientes en ambos grupos de tratamiento con ENTRESTO® y enalapril tuvo aumentos en la creatinina sérica mayores al 50%.
Potasio sérico: Dentro del estudio clínico, se observaron concentraciones de potasio mayores a 5.5 mEq/L en aproximadamente el 4% de los pacientes, tanto en el grupo tratado con enalapril como en el grupo tratado con ENTRESTO®. Durante el periodo a doble ciego, aproximadamente el 16% de los pacientes de ambos grupos de tratamiento presentaron concentraciones de potasio superiores a 5.5 mEq/L.
PRECAUCIONES GENERALES:
No deben usarse dosis de ENTRESTO® superiores a 400 mg/día.
Bloqueo Dual del Sistema Renina-Angiotensina-Aldosterona (SRAA):
• No debe administrarse ENTRESTO® junto con un IECA porque existe riesgo de angioedema. No debe iniciarse el tratamiento con ENTRESTO® hasta 36 horas después de haber tomado la última dosis del IECA. Si se suspende el tratamiento con ENTRESTO®, no se debe comenzar la administración de un IECA hasta 36 horas después de la última dosis de ENTRESTO®(ver Contraindicaciones, dosis y vía de administración e interacciones medicamentosas y de otro género).
• Se debe proceder con cautela cuando se administre junto ENTRESTO® con inhibidores directos de la renina, como el aliskiren (ver Contraindicaciones e interacciones medicamentosas y de otro género). No debe combinarse ENTRESTO®con aliskiren en pacientes con diabetes de tipo 2 (ver Contraindicaciones).
• Dado que ENTRESTO® actúa como antagonista de los receptores de la angiotensina II (ARA), no debe administrarse junto con un ARA (ver Dosis y vía de administración e interacciones medicamentosas y de otro género).
Hipotensión arterial: Se han notificado casos de hipotensión sintomática en pacientes tratados con ENTRESTO® en ensayos clínicos. Si se produce hipotensión, se debe ajustar la dosis de los diuréticos o los antihipertensivos que se estén administrando en combinación, además de considerar la posibilidad de tratar otras causas de hipotensión (como la hipovolemia). En caso de que la hipotensión persista pese a estas medidas, se debe reducir la dosis de ENTRESTO® o suspender transitoriamente su administración (ver Dosis y vía de administración). No suele ser necesario retirar definitivamente el tratamiento. Es más probable que aparezca hipotensión sintomática si el paciente sufre hipovolemia como consecuencia, por ejemplo, de tratamiento con diuréticos, dieta hiposódica, diarrea o vómitos. Antes de iniciar el tratamiento con ENTRESTO® deben corregirse la pérdida de sodio, la hipovolemia o ambas.
Enfermedad renal: Como ocurre con todos los fármacos que actúan sobre el sistema renina-angiotensina-aldosterona, el uso de ENTRESTO® puede asociarse con una disminución de la función renal. En el estudio PARADIGM-HF, la incidencia de enfermedad renal clínicamente significativa era baja y las interrupciones del tratamiento por esta causa fueron menos frecuentes entre los pacientes tratados con ENTRESTO® (0.65%) que entre los que recibieron enalapril (1.28%). Se debe estudiar la posibilidad de reducir la dosis de ENTRESTO® en pacientes que presenten una disminución clínicamente significativa de la función renal. Se debe proceder con cautela cuando se administre ENTRESTO® a pacientes con enfermedad renal grave (ver Dosis y vía de administración y farmacocinética y farmacodinamia).
Hiperpotasemia: Como ocurre con todos los fármacos que actúan sobre el sistema renina-angiotensina-aldosterona, el uso de ENTRESTO® puede asociarse con un mayor riesgo de hiperpotasemia. En el estudio PARADIGM-HF, la incidencia de hiperpotasemia clínicamente significativa era baja y determinó la interrupción del tratamiento en el 0.26% de los pacientes que recibían ENTRESTO® y el 0.35% de los tratados con enalapril. Los fármacos que elevan las concentraciones de potasio (como los diuréticos ahorradores de potasio y los suplementos de potasio) deberán utilizarse con precaución cuando se administren junto con ENTRESTO®. Si se presenta hiperpotasemia clínicamente significativa, se deben adoptar medidas tales como reducir el contenido de potasio en la dieta o ajustar la dosis de los medicamentos concomitantes. Se recomienda vigilar las concentraciones séricas de potasio especialmente en los pacientes en los que se den factores de riesgo tales como enfermedad renal severa, diabetes mellitus, hipoaldosteronismo o una dieta rica en potasio (ver Dosis y vía de administración).
Angioedema: Se han notificado casos de angioedema en pacientes tratados con ENTRESTO®. Si se presenta angioedema, se debe suspender de inmediato la administración de ENTRESTO® e instaurar un tratamiento adecuado y la vigilancia necesaria hasta la desaparición completa y permanente de los signos y síntomas. No se debe volver a administrar ENTRESTO®. En los casos en que el angioedema solamente afectó al rostro y los labios, el trastorno se resolvió en general sin tratamiento, así mismo, los antihistamínicos resultaron útiles para aliviar los síntomas.
El angioedema que se asocia con edema laríngeo puede ser mortal. Cuando se afectan la lengua, la glotis o la laringe con riesgo de obstrucción de las vías respiratorias, es indispensable administrar sin demora el tratamiento adecuado (por ejemplo: una solución de epinefrina al 1:1000 por vía subcutánea [entre 0.3 mL y 0.5 mL]), adoptar las medidas necesarias para mantener la permeabilidad de las vías respiratorias o ambas cosas.
ENTRESTO® no se ha estudiado en pacientes con antecedentes de angioedema y dado que estos pacientes pueden correr un mayor riesgo de angioedema, se recomienda proceder con precaución cuando se les administre ENTRESTO®. No debe utilizarse ENTRESTO® en pacientes con antecedentes conocidos de angioedema relacionado con un tratamiento anterior a base de un IECA o un ARA, o pacientes con angioedema hereditario (ver Contraindicaciones).
Los pacientes de raza negra pueden tener mayor propensión a desarrollar angioedema.
Pacientes con estenosis de la arteria renal: Al igual que otros fármacos que actúan sobre el sistema renina-angiotensina-aldosterona, ENTRESTO® puede elevar las concentraciones de urea y creatinina séricas en pacientes con estenosis bilateral o unilateral de la arteria renal. Se debe proceder con cautela en los pacientes con estenosis de la arteria renal; en estos casos se recomienda vigilar la función renal.
DOSIS Y VÍA DE ADMINISTRACIÓN:
Dosis. La dosis prevista de ENTRESTO® es de 200 mg dos veces al día.
La dosis inicial recomendada de ENTRESTO® es de 100 mg dos veces al día. Se recomienda una dosis inicial de 50 mg dos veces al día en los pacientes que no estén tomando un inhibidor de la enzima convertidora de la angiotensina (IECA) ni un antagonista del receptor de la angiotensina II (ARA), y debe considerarse en el caso de los pacientes que hayan tomado anteriormente dosis bajas de estos fármacos.
Se duplicará la dosis de ENTRESTO® cada 2 a 4 semanas hasta alcanzar la dosis prevista de 200 mg dos veces al día, según la tolerabilidad del paciente.
La administración de ENTRESTO® con un IECA puede entrañar riesgo de angioedema, por lo que no se debe empezar a utilizar ENTRESTO® hasta 36 horas después de haber interrumpido el tratamiento con el IECA (ver Contraindicaciones).
Dado que ENTRESTO® también antagoniza a los receptores de la angiotensina II, no debe coadministrarse con un ara (ver Precauciones generales e Interacciones medicamentosas y de otro género).
Si los pacientes presentan problemas de tolerabilidad (hipotensión arterial sintomática, hiperpotasemia, disfunción renal), se debe estudiar la posibilidad de ajustar la dosis de los medicamentos concomitantes o de reducir transitoriamente la dosis de ENTRESTO®.
No deben usarse dosis de ENTRESTO® superiores a 400 mg/día.
Poblaciones especiales:
Insuficiencia renal: Se recomienda una dosis inicial de 50 mg dos veces al día en los pacientes con insuficiencia renal grave (TFGe <30 mL/min/1.73 m2). Se recomienda proceder con precaución al utilizar ENTRESTO® en esta población, ya que la información disponible es escasa (ver Farmacocinética y farmacodinamia).
No es preciso ajustar la dosis en los pacientes con insuficiencia renal leve (TFGe = 60-90 mL/min/1.73 m2) o moderada (TFGe = 30-60 mL/min/1.73 m2).
Insuficiencia hepática: Se recomienda una dosis inicial de 50 mg dos veces al día en los pacientes con insuficiencia hepática moderada (clase B de la clasificación de Child-Pugh).
No es preciso ajustar la dosis de ENTRESTO® cuando se administre a pacientes con disfunción hepática leve (clase A de la clasificación de Child-Pugh).
No se han llevado a cabo estudios en pacientes con insuficiencia hepática grave (clase C de la clasificación de Child-Pugh), por lo que no se recomienda utilizar ENTRESTO® en esta población (ver Farmacocinética yb farmacodinamia).
Pacientes pediátricos (menores de 18 años): No se ha determinado ni la seguridad ni la eficacia de ENTRESTO® en pacientes menores de 18 años.
Pacientes geriátricos (mayores de 65 años): No es necesario ajustar la dosis en pacientes mayores de 65 años.
Vía de administración: Oral.
Modo de administración:
ENTRESTO® puede administrarse con o sin alimentos (ver Farmacocinética y farmacodinamia).
MANIFESTACIONES Y MANEJO DE LA SOBREDOSIFICACIÓN O INGESTA ACCIDENTAL:
Se dispone de pocos datos relativos a la sobredosis de ENTRESTO® en el ser humano. Se ha estudiado la administración de una dosis única de 1,200 mg de ENTRESTO® y de dosis repetidas de 900 mg (durante 14 días) a voluntarios sanos, que fueron bien toleradas.
Dado el efecto hipotensor de ENTRESTO®, el síntoma más probable de la sobredosis es la hipotensión arterial. Se debe administrar tratamiento sintomático.
Es improbable que ENTRESTO® se elimine mediante hemodiálisis, ya que muestra un alto grado de unión a proteínas.
PRESENTACIONES:
Caja con 30 comprimidos de 50 mg.
Caja con 30 o 60 comprimidos de 100 mg.
Caja con 60 comprimidos de 200 mg.
RECOMENDACIONES SOBRE ALMACENAMIENTO:
Consérvese a no más de 25 °C.
Consérvese la caja bien cerrada. Protéjase de la humedad.
LEYENDAS DE PROTECCIÓN:
Literatura exclusiva para médicos. Su venta requiere receta médica. No se deje al alcance de los niños. No se use en el embarazo ni en la lactancia.
Reporte las sospechas de reacción adversa al correo: farmacovigilancia@cofepris.gob.mx
Nombre y domicilio del laboratorio
Titular de Registro Sanitario:
NOVARTIS PHARMA AG
Lichtstrasse 35, 4056 Basel, Suiza.
Representante Legal:
Novartis farmacéutica, s.A. De c.V.
Avenida Insurgentes Sur No. 2475
Piso 3, Col. Loreto, C.P.
01090, Álvaro Obregón,
Ciudad de México, México.
Reg. Núm. 521M2015 SSA IV
CDS: 19.May.2021, 10.Jul.2017, 10.
Ago.2015 NPI: Oct.2023 TN.:
N/A, 2017-PSB/GLC-0871-s,
2015-PSB/GLC-0769-s
NPI: Oct.2023
®Marca registrada


