
ENBREL - Solución inyectable
Sustancia(s):
- Etanercept
Presentaciones:
- 1 Caja, 4 Solución inyectable , 25 mg,
FORMA FARMACÉUTICA Y FORMULACIÓN:
Fórmula para frasco ámpula:
El frasco ámpula con polvo liofilizado contiene:
Etanercept 25 mg
Excipiente cbp 1 frasco
La jeringa con diluyente contiene:
Agua inyectable 1.0 mL
Fórmula para jeringa prellenada:
La jeringa prellenada contiene:
Etanercept 25 mg
Vehículo cbp 0.5 mL
Cada almohadilla contiene:
Alcohol isopropílico cs
Fórmula para jeringa prellenada:
La jeringa prellenada contiene:
Etanercept 50 mg
Vehículo cbp 1.0 mL
Cada almohadilla contiene:
Alcohol isopropílico cs
Fórmula para pluma precargada (MYCLIC):
La pluma precargada contiene:
Etanercept 50 mg
Vehículo cbp 1.0 mL
Cada almohadilla contiene:
Alcohol isopropílico cs
Fórmula para cartucho dispensador de dosis: (SMARTCLIC)
El cartucho dispensador de dosis contiene:
Etanercept 25 mg
Vehículo cbp 0.5 mL
Cada almohadilla contiene:
Alcohol isopropílico cs
Fórmula para cartucho dispensador de dosis: (SMARTCLIC)
El cartucho dispensador de dosis contiene:
Etanercept 50 mg
Vehículo cbp 1.0 mL
Cada almohadilla contiene:
Alcohol isopropílico cs
Etanercept es una proteína de fusión del receptor del factor de necrosis tumoral humano (RFNT) p75 Fc, producida por tecnología de ADN recombinante en un sistema de expresión celular de mamífero de ovario de hámster chino (OHC). Etanercept es un dímero de una proteína quimérica creada por ingeniería genética al fusionar el dominio de unión del ligando extracelular del receptor del factor de necrosis tumoral humano-2 (RFNT2 /p75) con el dominio Fc de la IgG1 humana. El componente Fc de etanercept contiene la unión principal de la región, de CH2, y de CH3, pero no la región CH1 de la IgG1.
Solubilidad: Etanercept es soluble en agua
Peso molecular (aparente) 150 kilodaltons
Polvo y diluyente para preparar solución inyectable:
La solución reconstituida de ENBREL® es clara a ligeramente opalescente e incolora o ligeramente amarilla o marrón pálido, con un pH de 7.4 ± 0.3
Solución inyectable en jeringa prellenada, pluma precargada y cartucho dispensador de dosis:
La solución para inyección en la jeringa prellenada, pluma precargada y cartucho dispensador de dosis es clara a ligeramente opalescente, incolora a amarillo o marrón pálido, y el líquido puede contener pequeñas partículas amorfas de proteína blancas o translúcidas, con un pH de 6.3 ± 0.2
INDICACIONES TERAPÉUTICAS:
Artritis reumatoide:
ENBREL® está indicado para reducir signos y síntomas en pacientes con artritis reumatoide (AR) activa moderada a severa e inhibir la progresión del daño estructural de la articulación. ENBREL® puede iniciarse en combinación con metotrexato o utilizarse solo.
ENBREL® puede utilizarse solo o en combinación con metotrexato para el tratamiento de la AR activa en adultos cuando la respuesta a uno o más medicamentos antirreumáticos modificadores de la enfermedad (FARME), incluyendo metotrexato (a menos que esté contraindicado), ha demostrado ser inadecuada.
Artritis idiopática juvenil:
En el tratamiento de la artritis idiopática juvenil de curso poliarticular (AIJ) en niños y adolescentes desde los 2 años cuando la respuesta a uno o más FARME ha demostrado ser inadecuada.
Artritis psoriásica:
ENBREL® está indicado para reducir signos y síntomas e inhibir la progresión del daño estructural de la artritis activa en pacientes con artritis psoriásica. ENBREL® puede usarse en combinación con metotrexato en pacientes adultos que no responden adecuadamente a metotrexato como agente único.
Espondilitis anquilosante:
ENBREL® está indicado para reducir los signos y síntomas en pacientes con espondilitis anquilosante.
Psoriasis en placas:
ENBREL® está indicado en el tratamiento de psoriasis en placas de moderada a severa en adultos (18 años o mayores) que son candidatos para tratamiento sistémico o fototerapia.
Psoriasis en placas pediátrica:
ENBREL® está indicado para el tratamiento de la psoriasis en placas crónica severa en niños y adolescentes desde la edad de 6 años que son controlados inadecuadamente por, o son intolerantes a, otras terapias sistémicas o fototerapias.
FARMACOCINÉTICA Y FARMACODINAMIA:
Propiedades farmacodinámicas:
Grupo farmacoterapéutico
Inhibidor de FNT-alfa
Código ATC: L04AB01
Uso geriátrico:
No se recomiendan ajustes específicos de dosis de ENBREL® según la edad del paciente.
Mecanismo de acción:
Etanercept es una forma soluble dimérica del receptor p75 del FNT (factor de necrosis tumoral) que puede unirse a dos moléculas del FNT. Etanercept inhibe la unión tanto de FNT (FNTα) como de linfotoxina alfa [LTα] (FNTβ) a los receptores del FNT de la superficie celular, volviendo así biológicamente inactivo al FNT y previniendo las respuestas celulares mediadas por FNT. El FNT es una citocina dominante en el proceso inflamatorio de pacientes adultos con artritis reumatoide. El FNT y LTα son expresados en pacientes con artritis idiopática juvenil. Se encuentran niveles elevados de FNT en el líquido sinovial de pacientes con artritis reumatoide y artritis idiopática juvenil. También se han encontrado niveles elevados de FNT en el suero y el tejido sinovial de pacientes con espondilitis anquilosante. En la psoriasis en placas la infiltración por células inflamatorias incluyendo linfocitos T conlleva a niveles más altos del FNT en las lesiones psoriásicas comparado con los niveles en piel sin lesiones. Dos receptores del FNT diferentes (RFNTs), una proteína de 55 kilodaltons (p55) y una proteína de 75 kilodaltons (p75) existen de manera natural como moléculas monoméricas sobre las superficies celulares y en formas solubles. La actividad biológica del FNT depende de la unión a cualquiera de los receptores de la superficie celular. Etanercept también puede modular las respuestas biológicas controladas por moléculas adicionales (p.ej., citocinas, moléculas de adhesión o proteinasas) que son inducidas o reguladas por el FNT. Etanercept inhibe la actividad del FNT in vitro y ha demostrado que afecta varios modelos animales de inflamación, incluyendo la artritis inducida por colágeno en ratones.
Eficacia clínica:
Esta sección presenta datos de cuatro ensayos en adultos con artritis reumatoide, 3 estudios en artritis idiopática juvenil, un estudio en adultos con artritis psoriásica, 4 estudios en adultos con espondilitis anquilosante, 3 estudios en adultos con psoriasis en placas y 2 estudios en pacientes pediátricos con psoriasis en placas.
Pacientes adultos con artritis reumatoide:
La eficacia de ENBREL® se evaluó en un estudio aleatorizado, doble ciego, controlado con placebo. El estudio evaluó a 234 pacientes adultos con artritis reumatoide activa que no respondieron al tratamiento con por lo menos un FARME, pero no más de cuatro. Las dosis de 10 o 25 mg de ENBREL® o placebo se administraron por vía subcutánea dos veces a la semana durante 6 meses consecutivos. Los resultados de este estudio controlado se expresaron en porcentaje de mejoría en la artritis reumatoide utilizando los criterios de respuesta del Colegio Americano de Reumatología (ACR, por sus siglas en inglés).
Las respuestas de ACR 20 y 50 fueron mayores en pacientes tratados con ENBREL® a los 3 y 6 meses que en pacientes tratados con placebo (ACR 20; ENBREL® 62% y 59%, placebo 23% y 11% a los 3 y 6 meses respectivamente; ACR 50: ENBREL® 41% y 40%, placebo 8% y 5% a los 3 y 6 meses respectivamente; p < 0.01 ENBREL® frente al placebo en todas las mediciones de tiempo para ambas respuestas ACR 20 y ACR 50).
Aproximadamente 15% de los sujetos que recibieron ENBREL® alcanzaron una respuesta ACR 70 en el mes 3 y 6 comparado con menos del 5% de los sujetos en el grupo placebo. Entre los pacientes que recibieron ENBREL®, las respuestas clínicas por lo general aparecieron en 1 a 2 semanas después de iniciar el tratamiento y casi siempre ocurrieron a los 3 meses. Se observó una respuesta a la dosis; los resultados con 10 mg fueron intermedios entre placebo y 25 mg, ENBREL® fue significativamente mejor que el placebo en todos los componentes de los criterios ACR, así como otras medidas de la actividad de la artritis reumatoide no incluida en los criterios de respuesta ACR, como rigidez matutina. El Cuestionario de Evaluación de la Salud (HAQ, por sus siglas en inglés), que incluyó discapacidad, vitalidad, salud mental, estado de salud general y subdominios del estado de salud asociado con artritis se aplicó cada 3 meses durante el estudio. Todos los subdominios del HAQ mejoraron en los pacientes tratados con ENBREL® comparado con los controles a los 3 y 6 meses.
Después de suspender ENBREL®, por lo general los síntomas de artritis se volvieron a presentar en un mes. La reintroducción del tratamiento con ENBREL® después de haberlo suspendido hasta por 24 meses produjo las mismas respuestas que aquellas de los pacientes que recibieron ENBREL® sin interrupción del tratamiento con base en resultados de estudios abiertos. Se han observado respuestas duraderas continuas hasta por 10 años en ensayos de extensión de tratamiento abiertos en pacientes que recibieron ENBREL® sin interrupción.
La eficacia de ENBREL® se comparó con metotrexato en un segundo estudio aleatorizado, controlado con principio activo con evaluaciones radiográficas ciegas como criterio de valoración primario en 632 pacientes adultos con artritis reumatoide activa (< 3 años de duración) que nunca habían recibido tratamiento con metotrexato. Dosis de 10 mg o 25 mg de ENBREL® se administraron por vía SC dos veces a la semana hasta por 24 meses. Las dosis de metotrexato se escalaron de 7.5 mg/semana a un máximo de 20 mg/semana durante las primeras 8 semanas del ensayo y se continuaron hasta por 24 meses. La mejoría clínica que incluyó el inicio de acción en 2 semanas con ENBREL® 25 mg fue similar al observado en ensayos previos y se mantuvo hasta por 24 meses. Al inicio, los pacientes tuvieron un grado moderado de discapacidad, con puntuaciones en el HAQ promedio de 1.4 a 1.5. El tratamiento con ENBREL® 25 mg resultó una mejoría sustancial a los 12 meses, con aproximadamente 44% de los pacientes alcanzando una puntación HAQ normal (menor a 0.5). Este beneficio se mantuvo en el segundo año del estudio.
En este estudio, el daño articular estructural se evaluó radiográficamente y se expresó como el cambio en la Puntuación Total de Sharp (TSS, por sus siglas en inglés) y sus componentes, la puntuación de la erosión y puntuación del estrechamiento del espacio articular (JSN, por sus siglas en inglés). Las radiografías de manos/muñecas y pies se interpretaron en el inicio y a los 6, 12 y 24 meses. La dosis de ENBREL® de 10 mg presentó de forma consistente un efecto menor sobre el daño estructural que la dosis de 25 mg. ENBREL® 25 mg fue significativamente superior a metotrexato en las puntuaciones de erosión tanto a los 12 como a los 24 meses. Las diferencias en TSS y JSN no fueron estadísticamente significativas entre metotrexato y ENBREL® 25 mg. Los resultados se muestran en la figura que aparece a continuación.
Progresión radiográfica: comparación de ENBREL® frente a metotrexato en pacientes con AR de < 3 años de duración

En otro estudio doble ciego, aleatorizado, controlado con principio activo, la eficacia clínica, seguridad y progresión radiográfica en pacientes con AR tratados con ENBREL® solo (25 mg dos veces a la semana), metotrexato solo (7.5 a 20 mg a la semana, mediana de la dosis 20 mg) y la combinación de ENBREL® y metotrexato iniciada concomitantemente se compararon en 682 pacientes adultos con artritis reumatoide activa de 6 meses a 20 años de duración (mediana 5 años) que tuvieron una respuesta no satisfactoria al tratamiento con al menos 1 FARME distinto del metotrexato.
Los pacientes en el grupo de ENBREL® en combinación con metotrexato tuvieron respuestas ACR 20, ACR 50, ACR 70 significativamente mayores y mejoría en las puntuaciones DAS y HAQ a las 24 y 52 semanas que los pacientes de cualquiera de los grupos de tratamiento solo (los resultados se muestran en la tabla a continuación). Después de 24 meses de tratamiento, también se observaron ventajas significativas con la administración de ENBREL® en combinación con metotrexato, comparado con la monoterapia tanto de ENBREL® como de metotrexato.
Resultados de eficacia clínica a los 12 meses: comparación de ENBREL® versus metotrexato versus ENBREL® en combinación con metotrexato en pacientes con AR de 6 meses a 20 años de duración
|
Punto final |
Metotrexato (n = 228) |
ENBREL® (n = 223) |
ENBREL® + metotrexato (n = 231) |
|
Respuestas ACRa |
|||
|
ACR 20 |
58.8% |
65.5% |
74.5%†,f |
|
ACR 50 |
36.4% |
43.0% |
63.2%†,f |
|
ACR 70 |
16.7% |
22.0% |
39.8%†,f |
|
DAS |
|||
|
Puntuación basalb |
5.5 |
5.7 |
5.5 |
|
Puntuación semana 52b |
3.0 |
3.0 |
2.3†,f |
|
Remisiónc |
14% |
18% |
37%†,f |
|
HAQ |
|||
|
Basal |
1.7 |
1.7 |
1.8 |
|
Semana 25 |
1.1 |
1.0 |
0.8†,f |
a Los pacientes que no completaron los 12 meses en el estudio se consideraron como pacientes que no respondieron al tratamiento.
b Los valores de la Puntuación de la Actividad de la Enfermedad (DAS, por sus siglas en inglés) son medias.
c La remisión se define como DAS < 1.6
Valores p de comparación de pares: † = p < 0.05 para comparaciones de ENBREL® + metotrexato versus metotrexato y f = p < 0.05 para comparaciones de ENBREL® + metotrexato versus ENBREL®
La progresión radiográfica a los 12 meses fue significativamente menor en el grupo de ENBREL® que en el grupo de metotrexato, mientras que la combinación fue significativamente mejor que en cualquier grupo de monoterapia en la ralentización de la progresión radiográfica (ver la siguiente figura).
Progresión radiográfica: comparación de ENBREL® versus metotrexato versus ENBREL® en combinación con metotrexato en pacientes con ar de 6 meses a 20 años de duración (resultados a los 12 meses)
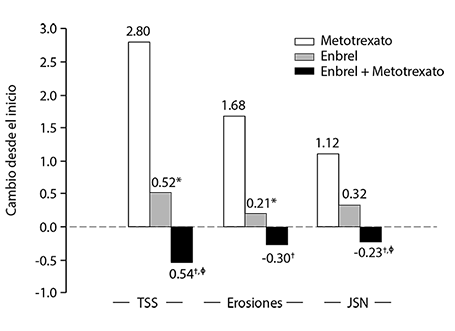
Valores p de comparación de pares: * = p <0.05 para comparaciones de ENBREL® versus metotrexato, † = p < 0.05 para comparaciones de ENBREL® + metotrexato versus metotrexato y Ø = p < 0.05 para comparaciones de ENBREL® + metotrexato versus ENBREL®
Después de 24 meses de tratamiento, también se observaron ventajas significativas con la administración de ENBREL® en combinación con metotrexato, comparado con la monoterapia tanto de ENBREL® como de metotrexato. De forma similar, también se observaron ventajas significativas del tratamiento con ENBREL® como monoterapia, comparado con la monoterapia con metotrexato después de 24 meses.
En un análisis, en el que se consideró que todos los pacientes que habían abandonado el estudio por cualquier motivo habían progresado, el porcentaje de pacientes sin progresión (cambio TSS ≤ 0.5) a los 24 meses era mayor en el grupo ENBREL® en combinación con metotrexato en comparación con los grupos de monoterapia con ENBREL® y metotrexato (62%, 50% y 36%, respectivamente; p < 0.05). La diferencia entre los grupos de ENBREL® en monoterapia y metotrexato en monoterapia también fue significativa (p<0.05). Las tasas de ausencia de progresión entre los pacientes del estudio que completaron el periodo total de 24 meses de tratamiento fueron de un 78%, 70% y 61%, respectivamente.
Se evaluaron la seguridad y eficacia de 50 mg de ENBREL® (dos inyecciones subcutáneas de 25 mg) administradas una vez a la semana en un estudio doble ciego, controlado con placebo con 420 pacientes con AR activa. En este estudio, 53 pacientes recibieron placebo, 214 recibieron 50 mg de ENBREL® una vez a la semana y 153 pacientes recibieron 25 mg de ENBREL® dos veces a la semana. Los perfiles de seguridad y eficacia de los dos esquemas de tratamiento de ENBREL® fueron comparables en la semana 8 en lo que se refiere a signos y síntomas de AR; los datos en la semana 16 no mostraron comparabilidad (no inferioridad) entre los dos esquemas.
Población pediátrica con artritis idiopática juvenil:
La seguridad y eficacia de ENBREL® se evaluaron en un estudio de dos partes en 69 niños con artritis idiopática juvenil de curso poliarticular que tenían una variedad de formas de inicio de artritis idiopática juvenil (poliartritis, pauciartritis, aparición sistémica). Se incluyeron pacientes de entre 4 y 17 años con artritis idiopática juvenil de curso poliarticular activa de moderada a severa refractaria o intolerante a metotrexato; los pacientes permanecieron con una dosis estable de un único fármaco antiinflamatorio no esteroideo y/o prednisona (≤ 0.2 mg/kg/día o máximo 10 mg). En la parte 1, todos los pacientes recibieron 0.4 mg/kg (máximo 25 mg por dosis) de ENBREL® por vía subcutánea, dos veces a la semana. En la parte 2, los pacientes con respuesta clínica en el día 90 fueron aleatorizados para continuar recibiendo ENBREL® o recibir placebo durante cuatro meses y se evaluaron para el brote de enfermedad. Las respuestas se midieron utilizando la ACR Pedi 30, definida como ≥ 30% de mejoría en al menos 3 de los 6 criterios y empeoramiento de ≥ 30% en no más de uno de los seis criterios principales de la JRA, incluyendo recuento de articulaciones activas, limitación de la movilidad, evaluaciones globales por un médico y el paciente/padre, evaluación funcional y VSG. El brote de la enfermedad se definió como un empeoramiento ≥ 30% en tres de los seis criterios principales de la JRA y mejoría ≥30% en no más de uno de los seis criterios principales de la JRA y un mínimo de 2 articulaciones activas.
En la parte 1 del estudio, 51 de 69 pacientes (74%) demostraron respuesta clínica y entraron en la parte 2. En la parte 2, 6 de 25 (24%) de los pacientes que continuaron con ENBREL® experimentaron un brote de la enfermedad comparado con 20 de 26 pacientes (77%) de los que recibieron placebo ( p = 0.007). Desde el inicio de la parte 2, la mediana del tiempo hasta el brote de la enfermedad fue ≥116 días para los pacientes que recibieron ENBREL® y 28 días para los que recibieron placebo. Cada componente de los criterios principales de la JRA empeoró en el grupo que recibió placebo y se mantuvo estable o mejoró en el grupo que continuó con ENBREL®. Los datos sugieren la posibilidad de una tasa de brote más alta entre aquellos pacientes con una VSG inicial más alta. De los pacientes que demostraron respuesta clínica a los 90 días y entraron en la parte 2 del estudio, algunos de los que continuaron con ENBREL® continuaron mejorando desde el mes 3 hasta el 7, mientras que los que recibieron placebo no mejoraron.
En un estudio de extensión de seguridad abierto, 58 pacientes pediátricos del estudio anterior (desde los 4 años al momento de la inscripción) continuaron recibiendo ENBREL® por hasta 10 años. Las tasas de eventos adversos serios e infecciones serias no aumentaron con la exposición a largo plazo.
En otro estudio abierto de un solo grupo (n =127), 60 pacientes con oligoartritis extendida (OE) (15 pacientes entre 2 y 4 años, 23 pacientes entre 5 y 11 años y 22 pacientes entre 12 y 17 años), 38 pacientes con artritis relacionada con entesitis (12 a 17 años), y 29 pacientes con artritis psoriásica (12 a 17 años) se trataron con ENBREL® a una dosis de 0.8 mg/kg (hasta un máximo de 50 mg por dosis) administrado semanalmente por 12 semanas. En cada uno de los subtipos de AIJ, la mayoría de los pacientes alcanzó los criterios ACR Pedi 30 y demostró mejoría clínica en los resultados secundarios tales como el número de articulaciones sensibles y la evaluación médica global. El perfil de seguridad fue consistente con el observado en otros estudios AIJ.
De los 127 pacientes del estudio original, 109 participaron en el estudio de extensión abierto y fueron seguidos durante 8 años adicionales para un total de hasta 10 años. Al final del estudio de extensión, 84/109 (77%) pacientes habían completado el estudio; 27 (25%) mientras tomaban activamente ENBREL®, 7 (6%) se habían retirado del tratamiento debido a enfermedad baja/inactiva; 5 (5%) habían reiniciado ENBREL® luego de una interrupción anterior del tratamiento; y 45 (41%) habían dejado de tomar ENBREL® (pero permanecían bajo observación); 25/109 (23%) pacientes abandonaron permanentemente el estudio. Las mejoras en el estado clínico logradas en el estudio principal generalmente se mantuvieron para todos los criterios de valoración de eficacia durante todo el periodo de seguimiento. Los pacientes que toman activamente ENBREL® podían entrar en un periodo opcional de retratamiento por interrupción una vez durante el estudio de extensión según el juicio del investigador sobre la respuesta clínica. 30 pacientes entraron en el periodo de interrupción. Se informó que 17 pacientes tuvieron un brote (definido como ≥ 30% de empeoramiento en al menos 3 de los 6 componentes de ACR Pedi con ≥ 30% de mejoría en no más de 1 de los 6 componentes restantes y un mínimo de 2 articulaciones activas); la mediana de tiempo hasta el brote después de la interrupción de ENBREL® fue de 190 días. 13 pacientes volvieron a tratarse y la mediana del tiempo hasta el retratamiento desde la interrupción se estimó en 274 días. Debido a la pequeña cantidad de puntos de datos, estos resultados deben interpretarse con precaución.
Se reportó una neoplasia maligna, enfermedad de Hodgkin, durante el primer año del estudio de extensión en un paciente de 18 años con JIA por OE. El número (tasa ajustada por exposición por 100 años-paciente [EP100PY]) de eventos adversos graves, neoplasias malignas e infecciones graves fue 40 (5.85 EP100PY), 1 (0.15 EP100PY) y 14 (2.05 EP100PY), respectivamente. El perfil de seguridad fue consistente con el observado en otros estudios de AIJ.
No se han realizado estudios en pacientes con artritis idiopática juvenil para evaluar los efectos del tratamiento continuo con ENBREL® en pacientes que no responden dentro de los 3 meses posteriores al inicio del tratamiento con ENBREL®. Además, no se han realizado estudios para evaluar los efectos de la reducción de la dosis recomendada de ENBREL® después de su uso a largo plazo en pacientes con AIJ.
Se evaluó la seguridad de ENBREL® a largo plazo como monoterapia (n = 103), ENBREL® más metotrexato (n = 294) o metotrexato como monoterapia (n = 197) hasta por 3 años en una población de 594 niños de 2 a 18 años con artritis idiopática juvenil, de los cuales 39 tenían de 2 a 3 años. En general, se reportaron infecciones más frecuentes en pacientes tratados con ENBREL® en comparación con metotrexato solo (3.8% versus 2%), y las infecciones asociadas con el uso de ENBREL® fueron de naturaleza más grave.
Pacientes adultos con artritis psoriásica:
La eficacia de ENBREL® se evaluó en un estudio aleatorizado, doble ciego, controlado con placebo con 205 pacientes con artritis psoriásica. Los pacientes tenían entre 18 y 70 años y tenían artritis psoriásica activa (≥ 3 articulaciones inflamadas y ≥ 3 articulaciones sensibles) en al menos una de las siguientes formas: (1) compromiso interfalángico distal (DIP, por sus siglas en inglés); (2) artritis poliarticular (ausencia de nódulos reumatoides y presencia de psoriasis); (3) artritis mutilante; (4) artritis psoriásica asimétrica; o (5) anquilosis similar a la espondilitis. Los pacientes también tenían psoriasis en placa con una lesión blanco calificada de ≥ 2 cm de diámetro. Los pacientes habían sido tratados previamente con AINE (86%), FARME (80%) y corticosteroides (24%). Los pacientes que en ese momento estaban siendo tratados con metotrexato (estables durante ≥ 2 meses) podían continuar con una dosis estable de ≤ 25 mg/semana de metotrexato. Se administraron dosis por vía subcutánea de 25 mg de ENBREL® (con base en los resultados de estudios de hallazgo de dosis de pacientes con artritis reumatoide) o placebo 2 veces a la semana durante 6 meses. Una vez finalizado el estudio doble ciego, los pacientes pudieron participar en un estudio de extensión abierto, a largo plazo durante un periodo máximo de 2 años.
Las respuestas clínicas se expresaron como porcentajes de pacientes que lograron una respuesta ACR 20, 50 y 70 y porcentajes con mejoría en los Criterios de Respuesta de Artritis Psoriásica (PsARC, por sus siglas en inglés). Los resultados se resumen en la siguiente tabla.
Respuestas de pacientes con artritis psoriásica
en un estudio controlado con placebo
|
Respuesta de la |
Porcentaje |
ENBREL® a |
|
Placebo |
||
|
ACR 20 |
||
|
Mes 3 |
15 |
59b |
|
Mes 6 |
13 |
50b |
|
ACR 50 |
||
|
Mes 3 |
4 |
38b |
|
Mes 6 |
4 |
37b |
|
ACR 70 |
||
|
Mes 3 |
0 |
11b |
|
Mes 6 |
1 |
9c |
|
PsARC |
||
|
Mes 3 |
31 |
72b |
|
Mes 6 |
23 |
70b |
a 25 mg ENBREL® S.C. dos veces a la semana.
b p < 0.001, ENBREL® frente a placebo.
c p < 0.01, ENBREL® frente a placebo.
Las respuestas clínicas entre pacientes con artritis psoriásica que recibieron ENBREL® fueron visibles en la primera visita (4 semanas) y se mantuvieron a lo largo de 6 meses de tratamiento. ENBREL® fue significativamente mejor que el placebo en todas las mediciones de actividad de la enfermedad (p < 0.001) y las respuestas fueron similares con y sin tratamiento concomitante con metotrexato. Se evaluó la calidad de vida en pacientes con artritis psoriásica en cada punto de medición utilizando el índice de discapacidad del HAQ. La puntuación del índice de discapacidad mejoró significativamente en todos los puntos de medición en los pacientes con artritis psoriásica tratados con ENBREL® en relación con los tratados con placebo (p < 0.001).
En el estudio de artritis psoriásica se evaluaron los cambios radiográficos. Las radiografías de manos y muñecas se interpretaron al inicio y a los 6, 12 y 24 meses. En la tabla que se presenta a continuación, se presenta la TSS modificada a los 12 meses. En un análisis, en el que se consideró que todos los pacientes que habían abandonado el estudio por cualquier motivo habían progresado, el porcentaje de pacientes que no presentó progresión (cambio TSS ≤ 0.5) a los 12 meses era mayor en el grupo de ENBREL® comparado con el grupo placebo (73% versus 47%, respectivamente; p ≤ 0.001). El efecto de ENBREL® sobre la progresión radiográfica se mantuvo en los pacientes que continuaron el tratamiento durante el segundo año.
En los pacientes con compromiso poliarticular simétrico se observó una ralentización del desarrollo de daño en las articulaciones periféricas.
Cambio medio (EE) anualizado a partir del valor inicial en la Puntuación Total de Sharp (TSS)
|
Tiempo |
Placebo (n = 104) |
ENBREL® (n = 101) |
|
Mes 12 |
1.00 (0.29) |
-0.03 (0.09)a |
EE = error estándar.
a p = 0.0001
El tratamiento con ENBREL® resultó en la mejoría de la función física durante el periodo doble ciego, y este beneficio se mantuvo durante la exposición a largo plazo hasta por 2 años.
No existe suficiente evidencia sobre la eficacia de ENBREL® en los pacientes que presentan artropatías tipo espondilitis anquilosante y artritis psoriásica mutilante debido al reducido número de pacientes estudiados.
No se ha realizado ningún estudio en pacientes con artritis psoriásica, utilizando el esquema de dosificación de 50 mg una vez a la semana. La evidencia sobre la eficacia del esquema de dosificación de una vez a la semana en esta población de pacientes se ha basado en los datos procedentes del estudio realizado en pacientes con espondilitis anquilosante.
Pacientes adultos con espondilitis anquilosante:
La eficacia de ENBREL® se evaluó en espondilitis anquilosante en 3 estudios aleatorizados, doble ciego, en los que se comparó la administración de 25 mg de ENBREL® dos veces a la semana con la administración de placebo. Un total de 401 pacientes se incluyeron de los cuales 203 se trataron con ENBREL®. El más grande de estos estudios (n = 277) incluyó a pacientes de edades de entre 18 y 70 años y tenían espondilitis anquilosante activa definida según las puntuaciones de la escala visual análoga (EVA) de ≥ 30 para un promedio de duración e intensidad de rigidez matutina más puntuaciones EVA de ≥ 30 para al menos 2 de los siguientes 3 parámetros: evaluación global del paciente; promedio de los valores de la EVA para dolor de espalda nocturno y dolor de espalda total; promedio de 10 preguntas sobre el Índice Funcional de Espondilitis Anquilosante (BASFI, por sus siglas en inglés). Los pacientes que recibieron FARME, AINE o corticosteroides pudieron continuar con ellos a dosis estables. No se incluyeron en el estudio pacientes con anquilosis completa de la columna. Se administraron a 138 pacientes dosis de 25 mg de ENBREL® (basadas en estudios de búsqueda de dosis en pacientes con artritis reumatoide) o placebo por vía subcutánea dos veces a la semana durante 6 meses.
La medida primaria de eficacia (ASAS 20) fue una mejoría ≥ 20% en al menos 3 de los 4 dominios de la evaluación en espondilitis anquilosante (ASAS) (evaluaciones globales del paciente, dolor de espalda, BASFI e inflamación) y ausencia de deterioro en los dominios restantes. Las respuestas ASAS 50 y 70 utilizaron los mismos criterios con una mejoría del 50% y del 70%, respectivamente.
Comparado con placebo, el tratamiento con ENBREL® dio como resultado mejorías significativas en la respuesta ASAS 20, ASAS 50 y ASAS 70, tan pronto como 2 semanas después del inicio del tratamiento.
Respuestas de pacientes con espondilitis anquilosante en un estudio controlado con placebo
|
Porcentaje de pacientes |
||
|---|---|---|
|
Respuesta de la Espondilitis anquilosante |
Placebo |
ENBREL® |
|
ASAS 20 |
||
|
2 semanas |
22 |
46a |
|
3 meses |
27 |
60a |
|
6 meses |
23 |
58a |
|
ASAS 50 |
||
|
2 semanas |
7 |
24a |
|
3 meses |
13 |
45a |
|
6 meses |
10 |
42a |
|
ASAS 70 |
||
|
2 semanas |
2 |
12b |
|
3 meses |
7 |
29b |
|
6 meses |
5 |
28b |
a p < 0.001, ENBREL® versus placebo
b p = 0.002, ENBREL® versus placebo
Entre los pacientes con espondilitis anquilosante que recibieron ENBREL®, las respuestas clínicas fueron evidentes en el momento de la primera visita (2 semanas) y se mantuvieron a lo largo de 6 meses de tratamiento. Las respuestas fueron similares en pacientes que estaban o no recibiendo tratamientos concomitantes al inicio del estudio.
En dos ensayos más pequeños de espondilitis anquilosante se obtuvieron resultados similares.
En un cuarto estudio, se evaluaron la seguridad y eficacia de 50 mg de ENBREL® (dos inyecciones subcutáneas de 25 mg) administradas una vez a la semana versus 25 mg de ENBREL® administrado dos veces a la semana en un estudio doble ciego, controlado con placebo en 356 pacientes con espondilitis anquilosante activa. Los perfiles de seguridad y eficacia de los esquemas de 50 mg una vez a la semana y 25 mg dos veces a la semana fueron similares.
Pacientes adultos con psoriasis en placas:
La seguridad y eficacia de ENBREL® en pacientes con psoriasis en placas se evaluaron en tres estudios aleatorizados, doble ciego, controlados con placebo. El criterio de valoración primario de eficacia en los tres estudios fue la proporción de pacientes en cada grupo de tratamiento que alcanzó un PASI de 75 (es decir, por lo menos un 75% de mejoría en el Índice de Área y Severidad de la Psoriasis desde el inicio) a las 12 semanas.
El estudio 1 fue un estudio de Fase 2 en pacientes con psoriasis en placas activa pero clínicamente estable que comprometía ≥ 10% de la superficie corporal y tenían ≥ 18 años. Ciento doce pacientes (112) se aleatorizaron para recibir una dosis de 25 mg de ENBREL® (n = 57) o placebo (n = 55) dos veces a la semana durante 24 semanas.
En el estudio 2 se evaluó a 652 pacientes con psoriasis en placas crónica utilizando los mismos criterios de inclusión que el estudio 1 con la adición de un Índice de Área y Severidad de la Psoriasis (PASI, por sus siglas en inglés) mínimo de 10 en el cribado. ENBREL® se administró a dosis de 25 mg una vez a la semana, 25 mg dos veces a la semana o 50 mg dos veces a la semana durante 6 meses consecutivos. Durante las primeras 12 semanas del periodo de tratamiento doble ciego, los pacientes recibieron placebo o una de las 3 dosis de ENBREL® mencionadas anteriormente. Después de 12 semanas de tratamiento, los pacientes del grupo placebo iniciaron tratamiento ciego con ENBREL® (25 mg dos veces por semana). Los pacientes en los grupos de tratamiento activo continuaron hasta la semana 24 bajo la misma dosis con la que originalmente se aleatorizaron.
En el estudio 3 se evaluaron a 583 pacientes y se aplicaron los mismos criterios de inclusión que el estudio 2. Los pacientes en este estudio recibieron una dosis de 25 o 50 mg de ENBREL®, o placebo dos veces a la semana durante 12 semanas y luego todos los pacientes recibieron en forma abierta 25 mg de ENBREL® dos veces a la semana durante otras 24 semanas.
En el estudio 1, el grupo tratado con ENBREL® tuvo una proporción significativamente más alta de pacientes con una respuesta PASI 75 en la semana 12 (30%) comparado con el grupo tratado con placebo (2%) (p < 0.0001). A las 24 semanas, 56% de los pacientes en el grupo tratado con ENBREL® había alcanzado PASI 75 comparado con 5% de los pacientes tratados con placebo. Los resultados clave de los estudios 2 y 3 se muestran a continuación.
Respuestas de los pacientes con psoriasis en los estudios 2 y 3
|
Estudio 2 |
Estudio 3 |
|||||||
|
ENBREL® |
ENBREL® |
|||||||
|
Placebo |
25 mg BIS |
50 mg BIS |
Placebo |
25 mg BIS |
50 mg BIS |
|||
|
n = 166 |
n = 162 |
n = 162 |
n = 164 |
n = 164 |
n = 193 |
n = 196 |
n = 196 |
|
|
Respuesta |
sem 12 |
sem 12 |
sem 24a |
sem 12 |
sem 24a |
sem 12 |
sem 12 |
sem 12 |
|
PASI 50% |
14 |
58* |
70 |
74* |
77 |
9 |
64* |
77* |
|
PASI 75% |
4 |
34* |
44 |
49* |
59 |
3 |
34* |
49* |
|
DSGA limpio o casi limpiob |
5 |
34* |
39 |
49* |
55 |
4 |
39* |
57* |
* p ≤ 0.0001 comparado con placebo.
a No se hicieron comparaciones estadísticas con placebo en la semana 24 en el Estudio 2 debido a que el grupo original de placebo empezó recibiendo ENBREL® 25 mg dos veces a la semana (BIS) de la semana 13 a la 24.
b La Evaluación Dermatológica Global Estática (DSGA, por sus siglas en inglés) limpia o casi limpia definida como 0 o 1 en una escala de 0 a 5.
Entre los pacientes con psoriasis en placas que recibieron ENBREL®, las respuestas significativas en relación con el placebo fueron evidentes en el momento de la primera visita (2 semanas) y se mantuvieron durante 24 semanas de tratamiento.
El estudio 2 tuvo también un periodo de suspensión del fármaco durante el cual a los pacientes que alcanzaron una mejoría de PASI de al menos un 50% en la semana 24 se les suspendió el tratamiento. Se observó a los pacientes fuera de tratamiento en cuanto a la posibilidad de recurrencia (PASI ≥ 150% del inicio) y en cuanto al tiempo para la recaída (definido como una pérdida de al menos la mitad de la mejoría alcanzada entre la visita inicial y la semana 24). Durante el periodo de suspensión volvieron gradualmente los síntomas de psoriasis con una mediana del tiempo de recurrencia de la enfermedad de 3 meses. No se observó ninguna exacerbación de la enfermedad y ningún evento adverso serio relacionado con la psoriasis. Hubo cierta evidencia que permite avalar un beneficio de la repetición del tratamiento con ENBREL® en pacientes que inicialmente respondían al tratamiento.
En el estudio 3, la mayoría de los pacientes (77%) que fueron aleatorizados inicialmente a ENBREL® 50 mg dos veces a la semana y que en la semana 12 se les redujo la dosis a 25 mg dos veces a la semana mantuvieron su respuesta PASI 75 a lo largo de la semana 36. Para pacientes que recibieron 25 mg de ENBREL® dos veces a la semana a lo largo del estudio, la respuesta PASI 75 continuó mejorando entre las semanas 12 a 36.
En estudios a largo plazo (hasta de 34 meses), abiertos en donde se administró ENBREL® sin interrupción, las respuestas clínicas fueron sostenidas y la seguridad fue comparable a los estudios de plazo más corto.
Pacientes pediátricos con psoriasis en placas:
La eficacia de ENBREL® se evaluó en un estudio aleatorizado, doble ciego, controlado con placebo en 211 pacientes pediátricos de 4 a 17 años con psoriasis en placas de moderada a severa (como se definió por una puntuación sPGA ≥ 3, que incluyó ≥ 10% de la SCA, y PASI ≥ 12). Los pacientes elegibles tenían antecedentes de haber recibido fototerapia o tratamiento sistémico o estaban inadecuadamente controlados con tratamiento tópico.
Los pacientes recibieron ENBREL® 0.8 mg/kg (hasta 50 mg) o placebo una vez a la semana por 12 semanas. En la semana 12, más pacientes aleatorizados a ENBREL® tuvieron respuestas de eficacia positivas (por ejemplo, PASI 75) que los aleatorizados a placebo.
Resultados de la psoriasis en placas en pacientes pediátricos a las 12 semanas
|
ENBREL® |
Placebo |
|
|
(n = 106) |
(n = 105) |
|
|
PASI 75, n (%) |
60 (57%)a |
12 (11%) |
|
PASI 50, n (%) |
79 (75%)a |
24 (23%) |
|
sPGA “limpio” o “mínimo”, n (%) |
56 (53%)a |
14 (13%) |
Abreviaturas: sPGA-Evaluación Global del Médico estática
a p < 0.0001 comparado con placebo
Después de las 12 semanas del periodo de tratamiento doble ciego, todos los pacientes que ingresaron al periodo abierto recibieron ENBREL® 0.8 mg/kg (hasta 50 mg) una vez a la semana durante otras 24 semanas. Las respuestas observadas durante el periodo abierto fueron similares a aquellas observadas en el periodo doble ciego.
Durante un periodo de suspensión aleatorizado, significativamente más pacientes re-aleatorizados a placebo, experimentaron recaída de la enfermedad (pérdida de respuesta de PASI 75) comparado con pacientes que se re-aleatorizaron a ENBREL®. Con el tratamiento continuo las respuestas se mantuvieron hasta por 48 semanas.
La seguridad y eficacia a largo plazo de ENBREL® a 0.8 mg/Kg (hasta 50 mg) una vez a la semana se evaluó en un estudio de extensión abierto de 181 pacientes pediátricos con psoriasis en placa hasta por 2 años después del estudio de 48 semanas mencionado arriba. La experiencia a largo plazo con ENBREL® fue generalmente similar a la del estudio original de 48 semanas y no reveló algún nuevo hallazgo de seguridad.
Propiedades farmacocinéticas:
Absorción:
Etanercept se absorbe lentamente desde el sitio de inyección SC, alcanzando su concentración máxima aproximadamente 48 horas después de una dosis única. La biodisponibilidad absoluta es del 76%.
Distribución:
Después de una dosis única SC de 25 mg de etanercept, la concentración sérica máxima promedio observada en voluntarios sanos fue de 1.65 ± 0.66 μg/mL, y el área bajo la curva fue de 235 ± 96.6 μg• h/mL. La proporcionalidad de la dosis no ha sido evaluada formalmente, pero no hay una saturación aparente de la depuración a lo largo del rango de dosificación.
El volumen de distribución en estado estable después de la administración subcutánea es de 13.9 ± 9.4 L.
Después de la dosificación continua a pacientes con AR (n = 25) con ENBREL® durante 6 meses con 25 mg dos veces por semana, la mediana observada fue de 3.0 μg/mL (rango 1.7 a 5.6 μg/mL). Con base en los datos disponibles, cada paciente puede presentar un aumento de dos a cinco veces los niveles séricos con la dosificación repetida.
Eliminación:
Etanercept es depurado lentamente del cuerpo. La vida media es de aproximadamente 80 horas.
La depuración es de aproximadamente 175 ± 116 mL/h en pacientes con artritis reumatoide y de 131 ± 81 mL/h en voluntarios sanos.
Después de la administración de etanercept radiomarcado a pacientes y voluntarios, la radioactividad se elimina por la orina.
Insuficiencia renal o hepática:
Aunque hay eliminación de radioactividad por la orina después de la administración de etanercept radiomarcado a pacientes y voluntarios sanos, no se observó un aumento de las concentraciones de etanercept en pacientes con insuficiencia renal o hepática aguda. La presencia de insuficiencia renal o hepática no requiere un cambio en la dosificación.
Género:
No hay ninguna diferencia farmacocinética aparente entre hombres y mujeres.
Relación concentración-efecto:
Concentraciones séricas en estado estable de 1 a 2 mg/L de etanercept se asocian con un efecto óptimo, y son obtenidas con dosis de 25 mg dos veces por semana. En un estudio cruzado, abierto, de dosis única y dos tratamientos, en 28 voluntarios sanos, se encontró que ENBREL®, administrado como una única inyección de 50 mg/mL, era bioequivalente a dos inyecciones simultáneas de 25 mg/mL.
Anticuerpos para ENBREL®:
Se detectaron anticuerpos para etanercept en el suero de algunos sujetos tratados con etanercept. Estos anticuerpos eran todos no neutralizantes y generalmente transitorios. No parece que exista correlación entre el desarrollo de anticuerpos y la respuesta clínica o los efectos adversos.
En sujetos tratados con dosis aprobadas de etanercept en los ensayos clínicos de hasta 12 meses, las tasas acumulativas de anticuerpos anti-etanercept fueron de aproximadamente el 6% de los sujetos con artritis reumatoide, el 7.5% de los sujetos con artritis psoriásica, el 2% de los sujetos con espondilitis anquilosante, el 7% de los sujetos con psoriasis, 9.7% de los sujetos con psoriasis pediátrica y el 4.8% de los sujetos con artritis idiopática juvenil.
La proporción de sujetos que desarrollaron anticuerpos para etanercept en ensayos a largo plazo (de hasta 3.5 años) aumentó con el tiempo, según lo esperado. Sin embargo, debido a su naturaleza transitoria, la incidencia de anticuerpos detectados en cada punto de evaluación fue normalmente inferior al 7% en sujetos con artritis reumatoide y sujetos con psoriasis.
En un ensayo de psoriasis a largo plazo en el que los pacientes recibieron 50 mg dos veces por semana durante 96 semanas, la incidencia de anticuerpos observada en cada punto de evaluación fue de hasta aproximadamente el 9%.
CONTRAINDICACIONES:
Hipersensibilidad a etanercept o a alguno de los excipientes de la fórmula.
Sepsis o riesgo de sepsis (Ver secciones Precauciones generales y Reacciones secundarias y adversas).
El tratamiento con ENBREL® no deberá iniciarse en pacientes con infecciones activas serias, incluyendo infecciones crónicas o localizadas.
RESTRICCIONES DE USO DURANTE EL EMBARAZO Y LA LACTANCIA:
Embarazo:
Los efectos de etanercept en los resultados del embarazo se han investigado en dos estudios de cohorte observacionales. Un registro de embarazos comparó las tasas de defectos congénitos mayores en bebés nacidos vivos de madres con enfermedades reumáticas o psoriasis expuestas a ENBREL® en el primer trimestre ( n = 319) versus aquellas no expuestas a ENBREL® durante el embarazo (n = 144). El cociente de posibilidades ajustado con todo incluido para los defectos de nacimiento mayores fue 2.77 (IC del 95%: 1.04-7.35) y cuando se eliminaron los trastornos cromosómicos y genéticos conocidos fue 2.49 (IC del 95%: 0.92-6.68). Los hallazgos mostraron que no hay un aumento en la tasa de malformaciones menores, ni un patrón de malformaciones menores o mayores. Además, no hubo un aumento en las tasas de deficiencias en el crecimiento postnatal o intrauterino o de desarrollo posnatal tardío.
En un segundo estudio observacional de registro multinacional que comparo el riesgo de resultados adversos de embarazos en mujeres expuestas a etanercept (n = 522) a aquellas expuestas a fármacos no biológicos (n = 3508), no se observó un aumento en el riesgo de defectos congénitos mayores (cociente de probabilidades ajustado 0.96, IC del 95%: 0.58-1.60). Este estudio también mostró que no hubo un aumento de riesgos de defectos congénitos menores, nacimiento prematuro, mortinato o infecciones en el primer año de vida de los neonatos nacidos de mujeres expuestas a etanercept durante el embarazo. ENBREL® debe utilizarse solamente durante el embarazo si el beneficio potencial para la madre supera los riesgos potenciales para el feto.
No están disponibles los datos preclínicos de la toxicidad peri y postnatal de etanercept, los efectos del etanercept en la fertilidad y en general el rendimiento reproductivo. Se han realizado estudios de toxicidad del desarrollo en ratas y conejos. Las exposiciones sistémicas basadas en las ABC de etanercept en ratas y conejos son 21 y 25 veces mayores que en humanos a la dosis terapéutica usual humana de 50 mg semanalmente y son aproximadamente 10 a 13 veces mayores que en humanos a una dosis humana máxima recomendada de etanercept de 50 mg dos veces a la semana (para psoriasis). No se observó evidencia de daño al feto en ratas o conejos o ratas neonatas debido al etanercept. Los estudios de reproducción animal no siempre son predictivos de la respuesta humana.
Etanercept atraviesa la placenta y se ha detectado en el suero de lactantes nacidos de pacientes de sexo femenino tratadas con ENBREL® durante el embarazo. Se desconoce el impacto clínico de esto, sin embargo, los lactantes pueden tener un mayor riesgo de infección. Generalmente no se recomienda la administración de vacunas vivas a lactantes por 16 semanas después de la última dosis de ENBREL® administrada a la madre.
Lactancia:
En ratas lactantes, después de la administración subcutánea, etanercept se excretó en la leche y se detectó en el suero de las crías. La información limitada de la literatura publicada indica que se han detectado niveles bajos de etanercept en la leche humana. Se podría considerar el uso de etanercept durante la lactancia teniendo en cuenta el beneficio de la lactancia para el niño y el beneficio del tratamiento para la mujer.
Si bien se espera que la exposición sistémica en el lactante sea baja porque el etanercept se degrada en gran medida en el tracto gastrointestinal, se dispone de datos limitados sobre la exposición sistémica en el lactante. Por lo tanto, la administración de vacunas vivas (p. ej., BCG) a un lactante amamantado cuando la madre está recibiendo etanercept podría considerarse 16 semanas después de interrumpir la lactancia (o antes si los niveles séricos de etanercept del lactante son indetectables).
REACCIONES SECUNDARIAS Y ADVERSAS:
Pacientes adultos:
La proporción de pacientes que interrumpieron el tratamiento debido a reacciones adversas en estudios clínicos controlados en pacientes con artritis reumatoide fue la misma tanto en el grupo de tratamiento de ENBREL® como en el de placebo.
Reacciones en el sitio de inyección:
Los pacientes en estudios clínicos controlados tratados con ENBREL® tuvieron una incidencia significativamente más alta de reacciones en el sitio de la inyección (eritema y/o comezón, dolor, o inflamación) comparados con los pacientes tratados con placebo. La frecuencia de reacciones en el sitio de la inyección fue más alta en el primer mes y subsecuentemente la frecuencia disminuyó. En ensayos clínicos, estas reacciones fueron generalmente transitorias con una duración media de 4 días. Algunos de los pacientes que experimentaron reacciones en el sitio de la inyección también experimentaron reacciones en sitios previos de inyección.
En la experiencia poscomercialización también se ha observado sangrado y equimosis en el sitio de inyección concomitantes con el tratamiento con ENBREL®.
Infecciones:
Se han reportado infecciones serias y mortales; los patógenos reportados incluyen bacterias, micobacterias (incluyendo tuberculosis), virus y hongos. Se han reportado también infecciones oportunistas incluyendo infecciones fúngicas invasivas, infecciones parasitarias (incluyendo protozoarios), virales (incluyendo herpes zóster), bacterianas (incluyendo Listeria y Legionella) e infecciones por micobacterias atípicas (Ver Sección Precauciones Generales). Las infecciones micóticas invasivas más comúnmente reportadas incluyeron Candida, Pneumocystis, Aspergillus, e Histoplasma.
En ensayos controlados en pacientes con artritis reumatoide, las tasas de infecciones serias (fatales, que ponen en riesgo la vida, o que requirieron hospitalización o antibióticos intravenosos) y no serias reportadas fueron similares para ENBREL® y placebo cuando se ajustó para la duración de la exposición. Las infecciones respiratorias de vías aéreas superiores fueron las infecciones no serias más comúnmente reportadas.
Los datos de un ensayo clínico en pacientes con sepsis establecida sugieren que el tratamiento con ENBREL® puede aumentar la mortalidad en estos pacientes.
Neoplasias malignas y trastornos linfoproliferativos:
Se han recibido reportes de neoplasias malignas que afectan varios sitios durante el periodo de poscomercialización.
Se han reportado casos de neoplasias malignas en un ensayos clínico de pacientes que estaban siendo tratados por granulomatosis de Wegener (ver sección Precauciones generales).
Enfermedad pulmonar Intersticial:
En ensayos clínicos controlados de etanercept para todas las indicaciones, la frecuencia (proporción de incidencia) de enfermedad pulmonar intersticial en pacientes que recibían etanercept sin metotrexato concomitante fue de 0.06% (frecuencia rara). En los ensayos clínicos controlados que permitieron el tratamiento concomitante con etanercept y metotrexato, la frecuencia (proporción de incidencia) de enfermedad pulmonar intersticial fue de 0.47% (frecuencia poco común). Ha habido informes posteriores a la comercialización de enfermedad pulmonar intersticial (incluida neumonitis y fibrosis pulmonar), algunos de los cuales han tenido desenlaces fatales.
Aumento de las enzimas hepáticas:
En los periodos doble ciego de los ensayos clínicos controlados de etanercept para todas las indicaciones, la frecuencia (proporción de incidencia) de eventos adversos de aumento de las enzimas hepáticas en pacientes que recibían etanercept sin metotrexato concomitante, fue del 0.54% (frecuencia poco común). En los periodos de doble ciego de los ensayos clínicos controlados que permitieron el tratamiento concomitante con etanercept y metotrexato, la frecuencia (proporción de incidencia) de eventos adversos de aumento de las enzimas hepáticas fue del 4.18% (frecuencia común).
Hepatitis autoinmune:
En ensayos clínicos controlados de etanercept para todas las indicaciones, la frecuencia (proporción de incidencia) de hepatitis autoinmune en pacientes que recibían etanercept sin metotrexato concomitante fue de 0.02% (frecuencia rara). En los ensayos clínicos controlados que permitieron el tratamiento concomitante con etanercept y metotrexato, la frecuencia (proporción de incidencia) de hepatitis autoinmune fue de 0.24% (frecuencia poco común).
Autoanticuerpos:
En ensayos controlados, el porcentaje de pacientes que desarrollaron nuevos anticuerpos antinucleares (AAN) positivos (≥ 1:40), nuevos anticuerpos anti-ADN de doble hélice positivos, y nuevos anticuerpos anticardiolipina, fue más alto comparado con en el grupo tratado con placebo. Se desconoce el impacto del tratamiento a largo plazo con ENBREL® sobre el desarrollo de enfermedades autoinmunes.
Se han descrito informes raros en pacientes, incluyendo aquellos con AR con factor reumatoide positivo, que han desarrollado autoanticuerpos adicionales junto con un síndrome similar al lupus o erupciones cutáneas compatibles con lupus cutáneo subagudo o lupus discoide según la presentación clínica y la biopsia.
Otras reacciones adversas:
La siguiente tabla de efectos no deseados sospechados se basa en ensayos clínicos y/o tasas de reportes espontáneos poscomercialización:
Reacciones adversas al medicamento (RAM) por clasificación por órganos y sistemas (SOC) y categoría de frecuencia del Consejo de organizaciones internacionales de las ciencias médicas (CIOMS) enumeradas en orden decreciente de gravedad médica o importancia clínica dentro de cada categoría de frecuencia y SOC
|
Clasificación por |
Muy común |
Común |
Poco común |
Raro |
Muy raro |
Frecuencia desconocida |
|---|---|---|---|---|---|---|
|
Infecciones e infestaciones |
Infección (incluida infección del tracto respiratorio superior, bronquitis, cistitis, infección cutánea) |
Infecciones serias (incluida neumonía, celulitis, artritis bacteriana, sepsis e infección parasitaria) |
Tuberculosis, infección oportunista (incluidas infecciones invasivas fúngicas, bacterianas, micobacterianas atípicas, infecciones virales y Legionella) (ver sección Precauciones generales) |
Reactivación de la Hepatitis B*, Listeria* |
||
|
Neoplasias benignas, malignas y no especificadas (incluidos quistes y pólipos) |
Cáncer de piel no melanoma (ver sección Precauciones generales) |
Melanoma maligno (ver sección Precauciones generales), linfoma*, leucemia* |
Carcinoma de células de Merkel* (ver sección Precauciones generales) |
|||
|
Trastornos de la sangre y del sistema linfático |
Trombocitopenia, anemia, leucopenia, neutropenia, |
Pancitopenia (ver sección Precauciones generales) |
Anemia aplásica* (ver sección Precauciones generales) |
Histiocitosis hematofágica (síndrome de activación de macrófagos)* |
||
|
Trastornos del sistema inmunitario |
Reacciones alérgicas (ver sección Trastornos de la piel y del tejido subcutáneo, más adelante), formación de autoanticuerpos |
Vasculitis (incluida vasculitis ANCA positivas) |
Reacciones alérgicas/anafilácticas serias (incluidos broncoespasmos), sarcoidosis |
|||
|
Trastornos del sistema nervioso |
Dolor de cabeza* |
Eventos desmielinizantes del SNC, incluida esclerosis múltiple y afecciones desmielinizantes localizadas tales como neuritis óptica y mielitis transversa (ver sección Precauciones generales), eventos desmielinizantes periféricos, incluido el síndrome de Guillain-Barré, polineuropatía desmielinizante inflamatoria crónica, polineuropatía desmielinizante, y neuropatía motora multifocal* (ver sección Precauciones generales), convulsiones |
||||
|
Trastornos del ojo |
Uveítis, escleritis |
|||||
|
Trastornos cardiacos |
Empeoramiento de insuficiencia cardiaca congestiva |
Insuficiencia cardiaca congestiva de nueva aparición |
||||
|
Trastornos respiratorios, torácicos, y mediastínicos |
Enfermedad pulmonar intersticial (incluida fibrosis pulmonar y neumonitis) |
|||||
|
Trastornos gastrointestinales |
Enfermedad inflamatoria intestinal* |
|||||
|
Trastornos hepatobiliares |
Aumento de las enzimas hepáticas (ver sección Aumento de enzimas hepáticas, más arriba) |
Hepatitis autoinmune |
||||
|
Trastornos de la piel y del tejido subcutáneo |
Prurito, erupción, |
Angioedema, psoriasis (nuevo comienzo o exacerbación, incluidos todos los subtipos), urticaria, erupción psoriasiforme* |
Síndrome de Stevens-Johnson*, vasculitis cutánea (incluida vasculitis por hipersensibilidad), eritema multiforme* |
Necrólisis epidérmica tóxica* |
||
|
Trastornos musculoesqueléticos y del tejido conectivo |
Lupus eritematoso cutáneo*, lupus eritematoso cutáneo subagudo*, síndrome similar al lupus |
|||||
|
Trastornos renales y urinarios |
Glomerulonefritis* |
|||||
|
Trastornos generales y afecciones en el lugar de la administración |
Reacciones en el lugar de la inyección (incluido sangrado, hematoma, eritema, comezón, dolor e hinchazón) |
Pirexia |
* RAM identificados posterior a la comercialización.
Población pediátrica:
En general, los eventos adversos en pacientes pediátricos fueron similares en frecuencia y tipo a los observados en pacientes adultos.
Efectos no deseados en pacientes pediátricos con artritis idiopática juvenil:
La infección fue el evento adverso más común reportado en pacientes pediátricos a quienes se les administró ENBREL® y ocurrió con una incidencia similar a la del placebo. Los tipos de infecciones reportadas en pacientes con artritis idiopática juvenil fueron por lo general leves y consistentes con los observados comúnmente en poblaciones pediátricas de pacientes ambulatorios.
En ensayos clínicos en pacientes con artritis idiopática juvenil tratados con ENBREL® se reportaron dos casos de infección por varicela con signos y síntomas sugestivos de meningitis aséptica.
Hubo cuatro reportes de síndrome de activación de macrófagos en ensayos clínicos de artritis idiopática juvenil.
Efectos no deseados en pacientes pediátricos con psoriasis en placas:
En un estudio de 48 semanas de 211 niños de 4 a 17 años con psoriasis en placas pediátrica, los eventos adversos reportados fueron similares a aquellos vistos en estudios previos en adultos con psoriasis en placas.
PRECAUCIONES EN RELACIÓN CON EFECTOS DE CARCINOGÉNESIS, MUTAGÉNESIS, TERATOGÉNESIS Y SOBRE LA FERTILIDAD:
Carcinogénesis:
No se han realizado estudios a largo plazo en animales para evaluar el potencial carcinógeno de etanercept. Los estudios a largo plazo en animales no son posibles debido a que estos pueden desarrollar anticuerpos contra etanercept, el cual es una proteína humana.
Mutagénesis:
Se realizaron estudios de mutagénesis in vitro e in vivo, y no se observó ninguna evidencia de actividad mutagénica.
Deterioro de la fertilidad:
No se han realizado estudios a largo plazo en animales para evaluar el efecto de etanercept sobre la fertilidad.
INTERACCIONES MEDICAMENTOSAS Y DE OTRO GÉNERO:
Tratamiento concurrente con anakinra:
Se observó que los pacientes tratados con ENBREL® y anakinra tenían una mayor tasa de infecciones severas cuando se compararon con pacientes que fueron tratados con ENBREL® solo (datos históricos). Además, en un ensayo doble ciego controlado con placebo en pacientes recibiendo de base metotrexato, se observó en los pacientes tratados con ENBREL® y anakinra que tenían una mayor tasa de infecciones serias y neutropenia que los pacientes tratados con ENBREL® solo (Ver sección Precauciones generales).
Tratamiento concurrente con abatacept:
En estudios clínicos, la administración concurrente de abatacept y la terapia con ENBREL® resultó un incremento en la incidencia de eventos adversos serios. Esta combinación no ha demostrado un mayor beneficio clínico; no se recomienda tal uso (ver sección Precauciones generales).
Tratamiento concurrente con sulfasalazina:
En un estudio clínico de pacientes que estaban recibiendo dosis establecidas de sulfasalazina, a los que se les añadió ENBREL®, los pacientes en el grupo de combinación experimentaron una disminución estadísticamente significativa en la cuenta media de células blancas en comparación con los grupos tratados con ENBREL® o sulfasalazina sola. La significancia clínica de esta interacción es desconocida.
No interacciones:
No se ha observado ninguna interacción cuando ENBREL® fue administrado con glucocorticoides, salicilatos (excepto sulfasalazina), fármacos antinflamatorios no esteroideos (AINE), analgésicos, o metotrexato en ensayos clínicos con pacientes adultos con artritis reumatoide.
El metotrexato no tiene ningún efecto sobre la farmacocinética de etanercept.
No se observaron interacciones medicamentosas clínicamente significativas de farmacocinética en estudios con digoxina y warfarina.
Compatibilidad e incompatibilidades:
En ausencia de estudios de incompatibilidad, este medicamento no debe ser mezclado con otros medicamentos.
ALTERACIONES EN LOS RESULTADOS DE PRUEBAS DE LABORATORIO:
No se han reportado hasta la fecha.
ENBREL® puede causar alteraciones hematológicas como anemia aplásica, leucopenia, trombocitopenia y neutropenia, así como alteraciones de las enzimas hepáticas. Ver tabla de eventos adversos.
PRECAUCIONES GENERALES:
Infecciones:
Se han reportado infecciones serias, incluyendo sepsis y tuberculosis (TB), con el uso de ENBREL® (Ver sección Reacciones secundarias y adversas). Algunas de estas infecciones han sido fatales. Estas infecciones son debidas a bacterias, micobacterias, hongos, virus y parásitos (incluyendo protozoarios). También se han reportado infecciones oportunistas (incluyendo listeriosis y legionelosis). Los pacientes que desarrollen una nueva infección mientras estén bajo tratamiento con etanercept deberán ser monitoreados de cerca. La administración de ENBREL® deberá suspenderse si el paciente desarrolla una infección seria. Deberá tenerse precaución al considerar el uso de ENBREL® en pacientes con antecedentes de infecciones recurrentes o crónicas o con padecimientos subyacentes que puedan predisponer a los pacientes a infecciones (Ver secciones Contraindicaciones y Reacciones secundarias y adveras).
Antes, durante y después del tratamiento con ENBREL® los pacientes deben de ser evaluados para infecciones, tomando en consideración que la vida media de eliminación del etanercept es de 80 horas (desviación estándar de 28 horas; rango de 7 a 300 horas).
Se han reportado infecciones oportunistas en pacientes que reciben ENBREL®, incluyendo infecciones fúngicas invasivas. En algunos casos, no se reconocen las infecciones por hongos u otras oportunistas, y esto ha dado por resultado retrasos en el tratamiento adecuado, algunas veces dando por resultado la muerte. En muchos de los reportes, los pacientes también han recibido medicamentos concomitantes incluyendo inmunosupresores. En la evaluación de los pacientes por infecciones, los médicos deberán considerar el riesgo del paciente para infecciones oportunistas relevantes (p. ej., exposición a micosis endémicas).
Tuberculosis (TB):
Se ha observado tuberculosis (incluyendo la presentación diseminada o extrapulmonar) en pacientes que reciben agentes bloqueadores de FNT, incluyendo ENBREL®. La tuberculosis puede ser debida a la reactivación de una infección latente de TB o a una nueva infección.
Antes de iniciar la terapia con ENBREL®, cualquier paciente con riesgo incrementado de TB deberá de ser evaluado para una infección activa o latente. Deberá de ser iniciada una profilaxis de infección latente de TB antes de la terapia con ENBREL®. Algunos pacientes que dieron prueba negativa para TB latente antes de recibir ENBREL® desarrollaron TB activa. Los médicos deben monitorear a los pacientes que reciben ENBREL® en búsqueda de signos y síntomas de TB activa, incluyendo pacientes que dieron prueba negativa para infección de TB latente. Deberán de consultarse las normas nacionales aplicables. Los pacientes con AR parecen tener una tasa incrementada de infección por TB.
Reactivación del virus de la hepatitis B:
Se ha reportado la reactivación de la hepatitis B en pacientes previamente infectados con el virus de la hepatitis B (VHB) y que han recibido concomitantemente agentes anti-FNT incluyendo ENBREL®. La mayoría de estos reportes han ocurrido en pacientes recibiendo concomitantemente otras medicaciones que suprimen el sistema inmune, las cuales podrían también contribuir a la reactivación de la hepatitis B. Los pacientes en riesgo de infección por VHB deberán ser evaluados en busca de una evidencia anterior de infección por VHB antes de iniciar la terapia anti-FNT.
Deberá de tenerse precaución cuando se administra ENBREL® a pacientes previamente infectados con VHB. Deberá monitorearse a estos pacientes para signos y síntomas de infección activa de VHB.
Empeoramiento de la hepatitis C:
Ha habido reportes de empeoramiento de hepatitis C en pacientes que reciben ENBREL®, a pesar de que no se ha establecido una relación causal con ENBREL®.
Tratamiento concurrente con anakinra:
La administración simultánea de ENBREL® y anakinra se ha asociado con un incremento en el riesgo de infecciones severas y neutropenia. Esta combinación no ha demostrado un mayor beneficio clínico; no se recomienda tal uso (Ver sección Interacciones medicamentosas y de otro género).
Tratamiento concurrente con abatacept:
En estudios clínicos, la administración simultánea de abatacept y la terapia con ENBREL® dieron como resultado un incremento en la incidencia de eventos adversos serios. Esta combinación no ha demostrado un mayor beneficio clínico; no se recomienda tal uso (Ver sección Interacciones medicamentosas y de otro género).
Granulomatosis de Wegener:
En un estudio controlado con placebo de 180 pacientes con granulomatosis de Wegener, la adición de ENBREL® al tratamiento convencional (incluyendo ciclofosfamida y esteroides de dosis altas) no fue más eficaz que el tratamiento convencional solo. El grupo de pacientes que recibieron ENBREL® experimentaron más neoplasias malignas no cutáneas de diversos tipos que el grupo de pacientes que recibió sólo el tratamiento convencional. No se recomienda el uso de ENBREL® para el tratamiento de la granulomatosis de Wegener.
Hepatitis alcohólica:
En un estudio de 48 pacientes hospitalizados tratados con ENBREL® o placebo para hepatitis alcohólica de moderada a severa [media del Modelo de Enfermedad Hepática en estado-terminal (MELD por sus siglas en inglés ) puntuación = 25], ENBREL® no fue eficaz y la tasa de mortalidad en pacientes tratados con ENBREL® fue significativamente mayor después de seis meses. Las infecciones fueron también mayores en el grupo tratado con ENBREL®. No se recomienda el uso del ENBREL® en pacientes para el tratamiento de hepatitis alcohólica. Los médicos deberán tener precaución cuando utilicen ENBREL® en pacientes que también tienen hepatitis alcohólica de moderada a severa.
Reacciones alérgicas:
Se han reportado reacciones alérgicas asociadas con la administración de ENBREL®. Si ocurre cualquier reacción alérgica seria o anafiláctica, interrúmpase la administración de ENBREL® inmediatamente (Ver sección Reacciones secundarias y adversas).
Contenido de látex:
La tapa de caucho de la jeringa con diluyente, la cubierta de la aguja para la jeringa prellenada, la tapa de la aguja de la pluma precargada y el cartucho dispensador de dosis contienen látex (caucho natural seco). Los pacientes o cuidadores deberán contactar a su médico antes de utilizar ENBREL® si la tapa de caucho de la jeringa del diluyente, la cubierta de la aguja para la jeringa prellenada, la tapa de la aguja de la pluma precargada o el cartucho dispensador de dosis serán manipulados por o si ENBREL® será administrado a alguien con una hipersensibilidad conocida o posible (alergia) al látex.
Inmunosupresión:
Las terapias con anti-FNT, incluyendo ENBREL®, pueden afectar las defensas del huésped en contra de infecciones y neoplasias malignas, dado que el FNT media la inflamación y regula las respuestas inmunes celulares.
Neoplasias malignas y trastornos linfoproliferativos:
Neoplasias malignas sólidas y hematopoyéticas (excluyendo cáncer de piel):
Se han recibido reportes en el periodo posterior a la comercialización de neoplasias malignas que afectan varios sitios. En las porciones controladas de estudios clínicos de antagonistas del FNT, se han observado más casos de linfoma entre pacientes recibiendo antagonistas del FNT comparados con pacientes control. Sin embargo, la frecuencia fue rara, y el periodo de seguimiento para los pacientes en placebo fue menor que para los pacientes que recibieron terapia de antagonistas del FNT. Se han reportado casos de leucemia en pacientes tratados con antagonistas del FNT. Hay un incremento en el riesgo de fondo de linfoma y leucemia en los pacientes con artritis reumatoide de larga duración con enfermedad inflamatoria, muy activa, lo que complica la estimación del riesgo. Los análisis Post hoc de los estudios clínicos de artritis reumatoide con ENBREL® no han confirmado o excluido un incremento en el riesgo de neoplasias malignas.
Se han reportado neoplasias malignas (particularmente linfomas Hodgkin y no Hodgkin), algunas mortales entre niños y adolescentes que recibieron tratamiento con antagonistas del FNT, incluyendo ENBREL®. Muchos de los pacientes estuvieron recibiendo inmunosupresores concomitantemente.
Basado en el conocimiento actual, no se puede excluir un posible riesgo de desarrollar linfomas u otras neoplasias malignas solidas o hematopoyéticas en los pacientes tratados con antagonistas del FNT.
Cánceres de piel:
Se ha reportado melanoma y cáncer de piel no melanoma (CPNM) en pacientes tratados con antagonistas del FNT incluyendo ENBREL®. Se han notificado con muy poca frecuencia casos poscomercialización de carcinoma de células de Merkel en pacientes tratados con ENBREL®. Se recomienda un examen de piel periódico para todos los pacientes que tienen un mayor riesgo de sufrir cáncer de piel.
Combinando los resultados de las porciones controladas de estudios clínicos de ENBREL®, se observaron más casos de CPNM en pacientes recibiendo ENBREL® comparado con los pacientes control, particularmente en pacientes con psoriasis.
Reacciones hematológicas:
Se han reportado casos raros de pancitopenia y casos muy raros de anemia aplásica, algunos con un resultado fatal en pacientes tratados con ENBREL®. Deberá tenerse precaución en pacientes que sean tratados con ENBREL® y que tengan historia previa de discrasias sanguíneas. Todos los pacientes deberán recibir instrucciones de que si desarrollan signos y síntomas que sugieran discrasias sanguíneas o infecciones (p. ej., fiebre persistente, dolor de garganta, equimosis, sangrado, palidez) mientras reciben ENBREL®, deberán buscar atención médica inmediata. Tales pacientes deberán ser evaluados urgentemente, incluyendo una cuenta sanguínea completa; etanercept deberá ser suspendido si se confirma una discrasia sanguínea.
Formación de autoanticuerpos:
El tratamiento con ENBREL® puede asociarse con la formación de anticuerpos autoinmunes (Ver sección Reacciones secundarias y adversas).
Vacunaciones:
En un estudio clínico doble ciego, controlado con placebo y aleatorizado en pacientes con artritis psoriásica, 184 pacientes también recibieron una vacuna neumocócica polisacárida multivalente en la semana 4. En este estudio la mayor parte de los pacientes con artritis psoriásica que recibieron ENBREL® fueron capaces de formar respuestas inmunes efectivas de células B a la vacuna neumocócica polisacárida, pero los títulos en los agregados fueron moderadamente menores y pocos pacientes tuvieron aumentos de dos veces comparados con los pacientes que no recibieron ENBREL®. La significancia clínica de este hallazgo es desconocida. No se deben administrar vacunas vivas concurrentemente con ENBREL®. Si es posible, actualice las vacunas de los pacientes pediátricos de acuerdo con las guías locales vigentes antes de comenzar el tratamiento con ENBREL®.
Trastornos neurológicos:
Aunque no se han realizado estudios clínicos que evalúen el tratamiento con ENBREL® en pacientes con esclerosis múltiple, los ensayos clínicos con otros antagonistas del FNT en pacientes con esclerosis múltiple han demostrado un aumento en la actividad de la enfermedad. Ha habido reportes raros de trastornos de desmielinización del sistema nervioso central (SNC) en pacientes tratados con ENBREL® (Ver sección Reacciones secundarias y adversas). Adicionalmente, se han reportado casos raros de polineuropatías desmielinizantes periféricas (incluyendo el síndrome de Guillain-Barré). Se recomienda una evaluación cuidadosa del riesgo /beneficio, incluyendo una evaluación neurológica, cuando se prescriba la terapia con ENBREL® a pacientes con enfermedad desmielinizante preexistente o de inicio reciente, o aquellos que se considera que tienen un riesgo incrementado de desarrollar una enfermedad desmielinizante.
Insuficiencia cardiaca congestiva:
Ha habido informes posteriores a la comercialización de empeoramiento de la insuficiencia cardíaca congestiva (ICC), con y sin factores precipitantes identificables, en pacientes que toman ENBREL®. También ha habido informes raros (< 0.1%) de ICC de nueva aparición, incluyendo la ICC en pacientes sin enfermedad cardiovascular preexistente conocida. Algunos de estos pacientes han sido menores de 50 años. Dos grandes ensayos clínicos que evaluaron el uso de ENBREL® en el tratamiento de ICC fueron suspendidos en forma temprana debido a la falta de eficacia. Aunque no es concluyente, los datos obtenidos de uno de los ensayos sugieren una posible tendencia respecto al agravamiento de la ICC en aquellos pacientes asignados para el tratamiento con ENBREL®. Además, un ensayo clínico evaluando el uso de infliximab (un anticuerpo monoclonal que se une al FNT alfa) en el tratamiento de ICC fue terminado tempranamente debido a un incremento en la mortalidad entre los pacientes tratados con infliximab. Los médicos deberán usar con precaución ENBREL® en pacientes que tengan también ICC.
Hipoglucemia en pacientes tratados por diabetes:
Ha habido reportes de hipoglucemia después del inicio de ENBREL® en pacientes recibiendo medicamentos para la diabetes, necesitando una reducción en la medicación antidiabética en algunos de estos pacientes.
Efectos en la capacidad para conducir y operar maquinaria:
No se han realizado estudios sobre la capacidad para conducir y usar máquinas.
DOSIS Y VÍA DE ADMINISTRACIÓN:
Uso en adultos:
Artritis reumatoide, artritis psoriásica y espondilitis anquilosante activa:
Pacientes de 18 años o mayores: 50 mg de ENBREL® a la semana administrados ya sea una vez a la semana (como inyección subcutánea utilizando una jeringa de 50 mg o como dos inyecciones de 25 mg administradas aproximadamente al mismo tiempo) o 25 mg de ENBREL® dos veces a la semana (separados por 72 a 96 horas) como inyección subcutánea.
La administración de: Metotrexato, glucocorticoides, salicilatos, fármacos antiinflamatorios no esteroideos (AINE), o analgésicos pueden continuarse durante el tratamiento con ENBREL® en adultos.
El uso de 25 mg administrados una vez por semana da una respuesta más lenta y puede ser menos efectivo.
Psoriasis en placas:
La dosis de ENBREL® es 50 mg una vez a la semana (como una inyección subcutánea utilizando una jeringa de 50 mg o como dos inyecciones de 25 mg administrados aproximadamente al mismo tiempo) o 25 mg administrado dos veces a la semana (con una separación de 72 a 96 horas) como una inyección subcutánea. Se pueden alcanzar respuestas más altas con el tratamiento inicial con una dosis de 50 mg dos veces a la semana durante un máximo de 12 semanas, seguida, si es necesario, por una dosis de 50 mg una vez a la semana o 25 mg dos veces a la semana.
Los pacientes adultos pueden ser tratados intermitente o continuamente, basados en el juicio del médico y de las necesidades individuales del paciente (ver sección Farmacocinética y farmacodinamia). El tratamiento deberá ser discontinuado en pacientes que no muestran respuesta después de 12 semanas. Con el uso intermitente, los ciclos de tratamiento subsecuentes al ciclo inicial deberán usar una dosis de 50 mg una vez a la semana o 25 mg dos veces a la semana.
Pacientes de edad avanzada (≥ 65 años):
No se requiere de ajuste en la dosis.
Insuficiencia renal:
No se requiere de ajuste en la dosis.
Insuficiencia hepática:
No se requiere de ajuste en la dosis.
Población pediátrica:
La dosis de ENBREL® está basada en el peso corporal de los pacientes pediátricos. Los pacientes que pesan menos de 62.5 kg deben ser dosificados adecuadamente en base a mg/kg usando ENBREL® 25 mg/mL polvo liofilizado y diluyente para la inyección (ver posteriormente las indicaciones específicas para dosificación). Los pacientes que pesan 62.5 kg o más pueden ser dosificados usando una jeringa prellenada, pluma precargada o cartucho dispensador de dosis con dosis fija.
Artritis idiopática juvenil (2 años y mayores):
Niños (≥ 2 a < 18 años): 0.4 mg/kg (hasta un máximo de 25 mg por dosis) dos veces a la semana (separados por 72 a 96 horas), o 0.8 mg/kg (hasta un máximo de 50 mg por dosis) una vez a la semana.
La administración de: glucocorticoides, fármacos antinflamatorios no esteroideos (AINE), o analgésicos pueden continuarse durante el tratamiento con ENBREL® en niños.
ENBREL® no ha sido estudiado en niños < 2 años.
Para obtener información de seguridad específica pediátrica relativa a neoplasias malignas y vacunas, ver secciones precauciones generales y reacciones secundarias y adversas.
Psoriasis en placas pediátrica (6 años y mayores):
Niños (≥ 6 a < 18 años): 0.8 mg/kg (hasta un máximo de 50 mg por dosis) una vez a la semana hasta por 24 semanas. El tratamiento debe ser descontinuado en pacientes que no muestran respuesta después de las 12 semanas.
Si se indica un retratamiento con ENBREL®, se deberá de seguir la guía de arriba en la duración del tratamiento. La dosis deberá ser 0.8 mg/kg (hasta un máximo de 50 mg por dosis) una vez a la semana.
Método de administración:
Aplique ENBREL® como inyecciones subcutáneas en el muslo, abdomen, o parte superior del brazo. Aplique cada nueva inyección a cuando menos a 3 cm de un sitio previo. NO se inyecte en áreas en las que la piel esté sensible, tenga hematomas, esté enrojecida o esté dura.
Los pacientes o cuidadores que deban administrar(se) ENBREL® deben recibir instrucciones sobre las técnicas de inyección. La primera inyección deberá realizarse bajo la supervisión de un médico calificado si etanercept va a ser administrado por un cuidador o por el mismo paciente.
Polvo y diluyente para preparar solución inyectable:
Los pacientes o cuidadores que administrarán ENBREL® deberán recibir instrucciones acerca del mezclado del polvo liofilizado con el líquido. ENBREL® reconstituido es una solución líquida incolora ligeramente amarilla o marrón pálido y transparente a ligeramente opalescente.
Solución inyectable en jeringa prellenada:
Antes de aplicar, la jeringa prellenada de un solo uso de ENBREL® deberá permitírsele alcanzar la temperatura ambiente (aproximadamente 15 a 30 minutos). La cubierta de la aguja no deberá ser retirada mientras se permite a la jeringa prellenada alcanzar la temperatura ambiente. La solución debe ser de clara a opalescente, incolora a amarilla o marrón pálido y el líquido puede contener trazas de partículas amorfas translúcidas a blancas.
Solución inyectable en pluma precargada:
Antes de aplicar, la pluma precargada de un solo uso de ENBREL® deberá permitírsele alcanzar la temperatura ambiente (aproximadamente 15 a 30 minutos). La cubierta de la aguja no deberá ser retirada mientras se permite a la pluma precargada alcanzar la temperatura ambiente. Al mirar a través de la ventana de inspección, la solución deberá de ser de clara a opalescente, incolora a amarilla o marrón pálido y el líquido puede contener trazas de partículas amorfas translúcidas a blancas.
Solución inyectable en cartucho dispensador de dosis:
El cartucho dispensador de dosis debe administrarse mediante el dispositivo multiusos para un solo paciente denominado SMARTCLIC. Antes de la inyección, se debe permitir que el cartucho dispensador de dosis de un solo uso de ENBREL® alcance la temperatura ambiente (aproximadamente de 15 a 30 minutos). No se debe quitar la cubierta de la aguja mientras se permite que el cartucho dispensador de dosis alcance la temperatura ambiente. Al mirar a través de la ventana de inspección, la solución deberá de ser de clara a ligeramente opalescente, de incolora a amarillo pálido o marrón pálido, y puede contener pequeñas partículas de proteína blancas o casi transparentes.
El contenido total (0.5 mL para la dosis de 25 mg o 1 ml para la dosis de 50 mg) del cartucho dispensador de dosis debe administrarse utilizando SMARTCLIC solo para inyección subcutánea. Después de un entrenamiento adecuado sobre la técnica de administración, los usuarios podrán administrar una dosis utilizando el dispositivo SMARTCLIC con el cartucho dispensador de dosis de un solo uso si su médico determina que es apropiado y con el seguimiento médico que sea necesario.
Dosis olvidadas:
Si se olvida una dosis, se debe advertir a los pacientes que se administren la dosis tan pronto como lo recuerden, a menos que la siguiente dosis programada sea al día siguiente, en cuyo caso se debe omitir la dosis olvidada. Los pacientes deben seguir inyectándose el medicamento en su(s) día(s) habitual(es). Si un paciente no recuerda hasta el día en que debe recibir la siguiente inyección, indíquele que no debe inyectarse una dosis doble.
Manejo:
Polvo y diluyente para preparar solución inyectable:
El polvo liofilizado de etanercept se reconstituye asépticamente inyectando 1 mL del diluyente lentamente en el frasco ámpula. Se debe rotar suavemente el contenido para evitar la formación excesiva de espuma. Ocurrirá cierta formación de espuma; esto es normal. No se debe agitar vigorosamente. La disolución usualmente toma menos de 10 minutos.
Jeringa con diluyente:
La tapa de caucho de la jeringa con diluyente contiene látex (hule natural seco). Los pacientes y los cuidadores deberán contactar a su médico antes de utilizar etanercept, si la tapa de caucho de la jeringa con diluyente será manejada por o si el etanercept será administrado a alguien con una hipersensibilidad (alergia) conocida o posible al látex.
Solución inyectable en jeringa prellenada, pluma precargada y cartucho dispensador de dosis:
La cubierta de la aguja de la jeringa prellenada, la tapa de la aguja de la pluma precargada y el cartucho dispensador de dosis contienen látex (caucho natural seco). Los pacientes o cuidadores deberán contactar a su médico antes de utilizar etanercept, si la cubierta de la aguja será manipulada o si etanercept será administrado a alguien con una hipersensibilidad (alergia) conocida o posible al látex.
Los pacientes o cuidadores que deben administrar etanercept deben recibir instrucciones sobre el desecho apropiado de la jeringa y la aguja, y debe advertírseles que no vuelvan a usar estos artículos.
MANIFESTACIONES Y MANEJO DE LA SOBREDOSIFICACIÓN O INGESTA ACCIDENTAL:
La dosis máxima tolerada de etanercept no ha sido establecida en humanos. Dosis intravenosas únicas de hasta 60 mg/m2 han sido administradas a voluntarios sanos en un estudio de endotoxemia sin evidencia de toxicidades limitantes de la dosis. El nivel de dosis más alto evaluado en pacientes con artritis reumatoide ha sido una dosis de carga intravenosa de 32 mg/m2 seguida por dosis subcutáneas de 16 mg/m2 (~25 mg) administradas dos veces por semana.
Etanercept no indujo letalidad o signos notables de toxicidad en ratones o ratas, posterior a una dosis única subcutánea de 2000 mg/kg o una dosis intravenosa única de 1000 mg/kg. El etanercept no provocó toxicidad limitante de la dosis ni toxicidad en órganos blanco en monos Cynomolgus después de la administración subcutánea dos veces por semana durante 4 o 26 semanas consecutivas a una dosis (15 mg/kg) que resultó en una concentración sérica del medicamento basada en el ABC que fue 27 veces mayor que la obtenida en humanos a la dosis humana recomendada de 25 mg.
No se observaron toxicidades limitantes de la dosis durante los ensayos clínicos en pacientes con artritis reumatoide.
No hay ningún antídoto conocido para etanercept. En caso de sobredosis, se recomienda que se vigilen los signos y síntomas de reacciones adversas. Los pacientes que desarrollen reacciones adversas deben recibir tratamiento sintomático adecuado de inmediato.
PRESENTACIONES:
Frasco ámpula con jeringa y adaptador:
Caja de cartón con 4 charolas, cada charola contiene: un frasco ámpula con 25 mg de polvo liofilizado, una jeringa con 1 mL de diluyente, una aguja de acero inoxidable, un adaptador del frasco ámpula y dos almohadillas impregnadas de alcohol isopropílico (al 70%).
Jeringa prellenada:
Caja de cartón con 4 charolas, cada charola contiene: una jeringa prellenada con 0.5 mL (25 mg/0.5 mL) y una almohadilla impregnada con alcohol isopropílico (al 70%).
Caja de cartón con 2 charolas, cada charola contiene: una jeringa prellenada con 1 mL (50 mg/mL) y una almohadilla impregnada con alcohol isopropílico (al 70%).
Pluma precargada MYCLIC:
Caja de cartón con 2 charolas, cada charola contiene: una pluma precargada MYCLIC con 1 mL (50 mg/mL) y una almohadilla impregnada con alcohol isopropílico (al 70%).
Cartucho dispensador de dosis (SMARTCLIC)
|
Dosis |
Concentración |
Presentación |
|
25 mg |
0.5 mL (25 mg/mL) |
Caja de cartón con 4 charolas* |
|
Caja de cartón con 8 charolas* |
||
|
Caja de cartón con 24 charolas* |
||
|
50 mg |
1 mL (50 mg/mL) |
Caja de cartón con 2 charolas* |
|
Caja de cartón con 4 charolas* |
||
|
Caja de cartón con 12 charolas* |
* Cada charola contiene: un cartucho dispensador de dosis y 2 almohadillas impregnadas con alcohol isopropílico (al 70%).
Todas las presentaciones con instructivo anexo.
RECOMENDACIONES SOBRE ALMACENAMIENTO:
No se use el producto después de la fecha de caducidad impresa en la caja y el frasco.
La charola que contenga ENBREL® debe conservarse en refrigeración entre 2 °C y 8 °C. NO SE CONGELE.
Se recomienda que la solución de ENBREL® sea administrada inmediatamente después de su reconstitución.
Si no se utiliza inmediatamente, la solución reconstituida de ENBREL® puede ser refrigerada en el frasco de 2 °C a 8 °C por hasta 6 horas. La solución debe ser desechada si no es utilizada en 6 horas. Después de la refrigeración, la solución debe alcanzar la temperatura ambiente antes de ser inyectada.
Consérvese en la caja de cartón la jeringa prellenada, la pluma precargada y el cartucho dispensador de dosis para protegerlos de la luz.
LEYENDAS DE PROTECCIÓN:
Literatura exclusiva para profesionales de la salud. Léase instructivo anexo. Su venta requiere receta médica. No se deje al alcance de los niños. No se use en el embarazo y la lactancia. Consérvese en refrigeración entre 2 °C y 8 °C. No se congele. No se use en menores de 2 años, ni en niños con esquema de vacunación incompleto. Si no se usa todo el producto, deséchese el sobrante. Deseche la jeringa, pluma o cartucho dispensador de dosis después de su uso. La solución reconstituida del frasco ámpula debe ser clara a ligeramente opalescente, incolora a amarillo o marrón pálido, y puede contener pequeñas partículas blancas a casi transparentes de proteína. La solución de la jeringa prellenada, pluma precargada o cartucho dispensador de dosis debe ser clara a ligeramente opalescente, incolora a amarillo o marrón pálido, y puede contener pequeñas partículas blancas a casi transparentes de proteína. No se use la solución si esta nubosa (turbia), o si contiene otras partículas además de las descritas anteriormente o si el cierre ha sido violado.
Reporte las sospechas de reacción adversa al correo: farmacovigilancia@cofepris.gob.mx,
MEX.AEReporting@pfizer.com
y a la línea Pfizer 800 401 2002.
PFIZER, S.A. de C.V.
Km. 63 Carretera México-Toluca,
Zona Industrial, C.P. 50140,
Toluca, México, México.
Reg. Núm. 557M99 SSA IV
Registro Dispositivo de Inyección
(SMARTCLIC) Número SSA: 1677E2022 SSA
®Marca Registrada

