
ELECSYS AFP
Para visualizar el contenido completo de la IPPA (información para prescribir amplia), deberá iniciar sesión.
VALORES TEÓRICOS:
Véase a continuación los resultados de los siguientes estudios efectuados con el test ELECSYS AFP:
a) Estudio multicéntrico efectuado con el analizador Elecsys 2010 de septiembre de 1997 y estudio de referencia realizado en Alemania y Francia, datos evaluados en septiembre de 1998.
Los siguientes valores de AFP se encontraron en las muestras séricas de 646 individuos sanos:
≤ 5.8 UI/mL o bien ≤ 7.0 ng/mL para el 95% de los resultados.
Valores de la mediana de AFP por semanas de embarazo completas (definidas como semanas completas contando a partir del comienzo del último periodo menstrual):
UI/mL. La línea gruesa de las diagramas de caja representa el valor de la mediana.
|
Semanas |
14 |
15 |
16 |
17 |
18 |
19 |
|
N |
382 |
1782 |
2386 |
975 |
353 |
146 |
|
UI/mL |
23.2 |
25.6 |
30.0 |
33.5 |
40.1 |
45.5 |
|
ng/mL |
27.9 |
30.9 |
36.1 |
40.4 |
48.3 |
54.8 |
b) Estudio multicéntrico de determinación de los valores de referencia para evaluar el riesgo de trisomía 21 en suero materno (estudio N° BO1P019).
Se evaluaron los valores séricos de un total de 1753 embarazadas (semanas de gestación relevantes: 14 a 18).
Las determinaciones efectuadas con el test Elecsys HCG+β y el test ELECSYS AFP se llevaron a cabo en 5 centros clínicos de Bélgica, Francia y Alemania.
Se indica la edad gestacional en días de cada muestra determinada por ultrasonido. A partir de un análisis de regresión log-lineal de los 1753 valores de AFP frente a la edad gestacional se calcularon los siguientes valores de la mediana para la mitad de las respectivas semanas (por ejemplo, semana 14 + 3 días):
|
Semanas |
14 |
15 |
16 |
17 |
18 |
|
UI/mL |
20.9 |
24.0 |
27.6 |
31.7 |
36.4 |
|
ng/mL |
25.3 |
29.0 |
33.3 |
38.3 |
44.0 |
Nota: Para el análisis prenatal se recomienda volver a evaluar los valores de la mediana periódicamente (cada 1 a 3 años) y siempre que se efectúen cambios metodológicos.
No se ha verificado la transferibilidad de los valores de referencia a muestras de plasma.
Cada laboratorio debería comprobar si los intervalos de referencia pueden aplicarse a su grupo de pacientes y, en caso necesario, establecer sus propios valores.
ELECSYS AFP como ayuda en el diagnóstico de CHC:
Se realizó un estudio prospectivo multicéntrico (estudio No. RD002542 y RD002543 de Roche) para evaluar el funcionamiento clínico del ensayo ELECSYS AFP como ayuda en el diagnóstico del CHC. Este estudio incluyó a 376 pacientes con enfermedad hepática de los cuales 168 tenían CHC y 208 una enfermedad hepática sin diagnóstico de CHC (grupo de control).
|
Edad mediana |
Sexo (masculino, %) |
Raza |
|||||
|
Asiática (%) |
Caucásica (%) |
Negra (%) |
Otros (%) |
Faltante (%) |
|||
|
Control |
53 |
60.6 |
47.6 |
48.6 |
1.4 |
0 |
2.4 |
|
CHC |
64 |
83.9 |
42.3 |
56.5 |
0 |
0.6 |
0.6 |
a) Intervalo de concentración de AFP en los casos de CHC en comparación con los controles:
La tabla y la gráfica siguientes indican el intervalo de concentración de AFP en las muestras de pacientes con CHC estadificados según la clasificación clínica de cáncer de hígado de Barcelona (BCLC)23 en comparación con controles. Para los 168 pacientes con un diagnóstico de CHC, la concentración de AFP aumentó con la progresión de la enfermedad especialmente en el estadio tardío. Las concentraciones de la tabla se indican en UI/mL y ng/mL y las concentraciones de la gráfica se indican en UI/mL. La línea gruesa de las diagramas de caja representa el valor de la mediana.
|
Estadio de la enfermedad |
N |
Mín./máx. |
Mediana ± DE |
Mediana |
Percentil b) 25-75 |
|
Control c) |
208 |
0.85/327.84 (1.03/396.69) |
5.33±23.15 (6.45±28.01) |
2.42 (2.92) |
1.86-3.89 (2.25-4.71) |
|
Temprano (estadios 0 + A) |
77 |
1.04/5224 (1.26/6322) |
252±799 (305±966) |
9.7 (11.7) |
3.72-39.6 (4.5-47.9) |
|
BCLC estadio 0 |
10 |
1.04/258 (1.26/312) |
58.4±98.9 (70.7±120) |
9.67 (11.7) |
- |
|
BCLC estadio A |
67 |
1.48/5224 (1.79/6322) |
281±852 (340±1031) |
9.7 (11.7) |
3.72-39.6 (4.5-47.9) |
|
Tardío (estadios B, C y D) |
91 |
1.3/44687 (1.57/54071) |
2874±8259 (3478±9994) |
119 (144) |
5.95-909 (7.2-1100) |
|
BCLC estadio B |
26 |
2.85/34944 (3.45/42282) |
1989±7301 (2407±8834) |
15.5 (18.8) |
5.33-132 (6.45-160) |
|
BCLC estadio C |
57 |
1.3/44687 (1.57/54071) |
2313±7079) (2798±8566) |
150 (182) |
6.14-959 (7.43-1160) |
|
BCLC estadio D |
8 |
557/34531 (674/41782) |
9751±15043 (11799±18201) |
999 (1209) |
- |
b) No calculado si el número de muestras era igual o inferior a 20
c) En la siguiente representación gráfica, este grupo se designa con una "X"
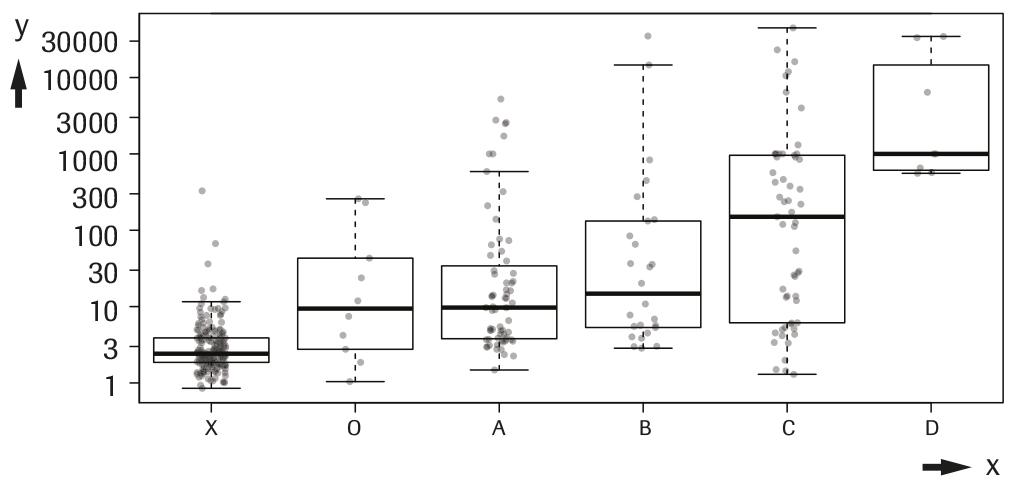
x ---> X: control; O: estadio 0; A: estadio A; B: estadio B; C: estadio C; D: estadio D.
y ---> AFP (UI/mL)
b) Concentración de AFP y etiología de la enfermedad:
La concentración de AFP según etiología para los dos grupos de pacientes (control, 1-A a 1-F y CHC, 2-A a 2F) se muestra en la tabla y gráfica siguientes. Las concentraciones de la tabla se indican en UI/mL y ng/mL y las concentraciones de la gráfica se indican en UI/mL. La línea gruesa de las diagramas de caja representa el valor de la mediana.
|
Grupo |
Etiología d) |
N |
Mín./máx. |
Mediana ± DE |
Mediana |
Percentil 25-75 |
|
1-A |
Cirrosis |
79 |
0.851/66.9 (1.03/80.9) |
4.92±8.59 (5.95±10.4) |
2.63 (3.19) |
1.85-4.34 (2.24-5.25) |
|
1-B |
Hepatitis B |
72 |
1.03/328 (1.25/397) |
7.4±38.3 (8.95±46.4) |
2.31 (2.79) |
1.73-3.11 (2.1-3.76) |
|
1-C |
Hepatitis C |
27 |
1.3/6.33 (1.57/7.66) |
3.23±1.43 (3.9±1.73) |
2.49 (3.01) |
2.21-4.73 (2.67-5.73) |
|
1-D |
NASH e) |
30 |
1.01/10.9 (1.22/13.2) |
3.36±2.36 (4.06±2.86) |
2.48 (3.00) |
1.74-3.96 (2.11-4.79) |
|
1-E |
ALD f) |
0 |
- |
- |
- |
- |
|
1-F |
Otras |
0 |
- |
- |
- |
- |
|
2-A |
Cirrosis |
139 |
1.04/44687 (1.26/54071) |
1536±6096 (1859±7377) |
16.6 (20.1) |
4.82-320 (5.84-387) |
|
2-B |
Hepatitis B |
14 |
2.25/1711 (2.73/2070) |
296±536 (358±649) |
38.2 (46.2) |
- |
|
2-C |
Hepatitis C |
3 |
1.86/999 (2.25/1209) |
348±564 (421±683) |
43.1 (52.1) |
- |
|
2-D |
NASH |
1 |
- |
3.55 (4.3) |
- |
- |
|
2-E |
ALD |
2 |
3.87/33288 (4.69/40278) |
16646±23535 (20141±28478) |
16646 (20141) |
- |
|
2-F |
Otras |
9 |
1.98/16115 (2.4/19499) |
3216±5924 (3891±7168) |
210 (254) |
- |
d) Todas las etiologías son no cirróticas, con excepción de la propia cirrosis
e) Esteatohepatitis no alcohólica
f) Enfermedad hepática alcohólica
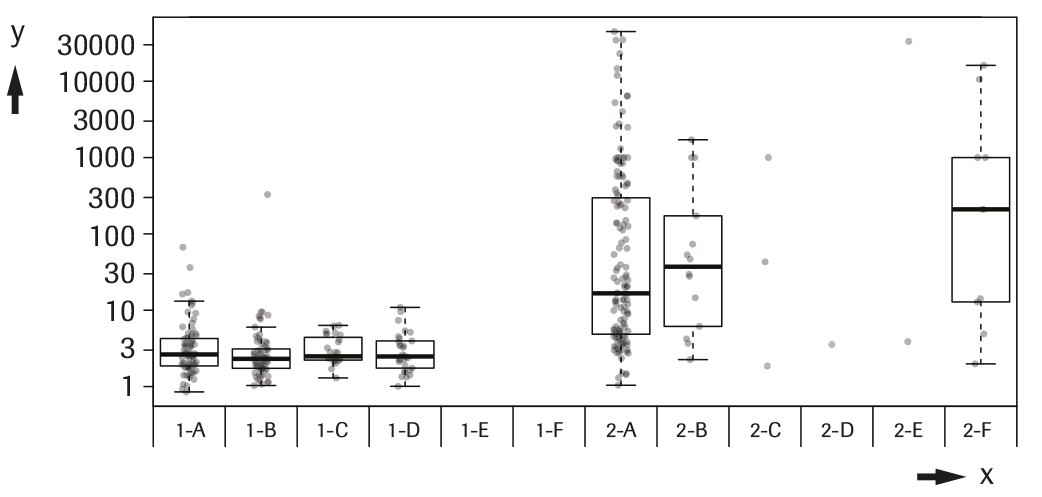
y ---> AFP (UI/mL)
C) Funcionamiento clínico del test ELECSYS AFP para la detección del CHC:
A continuación, se muestran la sensibilidad y especificidad del test ELECSYS AFP en la detección del CHC a un punto de corte de 165 UI/mL (200 ng/mL) y de 16.5 UI/mL (20 ng/mL) y los resultados del análisis ROC (receiver operating characteristic).
|
CHC, en total |
CHC, estadio temprano g) |
CHC, estadio tardío h) |
||
|
AFP punto de corte 200 ng/mL |
Sensibilidad IC del 95% i) |
31.5% (24.6%, 39.2%) |
15.6% (8.3%, 25.6%) |
45.1% (34.6%, 55.8%) |
|
Especificidad (IC del 95%) |
99.5% |
|||
|
(97.4%, 100%) |
||||
|
AFP punto de corte 20 ng/mL |
Sensibilidad (IC del 95%) |
51.8% (44%, 59.5%) |
36.4% (25.7%, 48.1%) |
64.8% (54.1%, 74.6%) |
|
Especificidad (IC del 95%) |
98.1% |
|||
|
(95.1%, 99.5%) |
||||
|
ROC ABCj) |
88% (84.5%, 91.5%) |
84.5% (79.3%, 89.7%) |
90.9% (86.8%, 95.1%) |
|
g) Estadios BCLC 0, A
h) Estadios BCLC B, C, D
i) Intervalo de confianza
j) Área bajo la curva
d) Valores de AFP en diferentes enfermedades benignas y malignas:
La tabla y la gráfica siguientes muestran la concentración de AFP en UI/mL (ng/mL) en un panel de muestras de pacientes con hepatopatía benigna, trastorno inmunológico o una enfermedad maligna que no sea CHC (N total 397; edad mediana 54 años, 58% femeninas, 39% asiáticos y 61% caucásicos).
|
Grupo |
Enfermedad |
N |
Mín./máx. |
Media ± (DE) |
Mediana |
Percentil 25-75 |
|
A |
Hepatopatías benignask) |
87 |
0.843/999 (1.02/1209) |
14.3±107 (17.3±129) |
2.20 (2.66) |
1.73-3.48 (2.10-4.21) |
|
B |
Artritis reumatoide |
38 |
1.11/11.7 (1.34/14.2) |
2.80±1.84 (3.39±2.22) |
2.28 (2.75) |
1.77-2.99 (2.14-3.62) |
|
C |
Enfermedad de Crohn |
37 |
0.676/10.0 (0.819/12.1) |
3.21±2.40 (3.88±2.90) |
2.42 (2.93) |
1.63-3.58 (1.97-4.34) |
|
D |
Colitis ulcerosa |
30 |
1.20/7.27 (1.45/8.80) |
2.58±1.35 (3.12±1.63) |
2.37 (2.86) |
1.63-2.94 (1.97-3.56) |
|
E |
Otras enfermedades autoinmunes l) |
26 |
0.909/7.93 (1.10/9.60) |
3.16±1.72 (3.83±2.08) |
2.62 (3.16) |
2.02-3.97 (2.44-4.80) |
|
F |
Cáncer pulmonar |
24 |
1.01/5.18 (1.22/6.27) |
2.50±0.978 (3.02±1.18) |
2.40 (2.90) |
1.90-3.03 (2.30-3.67) |
|
G |
Cáncer de mama |
27 |
0.859/7.67 (1.04/9.27) |
3.06±1.60 (3.70±1.93) |
2.59 (3.13) |
1.85-4.01 (2.24-4.85) |
|
H |
Cáncer renal |
10 |
0.58/6.43 (0.702/7.78) |
2.73±1.96 (3.30±2.37) |
2.21 (2.67) |
- |
|
I |
Colangiocarcinoma |
27 |
1.06/83.8 (1.28/101) |
7.48±15.9 (9.05±19.3) |
3.51 (4.25) |
2.15-4.82 (2.60-5.84) |
|
J |
Cáncer pancreático |
10 |
1.03/6.65 (1.25/8.05) |
2.83±2.02 (3.43±2.45) |
1.92 (2.32) |
- |
|
K |
Otros cánceres gastrointestinales m) |
55 |
0.512/13.1 (0.62/15.9) |
3.00±1.95 (3.63±2.35) |
2.68 (3.24) |
2.02-3.43 (2.44-4.15) |
|
L |
Cánceres ginecológicosn) |
26 |
0.999/9.19 (1.21/11.1) |
3.24±2.02 (3.92±2.44) |
2.62 (3.16) |
1.86-3.96 (2.25-4.79) |
k) Enfermedad poliquística hepática, quistes simples, hiperplasia nodular focal, hemangioma, adenoma hepatocelular, enfermedad hepática alcohólica no cirrótica.
l) Lupus eritematoso sistémico, tiroiditis autoinmune.
m) Cáncer colorrectal, gástrico y esofágico.
n) Cáncer ovárico, endometrial y cervical.
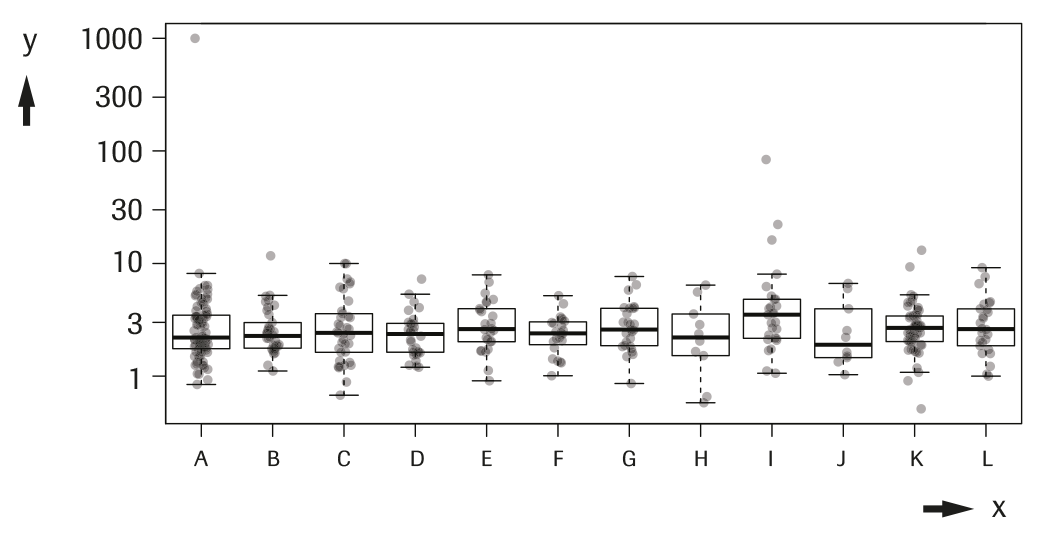
y ---> AFP (ng/mL)
MEDIDAS DE PRECAUCIÓN Y ADVERTENCIAS:
Para el uso diagnóstico in vitro por los profesionales de la salud. Observe las medidas de precaución usuales para la manipulación de reactivos de laboratorio.
Residuos infecciosos o microbiológicos:
Advertencia: Manipule los residuos como material biológico potencialmente peligroso. Deseche los residuos de acuerdo con las instrucciones y procedimientos de laboratorio aceptados.
Peligros ambientales: Aplique todas las normas locales de eliminación pertinentes para asegurar una eliminación segura.
Existe una ficha de datos de seguridad a disposición del usuario profesional que la solicite.
El presente estuche contiene componentes que han sido clasificados por la directiva CE No. 1272/2008 de la siguiente manera:

Atención:
|
H317 |
Puede provocar una reacción alérgica en la piel. |
|
Prevención: |
|
|
P261 |
Evitar respirar la niebla o el vapor. |
|
P272 |
Las prendas de trabajo contaminadas no podrán sacarse del lugar de trabajo. |
|
P280 |
Llevar guantes de protección. |
|
Respuesta: |
|
|
P333 + P313 |
En caso de irritación o erupción cutánea: consultar a un médico. |
|
P362 + P364 |
Quitar las prendas contaminadas y lavarlas antes de volver a usarlas. |
|
Eliminación: |
|
|
P501 |
Eliminar el contenido/el recipiente en una planta de eliminación de residuos aprobada. |
Compuestos peligrosos:
• Clorhidrato de 2-metil-2H-isotiazol-3-ona.
Las indicaciones de seguridad del producto corresponden a los criterios del sistema globalmente armonizado de clasificación y etiquetado de productos químicos (GHS por sus siglas en inglés) válidas en la UE.
Contacto telefónico internacional: +49-621-7590
Evite la formación de espuma en reactivos y muestras de todo tipo (especímenes, calibradores y controles).
CONSERVACIÓN Y ESTABILIDAD:
Conservar a 2-8°C. No congelar.
Conservar el cobas e pack en posición vertical para garantizar la disponibilidad total de las micropartículas durante la mezcla automática antes del uso.
|
Estabilidad: |
|
|
Sin abrir, a 2-8°C |
Hasta la fecha de caducidad indicada |
|
En los analizadores |
16 semanas |
DILUCIÓN:
Las muestras con concentraciones de AFP superiores al intervalo de medición pueden diluirse con Diluent Universal. Se recomienda una dilución a 1:50 (automáticamente por los analizadores o bien de forma manual). La concentración de la muestra diluida debe ser > 18 UI/mL (> 21.8 ng/mL).
Multiplicar los resultados obtenidos tras dilución manual por el factor de dilución.
El software de los analizadores tiene en cuenta la dilución automática al calcular la concentración de las muestras.
MATERIAL REQUERIDO ADICIONALMENTE (NO SUMINISTRADO):
•  09227261190, AFP CalSet II para 4 x 1.0 mL
09227261190, AFP CalSet II para 4 x 1.0 mL
• 11776452122, PreciControl Tumor Marker, para 4 x 3.0 mL o
• 11731416190, PreciControl Universal, para 4 x 3.0 mL o
• 08754551190, PreciControl HCC V2, para 4 x 1.0 mL
• 07299001190, Diluent Universal, 36 mL de diluyente para muestras
• Equipo usual de laboratorio
• Analizador cobas e
Materiales adicionales para los analizadores cobas e 402 y cobas e 801:
• 06908799190, ProCell II M, 2 x 2 L de solución del sistema
• 04880293190, CleanCell M, 2 x 2 L de solución de limpieza para la célula de medida
• 07485409001, Reservoir Cup, 8 recipientes para ProCell II M y CleanCell M
• 06908853190, PreClean II M, 2 x 2 L de solución de lavado
• 05694302001, Bandeja de Assay Tip/Assay Cup, 6 x 6 bandejas, cada una con 105 cubetas y 105 puntas de pipeta (3780 determinaciones), 3 cartones de residuos sólidos
• 07485425001, Liquid Flow Cleaning Cup, 2 recipientes para la solución de limpieza ISE Cleaning Solution/Elecsys SysClean para la unidad de detección Liquid Flow Cleaning Detection Unit
• 07485433001, PreWash Liquid Flow Cleaning Cup, 1 recipiente para la solución de limpieza ISE Cleaning Solution/Elecsys SysClean para la unidad de prelavado Liquid Flow Cleaning PreWash Unit
• 11298500316, ISE Cleaning Solution/Elecsys SysClean, 5 x 100 mL de solución de limpieza para el sistema
Para calcular el riesgo de trisomía 21:
• Un software adecuado, como p. ej.
• 05126193, SsdwLab (V5.0 o posterior), licencia monousuario
• 05195047, SsdwLab (V5.0 o posterior), licencia multiusuario
• 03271749190, HCG+β 100 pruebas
• 07251025190, Elecsys HCG+β, 300 pruebas
• 03302652190, HCG+β CalSet para 4 x 1.0 mL
MATERIAL SUMINISTRADO:
Consultar la sección "Reactivos - Soluciones de trabajo" en cuanto a los reactivos suministrados.
CALIBRACIÓN:
Trazabilidad: El presente método ha sido estandarizado frente al primer estándar de referencia IRP 72/225 de la OMS.
La curva máster predefinida es adaptada al analizador a través del CalSet correspondiente.
Intervalo de calibraciones: efectuar la calibración una vez por lote de reactivos con reactivos frescos de un cobas e pack registrado como máximo 24 horas antes en el analizador.
El intervalo de calibración puede ampliarse si el laboratorio asegura una verificación aceptable de la calibración.
Se recomienda repetir la calibración:
• Después de 12 semanas si se trata del mismo lote de reactivos.
• Después de 28 días (si se emplea el mismo cobas e pack en el analizador).
• En caso necesario: por ejemplo, si los valores del control de calidad están fuera del intervalo definido.
CONTROL DE CALIDAD:
Efectuar el control de calidad con PreciControl Tumor Marker, PreciControl Universal, PreciControl HCC V2 u otros controles adecuados para los controles de calidad de rutina.
Los controles de los diferentes intervalos de concentración deberían efectuarse junto con el test en determinaciones simples por lo menos 1 vez cada 24 horas, con cada cobas e pack y después de cada calibración.
Adaptar los intervalos y límites de control a los requisitos individuales del laboratorio. Los resultados obtenidos deben hallarse dentro de los límites definidos. Cada laboratorio debería establecer medidas correctivas a seguir en caso de obtener valores fuera del intervalo definido.
Si fuera necesario, repetir la medición de las muestras en cuestión.
Cumplir con las regulaciones gubernamentales y las normas locales de control de calidad pertinentes.
CÁLCULO:
El analizador calcula automáticamente la concentración de analito de cada muestra (en UI/mL, ng/mL, kUI/L o adicionalmente en UI/L).
|
Factores de conversión: |
UI/mL x 1.21 = ng/mL |
|
ng/mL x 0.83 = UI/mL |
INFORMACIÓN DEL SISTEMA:
Nombre abreviado: AFP
ACN (código de aplicación): AFP 10209
Nota: El valor del AFP de una muestra de paciente puede variar según el método de ensayo aplicado. Por lo tanto, el laboratorio siempre debe indicar el método de determinación de AFP empleado. Los valores de AFP de un paciente, obtenidos mediante diferentes procedimientos de test, no pueden compararse directamente entre sí y dan lugar a interpretaciones erróneas por parte del médico.
En caso de cambiar el método de determinación de AFP durante el control del tratamiento, los valores del mismo deben confirmarse en el periodo de transición mediante mediciones paralelas de ambos métodos.
LÍMITES E INTERVALOS:
Intervalo de medición: 0.75-1000 UI/mL o 0.908-1210 ng/mL (definido por el Límite de Blanco y el máximo de la curva máster). Los valores inferiores al Límite de Blanco se indican como < 0.75 UI/mL (< 0.908 ng/mL). Los valores superiores al intervalo de medición se indican como > 1000 UI/mL o > 1210 ng/mL (diluidos a 1:50 respectivamente hasta 50000 UI/mL o bien 60500 ng/mL).
Límites inferiores de medición:
Límite de Blanco, Límite de Detección y Límite de Cuantificación:
• Límite de Blanco = 0.75 UI/mL (o 0.91 ng/mL)
• Límite de Detección = 1.5 UI/mL (o 1.82 ng/mL)
• Límite de Cuantificación = 2.25 UI/mL (o 2.72 ng/mL)
El Límite de Blanco, el Límite de Detección y el Límite de Cuantificación fueron determinados cumpliendo con los requerimientos EP17-A2 del Instituto de Estándares Clínicos y de Laboratorio (CLSI-Clinical and Laboratory Standards Institute).
El Límite de Blanco es el valor del percentil 95 obtenido a partir de n ≥ 60 mediciones de muestras libres de analito en varias series independientes. El Límite de Blanco corresponde a la concentración por debajo de la cual se encuentran, con una probabilidad del 95%, las muestras sin analito.
El Límite de Detección se determina basándose en el Límite de Blanco y en la desviación estándar de muestras de baja concentración. El Límite de Detección corresponde a la menor concentración de analito detectable (valor superior al Límite de Blanco con una probabilidad del 95%).
El Límite de Cuantificación es la menor concentración de analito cuya medición puede reproducirse con un coeficiente de variación para la precisión intermedia de ≤ 20%.
PRECISIÓN:
La precisión ha sido determinada mediante reactivos Elecsys, una mezcla de sueros humanos y controles según un protocolo (EP05-A3) del CLSI (Clinical and Laboratory Standards Institute): 2 ciclos diarios por duplicado, cada uno durante 21 días (n = 84). Se obtuvieron los siguientes resultados:
|
Analizadores cobas e 402 y cobas e 801 |
|||||
|---|---|---|---|---|---|
|
Repetibilidad |
|||||
|
Muestra |
Media |
DE |
CV |
||
|
UI/mL |
ng/mL |
UI/mL |
ng/mL |
% |
|
|
Suero humano 1 |
2.23 |
2.70 |
0.078 |
0.095 |
3.5 |
|
Suero humano 2 |
1.80 |
2.18 |
0.073 |
0.088 |
4.1 |
|
Suero humano 3 |
5.51 |
6.67 |
0.088 |
0.107 |
1.6 |
|
Suero humano 4 |
16.0 |
19.4 |
0.379 |
0.459 |
2.4 |
|
Suero humano 5 |
156 |
189 |
2.81 |
3.40 |
1.8 |
|
Suero humano 6 |
458 |
554 |
7.18 |
8.69 |
1.6 |
|
Suero humano 7 |
927 |
1122 |
17.8 |
21.5 |
1.9 |
|
PC o) Tumor Marker1 |
7.93 |
9.60 |
0.116 |
0.140 |
1.5 |
|
PC Tumor Marker2 |
75.0 |
90.8 |
1.72 |
2.08 |
2.3 |
|
PC Universal1 |
10.8 |
13.1 |
0.226 |
0.273 |
2.1 |
|
PC Universal2 |
53.9 |
65.2 |
1.17 |
1.42 |
2.2 |
|
PC 1 HCC V2 p) |
3.20 |
3.87 |
0.049 |
0.059 |
1.5 |
|
PC 2 HCC V2 q) |
75.0 |
90.8 |
1.14 |
1.38 |
1.5 |
o) PC = PreciControl
p) PC 1 HCC V2 = PreciControl HCC V2, Nivel 1
q) PC 2 HCC V2 = PreciControl HCC V2, Nivel 2
|
Analizadores cobas e 402 y cobas e 801 |
|||||
|
Precisión intermedia |
|||||
|
Muestra |
Media |
DE |
CV |
||
|
UI/mL |
ng/mL |
UI/mL |
ng/mL |
% |
|
|
Suero humano 1 |
2.23 |
2.70 |
0.084 |
0.102 |
3.8 |
|
Suero humano 2 |
1.80 |
2.18 |
0.076 |
0.091 |
4.2 |
|
Suero humano 3 |
5.51 |
6.67 |
0.133 |
0.161 |
2.4 |
|
Suero humano 4 |
16.0 |
19.4 |
0.441 |
0.534 |
2.8 |
|
Suero humano 5 |
156 |
189 |
3.74 |
4.53 |
2.4 |
|
Suero humano 6 |
458 |
554 |
10.7 |
12.9 |
2.3 |
|
Suero humano 7 |
927 |
1122 |
22.4 |
27.1 |
2.4 |
|
PC Tumor Marker1 |
7.93 |
9.60 |
0.170 |
0.206 |
2.1 |
|
PC Tumor Marker2 |
75.0 |
90.8 |
2.23 |
2.70 |
3.0 |
|
PC Universal1 |
10.8 |
13.1 |
0.271 |
0.328 |
2.5 |
|
PC Universal2 |
53.9 |
65.2 |
1.40 |
1.69 |
2.6 |
|
PC 1 HCC V2 |
3.20 |
3.87 |
0.073 |
0.088 |
2.3 |
|
PC 2 HCC V2 |
75.0 |
90.8 |
1.89 |
2.29 |
2.5 |
CARACTERÍSTICAS:
La alfa1-fetoproteína (AFP) es una glucoproteína similar a la albúmina con un peso molecular de aproximadamente 70 kDa. Se sintetiza en la vesícula blastodérmica durante la vida fetal, en las células hepáticas no diferenciadas así como en el tracto gastrointestinal fetal.1,2
Los tumores que sintetizan AFP son principalmente tumores testiculares no seminomatosos de células germinales (NSGCT, por sus siglas en inglés), tumores del saco vitelino de ovario o carcinomas hepatocelulares. Junto con la hCG+β y otros parámetros, la AFP también es un parámetro importante en la evaluación de riesgo de la trisomía 21 en el segundo trimestre del embarazo.3
Cáncer de testículo: El seguimiento cuidadoso de los marcadores tumorales séricos AFP y gonadotropina coriónica humana (hCG) es esencial en el manejo de pacientes con tumores de células germinales porque estos marcadores constituyen importantes indicadores pronósticos para el diagnóstico, sirven para el seguimiento de la respuesta al tratamiento y se aplican para la detección precoz de recaídas.4 Asimismo, la hCG y la AFP son parámetros importantes en la estimación de la tasa de supervivencia de pacientes con NSGCT avanzados. También la National Academy of Clinical Biochemistry recomienda su uso en el manejo de pacientes con dicha enfermedad.5
Carcinoma hepatocelular: El carcinoma hepatocelular (CHC) frecuentemente es resultado de una enfermedad hepática avanzada que puede desarrollarse en los pacientes con y sin cirrosis.6 Desde hace mucho tiempo, la AFP es reconocida como un biomarcador para el CHC y desempeña un papel destacado en el diagnóstico del CHC. Los valores sustancialmente elevados de AFP pueden indicar un carcinoma hepatocelular primario. Se ha mostrado que la concentración de AFP aumenta según el tamaño del tumor.7 El diagnóstico del CHC se ha apoyado principalmente en la presencia de características típicas observadas en estudios de imágenes con contraste, la evaluación histopatológica y las concentraciones séricas de AFP.8 Si bien la AFP es elevada durante la carcinogénesis hepática, también puede encontrarse en otros tumores tales como el cáncer testicular, embrionario o gástrico.9,10 En los pacientes con CHC, la AFP tiene una sensibilidad del 39 al 65% y una especificidad del 76 al 94%.11 La divergencia en la sensibilidad y especificidad de la AFP en los estudios realizados se debe probablemente a una variedad de factores que incluyen etiologías, diseños de estudio y valores de corte diferentes. Ya que la concentración de AFP también aumenta durante el proceso de regeneración hepática, sus valores se encuentran moderadamente elevados en la cirrosis hepática por alcohol y en la hepatitis viral aguda.12 En varias guías de práctica clínica se recomienda la vigilancia de los pacientes con riesgo de desarrollar CHC mediante ultrasonografía abdominal en combinación con AFP.13,14,15
Trisomía 21: La medición de AFP contribuye a evaluar el riesgo de una trisomía 21 (síndrome de Down) en el segundo trimestre del embarazo si se la evalúa junto con la hCG+β y con otros parámetros tales como la exacta edad gestacional y el peso materno.3 En las embarazadas con trisomía 21, la concentración de AFP sérica está reducida, mientras que la concentración de hCG+β sérica constituye aproximadamente el doble de la mediana normal.16 El riesgo de trisomía 21 en el segundo trimestre del embarazo puede calcularse con un software apropiado (consultar la sección "Materiales requeridos adicionalmente, pero no suministrados") mediante el algoritmo descrito por Cuckle y cols.17 y los parámetros específicos del test empleado.18,19,20,21,22
USO PREVISTO:
Test inmunológico in vitro para la determinación cuantitativa de la α1-fetoproteína en suero y plasma humanos.
El test ha sido concebido:
• Como ayuda en el diagnóstico del carcinoma hepatocelular (CHC).
• Como ayuda en el manejo de pacientes con tumores no seminomatosos de células germinales.
• En combinación con otros parámetros a fin de evaluar el riesgo de trisomía 21 (síndrome de Down). El diagnóstico de aberraciones cromosómicas requiere de análisis adicionales.
Este inmunoensayo de electroquimioluminiscencia (electrochemiluminescence immunoassay) "ECLIA" está previsto para el uso en inmunoanalizadores cobas e.
LIMITACIONES DEL ANÁLISIS - INTERFERENCIAS:
Se analizaron los efectos de las siguientes sustancias endógenas y los siguientes compuestos farmacéuticos sobre el funcionamiento del test sin que se hayan observado interferencias.
Sustancias endógenas:
|
Compuesto |
Concentración analizada |
|
Bilirrubina |
≤ 1112 μmol/L o ≤ 65 mg/dL |
|
Hemoglobina |
≤ 1.37 mmol/L o ≤ 2200 mg/dL |
|
Intralipid |
≤ 1500 mg/dL |
|
Biotina |
≤ 4912 nmol/L o ≤ 1200 ng/mL |
|
Factores reumatoides |
≤ 1500 UI/mL |
Criterio: Recuperación ± 0.4 UI/mL del valor inicial para muestras ≤ 4 UI/mL, dentro de ± 10% del valor inicial para muestras > 4 UI/mL.
Sin efecto high-dose hook con concentraciones de AFP de hasta 1 millón de UI/mL o resp. 1.21 millón de ng/mL.
Compuestos farmacéuticos: Se analizaron in vitro 16 fármacos de uso extendido sin encontrar interferencias con el presente ensayo.
Adicionalmente se analizaron los siguientes fármacos anticancerígenos sin encontrar interferencias con el presente ensayo.
Fármacos anticancerígenos:
|
Fármaco |
Concentración analizada (mg/L) |
|
Doxorrubicina |
75 |
|
Ciclofosfamida |
1000 |
|
Cisplatino |
225 |
|
5-Fluorouracilo |
500 |
|
Metotrexato |
1000 |
|
Tamoxifeno |
50 |
|
Mitomicina |
25 |
|
Carboplatino |
1000 |
|
Etopósido |
400 |
|
Taxol |
5.5 |
En casos aislados pueden presentarse interferencias por títulos extremadamente altos de anticuerpos dirigidos contra anticuerpos específicos del analito, la estreptavidina o el rutenio. Estos efectos se han minimizado gracias a un adecuado diseño del test.
Para el diagnóstico, los resultados del test siempre deben interpretarse teniendo en cuenta la anamnesis del paciente, la exploración clínica así como los resultados de otros exámenes.
SÍMBOLOS:
Roche Diagnostics utiliza los siguientes símbolos y signos adicionalmente a los indicados en la norma ISO 15223-1 (para los EE. UU.: consulte navifyportal.roche.com para la definición de los símbolos usados):
|
CONTENT |
Contenido del kit |
|
SYSTEM |
Analizadores/instrumentos adecuados para los reactivos |
|
REAGENT |
Reactivo |
|
CALIBRATOR |
Calibrador |
|
|
Volumen para reconstitución |
|
GTIN |
Número Global de Artículo Comercial |
|
Rx only |
Para los EE.UU.: atención: según la ley federal estadounidense, este producto puede ser vendido exclusivamente por facultativos o por prescripción médica. |
La barra del margen indica suplementos, eliminaciones o cambios.
© 2024 Roche Diagnostics
ROCHE DIAGNOSTICS GMBH
Sandhofer Strasse 116
D-68305 Mannheim
+800 5505 6606
OBTENCIÓN Y PREPARACIÓN DE LAS MUESTRAS:
Sólo se ha analizado y considerado apto el tipo de muestras aquí indicado.
Suero recogido en tubos estándar de muestra o en tubos que contienen gel de separación.
Plasma tratado con heparina de litio o con EDTA di o tripotásico. Pueden emplearse tubos para plasma que contengan gel de separación.
Criterio: pendiente de 0.9-1.1 + coeficiente de correlación ≥ 0.95.
Estabilidad: 5 días a 20-25°C, 14 días a 2-8°C, 6 meses a -20°C (± 5°C). Las muestras pueden congelarse 3 veces.
No se ha evaluado si las muestras de plasma son apropiadas para estimar el riesgo de trisomía 21.
Los tipos de muestra aquí indicados fueron analizados con tubos de recogida de muestras seleccionados, comercializados en el momento de efectuar el análisis, lo cual significa que no fueron analizados todos los tubos de todos los fabricantes. Los sistemas de recogida de muestras de diversos fabricantes pueden contener diferentes materiales que, en ciertos casos, pueden llegar a afectar los resultados de los análisis. Si las muestras se procesan en tubos primarios (sistemas de recogida de muestras), seguir las instrucciones del fabricante de tubos.
Centrifugar las muestras que contienen precipitado antes de realizar el ensayo.
No emplear muestras inactivadas por calor.
No utilizar muestras ni controles estabilizados con azida.
Se debe garantizar una temperatura de 20-25°C para la medición de muestras y calibradores.
Para evitar posibles efectos de evaporación, determinar las muestras y los calibradores que se sitúan en los analizadores dentro de un lapso de 2 horas.
PREPARACIÓN DE LOS REACTIVOS:
Los reactivos incluidos en el kit están listos para el uso y forman una unidad inseparable.
La información necesaria para el correcto funcionamiento está disponible a través de cobas link.
COMPARACIÓN DE MÉTODOS:
a) Una comparación entre el test ELECSYS AFP, 09015124190 (analizador cobas e 801; y) y el test ELECSYS AFP 07026706190 (analizador cobas e 801; x) generó las siguientes correlaciones (en UI/mL):
Número de muestras de suero medidas: 188
|
Passing/Bablok24 |
Regresión lineal |
|
y = 0.974x - 0.092 |
y = 0.968x + 0.993 |
|
T= 0.985 |
r = 0.999 |
Las concentraciones de las muestras se situaron entre 1.23 y 954 UI/mL.
Una comparación entre el test ELECSYS AFP 09015124190 (analizador cobas e 402; y) y el test ELECSYS AFP 09015124190 (analizador cobas e 801; x) generó las siguientes correlaciones (en UI/mL):
Número de muestras de suero medidas: 190
|
Passing/Bablok24 |
Regresión lineal |
|
y = 0.986x - 0.121 |
y = 0.993x - 0.345 |
|
T = 0.991 |
r = 1.00 |
Las concentraciones de las muestras se situaron entre 1.08 y 999 UI/mL.
DATOS ESPECÍFICOS DEL FUNCIONAMIENTO DEL TEST:
A continuación, se indican los datos representativos del funcionamiento de los analizadores. Los resultados de cada laboratorio en particular pueden diferir de estos valores.
PRINCIPIO DEL TEST:
Técnica sándwich duración total del test: 18 minutos.
• 1.ª incubación: 6 μL de muestra, un anticuerpo monoclonal biotinilado anti-AFP y un anticuerpo monoclonal anti-AFP marcado con quelato de rutenioa) forman un complejo sándwich.
• 2.ª incubación: después de incorporar las micropartículas recubiertas de estreptavidina, el complejo formado se fija a la fase sólida por interacción entre la biotina y la estreptavidina.
• La mezcla de reacción es trasladada a la célula de medida donde, por magnetismo, las micropartículas se fijan a la superficie del electrodo. Los elementos no fijados se eliminan posteriormente con ProCell II M. Al aplicar una corriente eléctrica controlada se produce una reacción quimioluminiscente cuya emisión de luz se mide con un fotomultiplicador.
• Los resultados se determinan mediante una curva de calibración generada específicamente para el instrumento a partir de una calibración a 2 puntos y una curva máster suministrada a través de cobas link.
a) Quelato Tris (2,2’-bipiridina) rutenio(II) (Ru(bpy)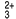 )
)
REACTIVOS - SOLUCIONES DE TRABAJO:
El cobas e pack está etiquetado como AFP.
M Micropartículas recubiertas de estreptavidina, 1 frasco, 14.1 mL: micropartículas recubiertas de estreptavidina: 0.72 mg/mL, conservante.
R1 Anticuerpo anti-AFP~biotina, 1 frasco, 19.7 mL:
Anticuerpos biotinilados monoclonales anti-AFP (ratón) 4.5 mg/L; tampón fosfato 100 mmol/L, pH 6.0; conservante.
R2 Anticuerpo anti-AFP~Ru(bpy), 1 frasco, 19.7 mL:
Anticuerpos monoclonales anti-AFP (ratón) marcados con quelato de rutenio 12.0 mg/L; tampón fosfato 100 mmol/L, pH 6.0; conservante.
REALIZACIÓN DEL TEST:
Para garantizar el funcionamiento óptimo del test, observe las instrucciones de la presente metódica referentes al analizador empleado. Consulte el manual del operador apropiado para obtener las instrucciones de ensayo específicas del analizador.
Las micropartículas se mezclan automáticamente antes del uso.
Colocar el cobas e pack refrigerado (a 2-8°C) en el gestor de reactivos (reagent manager). Evitar la formación de espuma. El analizador realiza automáticamente los procesos de atemperar, abrir y tapar el cobas e pack.
REFERENCIAS BIBLIOGRÁFICAS:
1. Taketa K. Alpha-Fetoprotein in the 1990s. In: Sell SS. Serological cancer markers. Humana Press 1992;31-46, ISBN: 0-89603-209-4
2. Terentiev AA., Moldogazieva NT. Alpha-fetoprotein: a renaissance. Tumor Biology 2013;34:2075-2091.
3. Wald NJ, Kennard A, Densem JW, et al. Antenatal maternal serum screening for Down’s syndrome: results of a demonstration project. BMJ 1992;305:391-394.
4. Klepp O. Serum tumor markers in testicular and extragonadal germ cell malignancies. Scand J Clin Lab Invest Suppl 1991;206:28-41.
5. Sturgeon CM, Duffy MJ, Stenman UH, et al. National Academy of Clinical Biochemistry Laboratory Medicine Practice Guidelines for Use of Tumor Markers in Testicular, Prostate, Colorectal, Breast, and Ovarian Cancers. Clin Chem 2008;54:12:e11-e79.
6. Llovet JM, Zucman-Rossi J, Pikarsky E, et al. Hepatocellular carcinoma. Nature Reviews Disease Primers. 2016;14:2:16018.
7. Toro A, Ardiri A, Mannino M, et al. Effect of pre- and post-treatment alpha-fetoprotein levels and tumor size on survival of patients with hepatocellular carcinoma treated by resection, transarterial chemoembolization or radiofrequency ablation: a retrospective study. BMC surgery 2014;14:40.
8. Gonzalez SA and Keeffe EB. Diagnosis of Hepatocellular Carcinoma: Role of Tumor Markers and Liver Biopsy. Clin Liver Dis 2011;15:297-306.
9. Gupta S, Bent S, Kohlwes J. Test characteristics of alpha-fetoprotein for detecting hepatocellular carcinoma in patients with hepatitis C. A systematic review and critical analysis. Ann. Intern. Med. 2003;139(1):46-50.
10. Chen J, Röcken C, Treiber G, et al. Clinical implications of alphafetoprotein expression in gastric adenocarcinoma. Dig Dis 2003;21(4):357-362.
11. Daniele B, Bencivenga A, Megna AS, et al. Alpha-fetoprotein and ultrasonography screening for hepatocellular carcinoma. Gastroenterology 2004;127:108-112.
12. Stuart KE, Anand AJ, Jenkins RL. Hepatocellular Carcinoma in the United States. Prognostic features, treatment outcome, and survival. Cancer 1996;77,11:2217-2222.
13. Heimbach JK, Kulik LM, Finn RS, et al. AASLD Guidelines for the Treatment of Hepatocellular Carcinoma. Hepatology 2018;67(1):358-80.
14. Kokudo N, Hasegawa K, Akahane M, et al. Evidence-based Clinical Practice Guidelines for Hepatocellular Carcinoma: The Japan Society of Hepatology 2013 update (3rd JSH-HCC Guidelines). Hepatol Res 2015; 45:123-127.
15. Omata M, Cheng AL, Kokudo N, et al. Asia-Pacific clinical practice guidelines on the management of hepatocellular carcinoma: a 2017 update. Hepatol Int 2017;11:317-370.
16. Schlebusch H. Prenatal screening for Down’s syndrome. In: Thomas L (ed.). Clinical Laboratory Diagnosis, TH-Books, Frankfurt, 1st English edition 1998:1124-1125.
17. Cuckle HS, Wald NJ, Thompson SG. Estimating a woman’s risk of having a pregnancy associated with Down’s syndrome using her age and serum alpha-fetoprotein level. Br J Obstet Gynaecol 1987;94:387-402.
18. Reynolds TM, Penney MD. The mathematical basis of multivariate risk screening: with special reference to screening for Down’s syndrome associated pregnancy. Ann Clin Biochem 1989;26:452-458.
19. Cuckle HS, Wald NJ, Nanchahal K, et al. Repeat maternal serum alpha-fetoprotein testing in antenatal screening programmes for Down’s syndrome. Br J Obstet Gynaecol 1989;96:52-60.
20. Dunstan FDJ, Gray JC, Nix ABJ, et al. Detection rates and false positive rates for Down’s Syndrome screening: How precisely can they be Estimated and what factors influence their value? Statistics Medicine 1997;16:1481-1495.
21. Lamson SH, Hook B. Comparison of Mathematical Models for the Maternal Age Dependence of Down’s Syndrome Rates. Hum Genet Vol 1981;59:232-234.
22.Cuckle HS. Improved parameters for risk estimation in Down’s syndrome screening. Prenat Diagn 1995;15:1057-1065.
23. Llovet JM, Brú C, Bruix J. Prognosis of hepatocellular carcinoma: the BCLC staging classification. Semin Liver Dis 1999;19(3):329-338.
24. Bablok W, Passing H, Bender R, et al. A general regression procedure for method transformation. Application of linear regression procedures for method comparison studies in clinical chemistry, Part III. J Clin Chem Clin Biochem 1988 Nov;26(11):783-790.
Para más información acerca de los componentes, consultar el manual del usuario o el manual del operador del analizador, las hojas de aplicación y las metódicas correspondientes (disponibles en su país).
En la presente metódica se emplea como separador decimal un punto para distinguir la parte entera de la parte fraccionaria de un número decimal. No se utilizan separadores de millares.
Todo incidente grave que se haya producido en relación con el producto se comunicará al fabricante y a la autoridad competente del Estado Miembro en el que se encuentre el usuario y/o el paciente.
Para el resumen del informe de seguridad y funcionamiento, consulte: https://ec.europa.eu/tools/eudamed



