
DUPIXENT
DUPILUMAB
Solución inyectable
1 Caja, 2 Jeringa prellenada, 300/2 mg/ml
1 Caja, 1 Jeringa prellenada, 200/1.14 mg/ml
1 Caja, 2 Jeringa prellenada, 200/1.14 mg/ml
1 Caja, 1 Jeringa prellenada, 300/2 mg/ml
FORMA FARMACÉUTICA Y FORMULACIÓN:
La jeringa prellenada contiene:
Dupilumab 200 mg, 300 mg
Vehículo cbp 1.14 mL, 2 mL
Anticuerpo monoclonal lgG4 humano de origen ADN recombinante expresado en células de Ovario de Hámster Chino (CHO)
INDICACIONES TERAPÉUTICAS:
Dermatitis atópica: DUPIXENT® está indicado para el tratamiento de pacientes a partir de los 6 años con dermatitis atópica moderada a grave, cuya enfermedad no está controlada adecuadamente por terapias de prescripción tópicas o cuando dichas terapias no están recomendadas. DUPIXENT® puede ser utilizado con o sin terapia tópica (corticosteroides tópicos).
Asma: DUPIXENT® está indicado como un tratamiento complementario de mantenimiento en pacientes con asma moderada a grave a partir de los 12 años, con un fenotipo eosinofílico o con asma dependiente de corticosteroides orales.
Limitación de uso: DUPIXENT® no está indicado para el alivio del broncoespasmo agudo o el estado asmático.
Rinosinusitis crónica con poliposis nasal: DUPIXENT® está indicado en el tratamiento complementario de mantenimiento en pacientes adultos con un control inadecuado de rinosinusitis crónica con poliposis nasal (RSCcPN).
FARMACOCINÉTICA Y FARMACODINAMIA:
Dupilumab, un antagonista de los receptores alfa de la interleucina 4, es un anticuerpo monoclonal humano de la subclase de lgG4 que se une a la sub-unidad IL-4Rα e inhibe la señalización de IL-4 e IL-13. Dupilumab tiene un peso molecular aproximado de 147 kDa. Dupilumab es producida por tecnología recombinante del ADN, en el cultivo de suspensión de células de Ovario de Hámster Chino.
Mecanismo de acción: Dupilumab es un anticuerpo monoclonal lgG4 humano, que inhibe la señalización de la interleucina-4 (IL-4) e interleucina-13 (IL-13), por medio de la unión específica a la sub-unidad IL-4Rα compartida por los complejos receptores de IL-4 e IL-13. Dupilumab inhibe la señalización de IL-4 a través del receptor tipo I y ambas señalizaciones de IL-4 e IL-13 a través del receptor tipo II.
El bloqueo de IL-4Rα con dupilumab inhibe las respuestas inducidas por las citocinas IL-4 e IL-13, incluyendo la liberación de citocinas proinflamatorias, quimiocinas e lgE.
La inflamación es un importante componente en la patogénesis del asma, la dermatitis atópica y RSCcPN. Múltiples tipos de células que expresan IL-4Rα (por ejemplo, mastocitos, eosinófilos, macrófagos, linfocitos, células epiteliales, células caliciformes) y mediadores de la inflamación (por ejemplo, histamina, eicosanoides, leucotrienos, quimiocinas y citocinas) están involucradas en el proceso de inflamación. El bloqueo de IL-4Rα con dupilumab inhibe la respuesta inflamatoria inducida por las citocinas IL-4 e IL-13, incluyendo la liberación de citocinas proinflamatorias, quimiocinas, óxido nítrico e lgE, sin embargo, el mecanismo de acción de dupilumab en el asma no ha sido definitivamente establecido.
Farmacodinamia: De manera consistente con la inhibición de la señalización de IL-4 e IL-13 el tratamiento con dupilumab disminuyó notablemente la fracción exhalada de óxido nítrico (FeNO) y las concentraciones circulantes de eotaxina-3, lgE total, lgE alergeno específica, TARC y periostina en sujetos con asma en relación con el placebo. Estas reducciones en los biomarcadores de inflamación fueron similares para los regímenes de 300 mg cada 2 semanas y 200 mg cada 2 semanas. Estos marcadores estuvieron cerca de la supresión máxima después de 2 semanas de tratamiento, a excepción de la lgE que disminuyó más lentamente. Estos efectos se mantuvieron durante todo el tratamiento. La mediana del porcentaje de reducción desde los valores iniciales en las concentraciones totales de lgE con los tratamientos con dupilumab fue del 52% en la semana 24 (estudio AS 1) y del 70% en la semana 52 (estudio AS 2). Para FeNO, la reducción porcentual media desde el inicio en la Semana 2 fue de 35% y 24% en los Ensayos 1 y 2 de AS, respectivamente, y en la población general de seguridad, el nivel medio de FeNO disminuyó a 20 ppb.
Farmacocinética: La farmacocinética de dupilumab es similar en sujetos con dermatitis atópica, asma y RSCcPN.
Absorción: Posterior a la administración de una dosis inicial por vía subcutánea (SC) de 600 mg, 400 mg o 300 mg, dupilumab alcanzó sus concentraciones máximas (Cmáx) en promedio ± DE de 70.1 ± 24.1 μg/mL, 41.8 ± 12.4 μg/mL o 30.5 ± 9.39 μg/mL, respectivamente, aproximadamente 1 semana después de la dosis.
Las concentraciones constantes se alcanzaron hacia la semana 16 después de la administración de la dosis de inicio de 600 mg y la dosis de 300 mg semanalmente (el doble de la frecuencia recomendada de dosificación) o cada 2 semanas, o dosis inicial de 400 mg y dosis de 200 mg cada 2 semanas, o 300 mg cada dos semanas (Q2W) sin dosis de carga. A través de los estudios clínicos, la media ± DE de las concentraciones mínimas constantes estuvieron en el rango desde 60.3 ± 35.1 μg/mL hasta 80.2 ± 35.3 μg/mL con 300 mg administradas cada 2 semanas, desde 173 ± 75.9 μg/mL hasta 193 ± 77.0 μg/mL con 300 mg administrada semanalmente y desde 29.2 ± 18.7 a 36.5 ± 22.2 mg/L con 200 mg administrados cada dos semanas (Q2W).
La biodisponibilidad de dupilumab posterior a una dosis SC es similar entre pacientes con dermatitis atópica, asma y RSCcPN, que varía entre 61% y 64%.
Distribución:
El volumen de distribución total estimado fue aproximadamente de 4.8 ± 1.3 L.
Eliminación: La vía metabólica de dupilumab no ha sido caracterizada. Como anticuerpo monoclonal humano lgG4, se espera que dupilumab sea degradado hacia péptidos y aminoácidos pequeños por catabolismo, en la misma forma que la lgG endógena. Después de la última dosis del estado estable de 300 mg cada 2 semanas. 300 mg cada semana (QW) o 200 mg cada 2 semanas de dupilumab, la mediana de los tiempos hasta las concentraciones no detectables (< 78 ng/mL) fueron de 10-12, 13 y 9 semanas, respectivamente.
Linealidad de la dosis: Dupilumab exhibió una farmacocinética no lineal mediada por el objetivo, con exposiciones que se incrementan en mayor proporción a la dosis. La exposición sistémica se incrementó en 30 veces cuando la dosis fue aumentada 8 veces después de una dosis única de dupilumab desde 75 mg hasta 600 mg (es decir, 0.25 veces a 2 veces la dosis recomendada).
Peso: Las concentraciones mínimas de dupilumab fueron menores en los sujetos con peso corporal más alto.
Edad: Según el análisis farmacocinético de la población, la edad no afectó la eliminación de dupilumab.
lnmunogenicidad: El desarrollo de anticuerpos hacia dupilumab fue asociado con concentraciones menores de dupilumab en el suero. Pocos sujetos que tuvieron títulos altos de anticuerpos también tuvieron concentraciones no detectables de dupilumab en el suero.
Poblaciones específicas:
Pacientes geriátricos: En sujetos que tienen 65 años y mayores, el promedio ± DE de las concentraciones mínimas continuas de dupilumab fueron de 69.4 ± 31.4 μg/mL y 166 ± 62.3 μg/mL, respectivamente, con 300 mg administrados cada 2 semanas y semanalmente y 39.7 ± 21.7 μg/mL para 200 mg administrados cada 2 semanas.
Pacientes pediátricos:
Dermatitis atópica: Para adolescentes de 12 a 17 años de edad con dermatitis atópica que fueron administrados con 200 mg (< 60 Kg) o 300 mg (≥ 60 Kg) cada dos semanas, la media ± DE de la concentración mínima en estado estable de dupilumab fue 54.5 ± 27.0 μg/mL.
Para niños de 6 a 11 años con dermatitis atópica que reciben dosis en semanas alternas (cada dos semanas) con 200 mg (≥ 30 kg) o dosis de 300 mg cada cuatro semanas (Q4W) (< 30 kg), la media ± DE de la concentración mínima en estado estable fue de 86.0 ± 34.6 μg/mL y 98.7 ± 33.2 μg/mL, respectivamente.
Asma:
Un total de 107 adolescentes de 12 a 17 años con asma fueron reclutados en el estudio AS 2, la media ± DE de las concentraciones mínimas en estado estable de dupilumab fueron 107 ± 51.6 μg/mL y 46.7 ± 26.9 μg/mL, respectivamente, para 300 mg o 200 mg administrados cada 2 semanas.
Se evaluó la seguridad y eficacia a largo plazo de DUPIXENT® en un estudio abierto con 89 pacientes adolescentes con asma de moderada a grave (estudio TRAVERSE). En este estudio, los pacientes tuvieron seguimiento durante un máximo de 96 semanas, lo que resultó en 99 pacientes-años de exposición acumulada a DUPIXENT®. El perfil de seguridad de DUPIXENT® en el estudio TRAVERSE fue coherente con el perfil de seguridad observado en estudios pivotales para el asma durante un máximo de 52 semanas de tratamiento. No se identificaron reacciones adversas adicionales. En este estudio, el beneficio clínico de DUPIXENT®, incluyendo la reducción de las exacerbaciones y en la mejora de la función pulmonar observada en estudios pivotales para de asma, se mantuvo hasta las 96 semanas.
Insuficiencia renal o hepática: No se han realizado estudios formales del efecto de la insuficiencia hepática o renal sobre la farmacocinética de dupilumab.
Estudios de interacción de medicamentos: No se espera un efecto de dupilumab sobre la farmacocinética de medicamentos administrados conjuntamente. Con base en el análisis de la población, los medicamentos comúnmente administrados conjuntamente no tuvieron efecto sobre la farmacocinética de DUPIXENT® en pacientes con asma moderado a grave.
Sustratos del citocromo P450: Los efectos de dupilumab en la farmacocinética de midazolam (metabolizado por CYP3A4), warfarina (metabolizada por CYP2C9), omeprazol (metabolizado por CYP2C19), metoprolol (metabolizado por CYP2D6) y cafeína (metabolizado por CYP1A2) se evaluaron en un estudio con 12-13 sujetos evaluables con dermatitis atópica (una dosis administrada vía SC de 600 mg seguida de 300 mg semanalmente durante seis semanas). No se observaron cambios clínicamente significativos en el ABC (área bajo la curva). El mayor efecto se observó para el metoprolol (CYP2D6) con un aumento en el ABC del 29%.
Estudios clínicos:
Dermatitis atópica en adultos: Tres estudios aleatorizados, doble ciego, controlados con placebo (Estudios 1, 2 y 3) reclutaron a un total de 2119 sujetos de 18 años de edad y mayores, con dermatitis atópica (DA) moderada a grave, no controlados adecuadamente con medicamento(s) tópico(s). La severidad de la enfermedad fue definida por una calificación de ≥ 3 en la escala de Evaluación Global del Investigador (IGA) en la evaluación global de las lesiones de DA, usando una escala de 0 a 4, así como una calificación > 16 en la escala del Índice de Severidad y Área del Eccema (EASI) usando una escala de 0 a 72, y un área de superficie corporal mínima afectada de ≥ 10%. En el registro basal, 59% de los sujetos eran hombres, 67% eran blancos, 52% de los sujetos tenían una calificación basal de 3 en la IGA (DA moderada) y 48% de los sujetos tenían una IGA basal de 4 (DA grave). La media basal de la calificación de EASI fue de 33 y la Escala de Clasificación Numérica (NRS) del prurito máximo basal, promediada semanalmente fue de 7 de una escala de 0-10.
En los tres estudios, los sujetos en el grupo de DUPIXENT® recibieron inyecciones subcutáneas de DUPIXENT® en dosis de 600 mg en la semana 0, seguido por 300 mg cada dos semanas. En los estudios de monoterapia (Estudios 1 y 2), los sujetos recibieron DUPIXENT® o placebo durante 16 semanas.
En el estudio de terapia concomitante (Estudio 3), los sujetos recibieron DUPIXENT® o placebo concomitantemente con corticoesteroides tópicos (TCS) y los inhibidores de calcineurina tópicos se usaron según fuese necesario únicamente para las áreas problema, tales como la cara, cuello, áreas intertriginosas y genitales, durante 52 semanas.
Los tres estudios evaluaron el desenlace primario consistente en el cambio desde el registro basal hasta la semana 16 en la proporción de sujetos con una calificación de 0 (limpio) o 1 (casi limpio) en la escala IGA y por lo menos 2 puntos de mejoría. Otros parámetros de evaluación incluyeron la proporción de sujetos con EASl-75 (mejoría de por lo menos 75% en la calificación de EASI desde el registro basal), y una reducción en el prurito, definido por una mejoría de por lo menos 4 puntos en la NRS del prurito máximo desde el registro basal hasta la semana 16.
Respuesta clínica en la semana 16 (Estudios 1, 2 y 3): Los resultados de los estudios de monoterapia de DUPIXENT® (Estudios 1 y 2) y del estudio de DUPIXENT® con corticoesteroides tópicos concomitantes (TCS) (Estudio 3) se presentan en la Tabla 1.
Tabla 1. Resultados de eficacia de DUPIXENT® con o sin corticoesteroides tópicos concomitantes en la semana 16 (FAS)
|
Estudio 1 |
Estudio 2 |
Estudio 3 |
||||
|
DUPIXENT® 300 mg cada 2 semanas |
Placebo |
DUPIXENT® 300 mg cada 2 semanas |
Placebo |
DUPIXENT® 300 mg cada 2 semanas + TCS |
Placebo + TCS |
|
|
Número de sujetos aleatorizados (FAS)ª |
224 |
224 |
233 |
236 |
106 |
315 |
|
IGA 0 o 1b,c |
38% |
10% |
36% |
9% |
39% |
12% |
|
EASl-75C |
51% |
15% |
44% |
12% |
69% |
23% |
|
EASI-90c |
36% |
8% |
30% |
7% |
40% |
11% |
|
Número de sujetos con calificación ≥ 4 en la escala NRS de prurito |
213 |
212 |
225 |
221 |
102 |
299 |
|
Evaluación del prurito en la escala NRS (mejoría ≥ 4 puntos)c |
41% |
12% |
36% |
10% |
59% |
20% |
a Serie de análisis completo (FAS) incluye a todos los sujetos aleatorizados.
b El paciente respondedor fue definido como un sujeto con una calificación de 0 o 1 en la escala IGA ("limpio" o "casi limpio"), con una reducción de ≥ 2 puntos en la escala de 0 a 4 de IGA.
Los sujetos quienes recibieron tratamiento de rescate o con datos faltantes fueron considerados como no respondedores NRS: Numerical-Rating Scale (Escala de Clasificación Numérica).
Figura 1. Proporción de sujetos con ≥ 4 puntos de mejoría en la escala NRS de prurito máximo, en los Estudios 1a y 2a (FAS)b
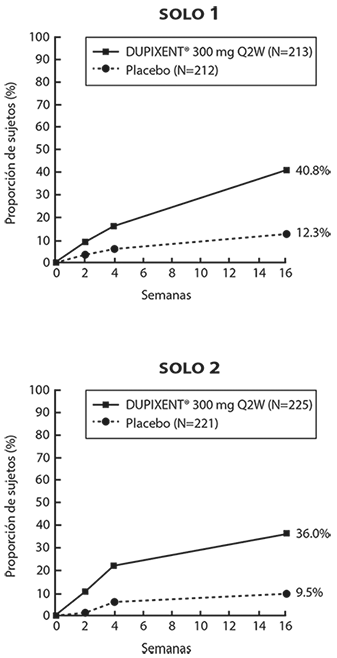
a En el análisis primario de los parámetros de eficacia, los sujetos quienes recibieron tratamiento de rescate o con datos faltantes fueron considerados como no respondedores.
b La serie de análisis completa (FAS) incluye a todos los sujetos aleatorizados.
En el Estudio 3, de los 421 sujetos, 353 habían estado en el estudio durante 52 semanas en el momento del análisis de datos. De estos 353 sujetos, los respondedores en la semana 52 representan una mezcla de sujetos quienes mantuvieron su eficacia desde la semana 16 (es decir, el 53% de los sujetos respondedores a DUPIXENT® con IGA 0 o 1 en la semana 16 permanecieron como respondedores en la semana 52) y los sujetos que no fueron respondedores en la semana 16, quienes posteriormente respondieron al tratamiento (es decir, 24% de los sujetos no respondedores a DUPIXENT® con lGA 0 o 1 en la semana 16, se hicieron respondedores en la semana 52). Los resultados de los análisis de soporte de los 353 sujetos en el estudio de DUPIXENT® con TCS (Estudio 3) se presentan en Tabla 2.
Tabla 2. Resultados de eficacia (IGA 0 o 1) de DUPIXENT® con TCS concomitantes en la semana 16 y 52
|
DUPIXENT® 300 mg cada 2 semanas + TCS |
Placebo + TCS |
|
|
Número de sujetosa |
89 |
264 |
|
Respondedoresb,c en la semana 16 y 52 |
22% |
7% |
|
Respondedores en la semana 16, pero no respondedores en la semana 52 |
20% |
7% |
|
No respondedores en la semana 16 y respondedores en la semana 52 |
13% |
6% |
|
No respondedores en la semana 16 y 52 |
44% |
80% |
|
Tasa global de respondedoresb,c en la semana 52 |
36% |
13% |
a En el Estudio 3, de los 421 sujetos aleatorizados y tratados, 68 sujetos (16%) no habían estado en el estudio durante 52 semanas en el momento del análisis de datos.
b El respondedor fue definido como un sujeto con una calificación de lGA de 0 o 1 ("limpio" o "casi limpio") con una reducción de ≥ 2 puntos en una escala de 0-4 de IGA.
c Los sujetos quienes recibieron tratamiento de rescate con datos omitidos fueron considerados como no respondedores.
Los efectos del tratamiento por subgrupos (peso, edad, género, raza y tratamiento previo, incluyendo inmunosupresores) en Estudios 1, 2 y 3 fue generalmente consistente con los resultados de la población global del estudio.
En los Estudios 1, 2 y 3, un tercer brazo de tratamiento aleatorizado de DUPIXENT® en dosis de 300 mg cada semana no demostró beneficio adicional del tratamiento sobre la dosis de DUPIXENT® de 300 mg cada 2 semanas.
Los sujetos del Estudio 1 (SOLO 1) y Estudio 2 (SOLO 2) que tenían un IGA 0 o 1 con una reducción de ≥ 2 puntos se volvieron a asignar al estudio 5. El Estudio 5 (SOLO CONTINUE) evaluó múltiples regímenes de dosis de monoterapia con DUPIXENT® para mantener la respuesta al tratamiento. El estudio incluyó sujetos aleatorizados para continuar con DUPIXENT® 300 mg cada 2 semanas (62 sujetos) o cambiar a placebo (31 sujetos) durante 36 semanas. Las respuestas IGA 0 o 1 en la semana 36 fueron las siguientes: 33 (53%) en el grupo cada 2 semanas y 3 (10%) en el grupo placebo.
Respuesta clínica en pacientes no controlados adecuadamente con ciclosporinas, intolerantes a las ciclosporinas o para quienes el tratamiento con ciclosporinas no fue recomendable (estudio CAFE):
El estudio CAFE evaluó la eficacia de DUPIXENT® en comparación con placebo durante un periodo de tratamiento de 16 semanas, administrado con TCS concomitantes, en pacientes adultos con DA que no fueron controlados adecuadamente con ciclosporina oral o que son intolerantes a ésta o para quienes este tratamiento está actualmente contraindicado o no es médicamente recomendable.
Se reclutó un total de 325 pacientes, con 210 pacientes que habían sido expuestos previamente a ciclosporina y 115 pacientes que nunca habían sido expuestos a ciclosporina debido a que el tratamiento con ciclosporina no era médicamente recomendable. La edad media fue 38.4 años, 38.8% fueron mujeres, la puntuación EASI media basal fue 33.1, el BSA media fue 55.7, la NRS de prurito semanal basal promedio fue 6.4, la puntuación SCORAD media basal fue 67.2 y el DLQI medio basal fue 13.8.
El criterio de valoración primario fue la proporción de pacientes con EASI-75 a la semana 16. Los criterios de valoración primarios y secundarios para el estudio CAFE de 16 semanas se resumen en la Tabla 3.
Tabla 3. Resultados de los criterios de valoración primarios y secundarios en el estudio CAFÉ.
|
Placebo + TCS |
DUPIXENT® 300 mg cada 2 semanas + TCS |
DUPIXENT® 300 mg cada semanas + TCS |
|
|
Pacientes aleatorizados |
108 |
107 |
110 |
|
EASl-75% de respondedores |
29.6% |
62.6% |
59.1% |
|
EASl, media LS del % de cambio desde la basal (+/-SE) |
-46.6 (2.76) |
-79.8 (2.59) |
-78.2 (2.55) |
|
NRS de prurito, media LS del % de cambio desde la asal (+/-SE) |
-25.4% (3.39) |
-53.9% (3.14) |
-51.7% (3.09) |
|
SCORAD, media LS del % de cambio desde la base (+/-SE) |
-29.5% (2.55) |
-62.4% (2.48) |
-58.3% (2.45) |
|
DLQI, media LS del cambio desde la basal (SE) |
-4.5 (0.49) |
-9.5 (0.46) |
-8.8 (0.45) |
En el subgrupo de pacientes que se asemejó a la población del estudio CAFE dentro del estudio CHRONOS de 52 semanas, 69.6% de los pacientes tratados con DUPIXENT® 300 mg cada 2 semanas alcanzó la EASI-75 contra 18.0% de los pacientes tratados con placebo en la semana 16 y 52.5% de los pacientes tratados con DUPIXENT® 300 mg cada 2 semanas contra 18.6% de los tratados con placebo en la semana 52.
En este subgrupo, el porcentaje de cambio en la NRS de prurito desde la basal fue -51.4% contra -30.2% a la semana 16 y -54.8% contra -30.9% a la semana 52, para los grupos de DUPIXENT® 300 mg cada 2 semanas y placebo, respectivamente.
Dermatitis atópica en adolescentes (12 a 17 años de edad): La eficacia y seguridad de la monoterapia con DUPIXENT® en sujetos adolescentes se evaluó en un estudio multicéntrico, aleatorizado, doble ciego, controlado con placebo [estudio 6 (AD-1526)] en 251 sujetos adolescentes de 12 a 17 años de edad con DA moderada a grave definida por una puntuación de IGA ≥ 3 en la evaluación general de las lesiones por DA una escala de gravedad de 0 a 4, una puntuación EASI ≥ 16 en una escala de 0 a 72 y un mínimo de BSA afectada de ≥ 10%. Los sujetos elegibles inscritos en este ensayo tuvieron una respuesta previa inadecuada a la medicación tópica.
Los sujetos del grupo DUPIXENT® recibieron una dosis inicial de 400 mg en la semana 0, seguidos de 200 mg cada 2 semanas para sujetos con un peso inicial de < 60 kg o una dosis inicial de 600 mg en la semana 0, seguidos de 300 mg cada 2 semanas para sujetos con un valor inicial de peso ≥ 60 kg durante 16 semanas. DUPIXENT® se administró mediante inyección subcutánea. Si era necesario para controlar los síntomas intolerables, los sujetos podían recibir tratamiento de rescate a discreción del investigador. Los sujetos que recibieron tratamiento de rescate se consideraron no respondedores.
En el estudio 6, la edad promedio fue de 14.5 años, la mediana del peso fue de 59.4 kg, el 41% de los sujetos eran mujeres, el 63% eran blancos, el 15% eran asiáticos y el 12% eran negros. Al inicio del estudio, el 46% de los sujetos tenía una puntuación IGA de 3 (DA moderada), el 54% tenía una puntuación IGA de 4 (DA grave), la BSA afectada promedio fue del 57% y el 42% había recibido inmunosupresores sistémicos anteriormente. Además, al inicio del estudio, la puntuación media de EASI fue de 36, y el promedio semanal de prurito máximo NRS fue de 8 en una escala de 0-10. En general, el 92% de los sujetos tenía al menos una afección alérgica comórbida, el 66% tenía rinitis alérgica, el 54% tenía asma y el 61% tenía alergias alimentarias.
Los criterios de valoración primarios fueron la proporción de sujetos con IGA 0 (claro) o 1 (casi claro) con al menos una mejora de 2 puntos, y la proporción de sujetos con EASl-75 (mejora de al menos el 75% en EASI), desde el inicio hasta la semana 16. Otros resultados evaluados incluyeron la proporción de sujetos con EASl-50 o EASl-90 (mejora de al menos 75% o 90% en EASI desde el inicio, respectivamente) y reducción de la picazón según lo medido por el pico prurito NRS.
Los resultados de eficacia en la semana 16 para el estudio 6 son presentados en la Tabla 4.
Tabla 4. Resultados de eficacia de DUPIXENT® en el estudio 6 en la semana 16 (FAS)
|
Placebo N=85a |
DUPIXENT®d 200 mg (< 60 Kg) o 300 mg (≥ 60 Kg) cada 2 semanas N=82a |
|
|
IGA 0 o 1b,c |
2% |
24% |
|
EASl-50c |
13% |
61% |
|
EASl-75c |
8% |
42% |
|
EASl-90c |
2% |
23% |
|
NRS de Prurito, media LS % del cambio desde el valor inicial (+/-DE) |
-19% (4.1) |
-48% (3.4) |
|
Pico del NRS de prurito (> 4-puntos de meiora)c |
5% |
37% |
ª El conjunto de análisis completo (FAS) incluye todos los sujetos asignados al azar.
b El respondedor se definió como un sujeto con IGA 0 o 1 ("claro" o "casi claro") con una reducción de ≥ 2 puntos en una escala de 0-4 IGA.
c Los sujetos que recibieron tratamiento de rescate o con datos faltantes se consideraron como no respondedores (59% y 21% en los grupos placebo y DUPIXENT®, respectivamente).
d En la semana 0, los sujetos recibieron 400 mg (peso basal < 60 kg) o 600 mg (peso basal ≥ 60 kg) de DUPIXENT®.
Un porcentaje mayor de sujetos asignados al azar a placebo necesitó tratamiento de rescate (corticosteroides tópicos, corticosteroides sistémicos o inmunosupresores sistémicos no esteroideos) en comparación con el grupo DUPIXENT® (59% y 21%, respectivamente).
Una proporción significativamente mayor de sujetos asignados al azar a DUPIXENT® logró una rápida mejoría en la NRS de prurito en comparación con el placebo (definida como una mejora > 4 puntos tan pronto como en la Semana 4; nominal de p<0.001) y la proporción de sujetos que respondieron en la NRS de prurito continuó aumentando a lo largo del periodo de tratamiento (ver Figura 2). La mejora en la NRS de prurito se produjo junto con la mejora de los signos objetivos de la dermatitis atópica.
Figura 2. Proporción de sujetos adolescentes con una mejora de ≥ 4 puntos en la NRS de prurito en el estudio 6a (FAS)
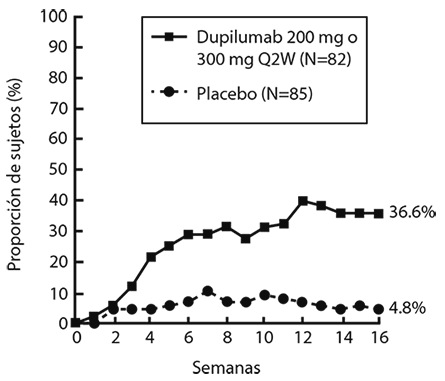
a En los análisis primarios de los criterios de valoración de eficacia, los sujetos que recibieron tratamiento de rescate o con datos faltantes se consideraron no respondedores.
b El conjunto de análisis completo (FAS) incluye todos los sujetos asignados al azar.
La eficacia a largo plazo de DUPIXENT® en pacientes adolescentes con DA de moderada a grave que habían participado en estudios clínicos previos de DUPIXENT® se evaluó en un estudio de extensión de etiqueta abierta (estudio 7). Los datos de eficacia de este estudio sugieren que el beneficio clínico proporcionado en la semana 16 se mantuvo hasta la semana 52.
Dermatitis atópica en niños (6 a 11 años de edad): Se evaluó la eficacia y seguridad del uso concomitante de DUPIXENT® con TCS en sujetos pediátricos en un estudio multicéntrico, aleatorizado, doble ciego y controlado con placebo (Estudio 8) en 367 sujetos de 6 a 11 años de edad, con DA grave definida por un Puntuación IGA de 4 (escala de 0 a 4), puntuación EASI ≥ 21 (escala de 0 a 72) y afectación mínima de BSA de ≥ 15%. Los sujetos elegibles a reclutamiento en este estudio tenían una respuesta inadecuada al tratamiento tópico previo. La inscripción se estratificó por peso inicial (< 30 kg; ≥ 30 kg).
Los sujetos del grupo DUPIXENT® cada 4 semanas + TCS recibieron una dosis inicial de 600 mg el día 1, seguida de 300 mg cada 4 semanas, desde la semana 4 hasta la semana 12, independientemente del peso.
Los sujetos del grupo DUPIXENT cada 2 semanas + TCS con un peso inicial de < 30 kg recibieron una dosis inicial de 200 mg el día 1, seguida de 100 mg cada 2 semanas, desde la semana 2 hasta la semana 14, y los sujetos con un peso inicial de ≥ 30 kg recibieron una dosis inicial dosis de 400 mg el día 1, seguida de 200 mg cada 2 semanas desde la semana 2 hasta la semana 14. Se permitió que los sujetos recibieran tratamiento de rescate a discreción del investigador.
Los sujetos que recibieron tratamiento de rescate se consideraron no respondedores.
En el Estudio 8, la edad media fue de 8.5 años, la mediana del peso fue de 29.8 kg, el 50% de los sujetos eran mujeres, el 69% eran blancos, el 17% eran negros y el 8% eran asiáticos.
Al inicio del estudio, la afectación media del promedio de superficie corporal afectada (BSA) fue del 58% y el 17% había recibido previamente inmunosupresores sistémicos no esteroideos. Además, al inicio del estudio, la puntuación media de EASI fue de 37.9 y la media semanal de la peor puntuación diaria de prurito fue de 7.8 en una escala de 0 a 10. En general, el 92% de los sujetos tenían al menos una condición alérgica comórbida; el 64% tenía alergias alimentarias, el 63% tenía otras alergias, el 60% tenía rinitis alérgica y el 47% tenía asma. La variable principal fue la proporción de pacientes con IGA 0 (claro) o 1 (casi claro) en la semana 16. Otros resultados evaluados incluyeron la proporción de pacientes con EASI-75 o EASI-90 (mejora de al menos 75% o 90% en EASI desde el inicio, respectivamente), el porcentaje de cambio en EASI desde el inicio hasta la semana 16, y reducción de la picazón medida por el pico de prurito NRS (mejora de ≥ 4 puntos).
La Tabla 5 presenta los resultados por estrato de peso inicial para los regímenes de dosis aprobados.
Tabla 5. Resultados de eficacia de DUPIXENT® con TCS concomitante en el ensayo 8 en la semana 16 (FAS)ª.
|
DUPIXENT® 300 mg cada 4 semanas (Q4W) + TCSd (N = 61) |
Placebo + TCS (N = 61) |
DUPIXENT®d 200 mg cada 2 semanas (Q2W) + TCSe (N = 59) |
Placebo + TCS (N = 61) |
|
|
< 30 Kg |
< 30 Kg |
≥ 30 Kg |
≥ 30 Kg |
|
|
IGA 0 o 1b,c |
30% |
13% |
39% |
10% |
|
EASl-75c |
75% |
28% |
75% |
26% |
|
EASl-90c |
46% |
7% |
36% |
8% |
|
Prurito pico NRS (mejoría ≥ 4 puntos)c |
54% |
12% |
61% |
13% |
ª El conjunto de análisis completo (FAS) incluye todos los sujetos asignados al azar.
b El respondedor se definió como un sujeto con IGA 0 o 1 ("limpio" o "casi limpio").
c Los sujetos que recibieron tratamiento de rescate o con datos faltantes se consideraron como no respondedores.
d En el día 1, los sujetos recibieron 600 mg de DUPIXENT®.
e En el día 1, los sujetos recibieron 200 mg (peso inicial < 30 kg) o 400 mg (peso inicial ≥ 30 kg) de DUPIXENT®.
Una mayor proporción de sujetos aleatorizados a DUPIXENT® + TCS logró una mejora en el pico de prurito NRS en comparación con placebo + TCS (definido como una mejoría de ≥ 4 puntos en la semana 16), ver figura 3.
Figura 3. Proporción de sujetos pediátricos con una mejora de ≥ 4 puntos en la escala numérica de prurito (NRS) a la semana 16 en el estudio 8a (FAS).b
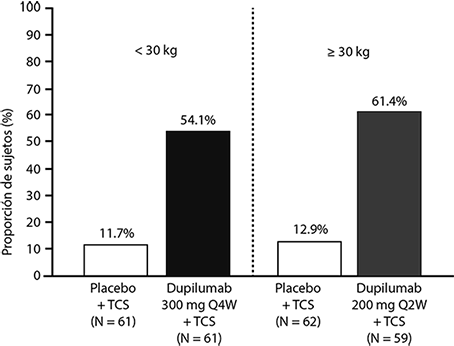
a En los análisis primarios de los criterios de valoración de eficacia, los sujetos que recibieron tratamiento de rescate o con datos faltantes se consideraron no respondedores.
b El conjunto de análisis completo (FAS) incluye todos los sujetos asignados al azar.
Asma: El programa de desarrollo de asma incluyó tres estudios aleatorizados, doble ciego, controlados por placebo, de grupos paralelos y multicéntricos [Estudios AS 1 (DRl12544), AS 2 (QUEST) y AS 3 (VENTURE)] de 24 a 52 semanas de duración del tratamiento, en los que se reclutó un total de 2888 sujetos (12 años de edad y mayores). Para los sujetos reclutados en los estudios AS 1 y 2 se requirió que tuvieran antecedentes de 1 o más exacerbaciones de asma que requirieron tratamiento con corticosteroides sistémicos o visita al departamento de urgencias u hospitalización para el tratamiento del asma en el año previo al ingreso al estudio. Los sujetos reclutados en el Estudio AS 3 requirieron dependencia de corticosteroides orales diarios además de uso regular de dosis altas de corticosteroides inhalados más un controlador adicional. En los 3 estudios, los sujetos fueron reclutados sin requerir un mínimo en el conteo de eosinófilos sanguíneos basales. En los estudios AS 2 y 3, se excluyeron los sujetos con niveles basales de eosinófilos sanguíneos de > 1500 células/μL (< 1.3%). DUPIXENT® fue administrado como adyuvante al tratamiento de asma inicial. Los sujetos que continuaron con tratamiento de asma inicial durante todo el tiempo de los estudios, excepto en el estudio AS 3 en el que la dosis de OCS se redujo como se describe a continuación.
Estudio AS 1: El estudio AS 1 fue un estudio de rango de dosis de 24 semanas que incluyó 776 sujetos (18 años de edad y mayores). DUPIXENT® en comparación con placebo fue evaluado en sujetos adultos con asma de moderada a grave con media o alta dosis de corticosteroide inhalado y un beta agonista de acción prolongada. Los sujetos fueron aleatorizados para recibir 200 mg (N=150) o 300 mg (N=157) de DUPIXENT® cada dos semanas o 200 mg (N=154) o 300 mg (N=157) de DUPIXENT® cada 4 semanas después de una dosis inicial de 400 mg, 600 mg o placebo (N=158), respectivamente. El criterio de valoración primario fue la media del cambio desde el valor basal hasta la semana 12 en FEV1 (L) en sujetos con un recuento basal de eosinófilos sanguíneos (≥ 300 células/μL). Otros criterios de valoración incluyeron el cambio porcentual desde el valor basal en el FEV1 y la tasa anualizada de eventos de exacerbación de asma grave durante el periodo de tratamiento controlado con placebo de 24 semanas. Los resultados se evaluaron en la población total y los subgrupos basados en el recuento basal de eosinófilos sanguíneos (≥ 300 células/μL y < 300 células/μL). Los criterios de valoración secundarios adicionales incluyeron la media del cambio de las tasas basales y del respondedor en los pacientes reportados en el Cuestionario de Control del Asma (ACQ-5) y del Cuestionario de Calidad de Vida en Asma, puntajes de versión estandarizada [AQLQ (S)].
Estudio AS 2: El estudio AS 2 fue un estudio de 52 semanas que incluyó 1902 sujetos (12 años de edad y mayores). DUPIXENT® en comparación con placebo fue evaluado en 107 adolescentes y 1795 adultos con asma moderada a grave con media o alta dosis de corticosteroide inhalado (lCS) y un mínimo de uno y hasta dos medicamentos controladores adicionales. Los sujetos fueron aleatorizados para recibir 200 mg (N=631) o 300 mg (N=633) de DUPIXENT® cada 2 semanas (o un placebo correspondiente para 200 mg [N=317] o 300 mg [N=321] cada 2 semanas) después de una dosis inicial de 400 mg, 600 mg o placebo respectivamente. Los criterios de valoración primarios fueron la tasa anualizada de eventos de exacerbación grave durante el periodo controlado por placebo de 52 semanas y el cambio desde el basal en FEV1 pre-broncodilatador en la semana 12 en la población total (sin restricción por conteos mínimos basales de eosinófilos en sangre). Los criterios de valoración secundarios adicionales incluyeron las tasas anualizadas de exacerbación grave y FEV1, en pacientes con diferentes niveles basales de eosinófilos en sangre, así como la tasa del respondedor en los puntajes ACQ-5 y AQLQ(S).
Estudio AS 3: El estudio AS 3 fue un estudio de reducción de corticosteroides orales de 24 semanas en 210 sujetos con asma que requirieron corticosteroides orales diarios además del uso regular de altas dosis de corticosteroides inhalados más un controlador adicional. Después de optimizar la dosis de OCS durante el periodo de selección, los sujetos recibieron 300 mg de DUPIXENT® (N=103) o placebo (N=107) una vez cada 2 semanas durante 24 semanas después de una dosis inicial de 600 mg o placebo. Los sujetos continuaron recibiendo su medicamento existente para el asma durante el estudio; sin embargo, su dosis de OCS, la cual se redujo cada 4 semanas durante la fase de reducción de OCS (semana 4-20), siempre que se mantuviera el control del asma. El criterio de valoración primario fue la reducción porcentual de la dosis de corticosteroides orales en las semanas 20 a 24 en comparación con la dosis basal, mientras se mantiene el control del asma en la población total (sin restricción por conteos mínimos basales de eosinófilos en sangre). Los criterios de valoración secundarios adicionales incluyeron la tasa anualizada de episodios de exacerbación grave durante el periodo de tratamiento y la tasa de respondedores en los puntajes ACQ-5 y AQLQ (S).
La demografía y las características basales de estos 3 ensayos se proporcionan en la Tabla 6 a continuación.
Tabla 6. Características demográficas y basales de los ensayos de asma
|
Parámetro |
Estudio 1 (N=776) |
Estudio 2 (N=1902) |
Estudio 3 (N=210) |
|
Media de edad (años) (DE) |
49 (13) |
48 (15) |
51 (13) |
|
% Mujeres |
63 |
63 |
61 |
|
% Blancos |
78 |
83 |
94 |
|
Duración del asma (años), media (± DE) |
22 (15) |
21 (15) |
20 (14) |
|
No fumadores (%) |
77 |
81 |
81 |
|
Media de exacerbaciones del año previo (± DE) |
2.2 (2.1) |
2.1 (2.2) |
2.1 (2.2) |
|
Uso de alta dosis de ICS (%) |
50 |
52 |
89 |
|
FEV1 (L) antes de la dosis al inicio (± DE) |
1.84 (0.54) |
1.78 (0.60) |
1.58 (0.57) |
|
Media del porcentaje de FEV1 predicho al inicio (%) (± DE) |
61 (11) |
58 (14) |
52 (15) |
|
% Reversibilidad (± DE) |
27 (15) |
26 (22) |
19 (23) |
|
% Total de antecedentes médicos atópicos |
73 |
78 |
72 |
|
(%AD, % NP, % AR) |
(8, 11, 62) |
(10, 13, 69) |
(8, 21, 56) |
|
Media de ppb de FeNO (± DE) |
39 (35) |
35 (33) |
38 (31) |
|
Media de lgE total IU/mL (± DE) |
435 (754) |
432 (747) |
431 (776) |
|
Media del conteo basal de eosinófilos (± DE) células/μL |
350 (430) |
360 (370) |
350 (310) |
ICS=corticosteroide inhalado; FEV1=volumen espiratorio forzado en 1 segundo; AD=dermatitis atópica; NP=poliposis nasal; AR=rinitis alérgica; FeNO=fracción de óxido nítrico exhalado.
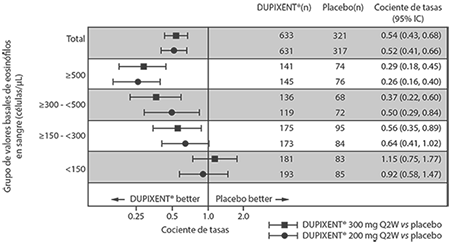
Exacerbaciones: Los estudios AS 1 y 2 evaluaron la frecuencia de las exacerbaciones de asma grave definidas como el deterioro del asma que requiere el uso de corticosteroides sistémicos durante al menos 3 días o la hospitalización o la visita a la sala de urgencias debido al asma que requirió corticosteroides sistémicos. En la población del análisis primario (sujetos con un recuento basal de eosinófilos en sangre de ≥ 300 células/μL en el estudio AS 1 y la población total del estudio AS 2), los sujetos que recibieron DUPIXENT® 200 mg o 300 mg cada 2 semanas tuvieron reducciones significativas en la tasa de exacerbaciones de asma severas en comparación con el placebo. En la población total del estudio AS 2, la tasa de exacerbaciones graves fue de 0.46 y 0.52 para DUPIXENT® 200 mg cada 2 semanas y 300 mg cada 2 semanas, respectivamente, en comparación con las tasas de placebo de 0.87 y 0.97, respectivamente. La proporción de exacerbaciones graves en comparación con el placebo fue de 0.52 (95% IC: 0.41, 0.66) y de 0.54 (95% IC: 0.43, 0.68) para DUPIXENT® 200 mg cada 2 semanas y 300 mg cada 2 semanas, respectivamente. Los resultados en sujetos con recuento basal de eosinófilos en sangre ≥ 300 células/μL en los estudios AS 1 y 2 se muestran en la Tabla 7.
Las tasas de respuesta de los valores iniciales de eosinófilos en sangre basal para el estudio AS 2 se muestran en la Figura 4. Los análisis de subgrupos preespecificados de los estudios AS 1 y 2 demostraron que hubo mayores reducciones en las exacerbaciones graves en los sujetos con niveles basales más altos de eosinófilos en sangre. En el estudio AS 2, las reducciones en las exacerbaciones fueron significativas en el subgrupo de sujetos con eosinófilos basales ≥ 150 células/μL. En sujetos con un recuento basal de eosinófilos en sangre < 150 células/μL, se observaron tasas de exacerbación severas similares entre DUPIXENT® y el placebo.
En el estudio AS 2, el cociente de tasa estimado de exacerbaciones que dieron lugar a hospitalizaciones y/o visitas a la sala de emergencia respecto al placebo fue de 0.53 (95% IC: 0.28, 1.03) y 0.74 (95% lC: 0.32, 1.70) con DUPIXENT® 200 mg o 300 mg cada 2 semanas, respectivamente.
Tabla 7. Tasa de exacerbaciones graves en los estudios AS 1 y 2
|
Estudio |
Tratamiento |
EOS sanguíneos basales ≥ 300 células/μL (análisis primario de población, estudio 1) |
||
|
N |
Tasa (IC 95%) |
Relación proporcional (IC 95%) |
||
|
Estudio AS 1 |
DUPIXENT® 200 mg cada 2 semanas |
65 |
0.30 (0.13, 0.68) |
0.29 (0.11, 0.76) |
|
DUPIXENT® 300 mg cada 2 semanas |
64 |
0.20 (0.08, 0.52) |
0.19 (0.07, 0.56) |
|
|
Placebo |
68 |
1.04 (0.57, 1.90) |
||
|
Estudio AS 2 |
DUPIXENT® 200 mg cada 2 semanas |
264 |
0.37 (0.29, 0.48) |
0.34 (0.24, 0.48) |
|
Placebo |
148 |
1.08 (0.85, 1.38) |
||
|
DUPIXENT® 300 mg cada 2 semanas |
277 |
0.40 (0.32, 0.51) |
0.33 (0.23, 0.45) |
|
|
Placebo |
142 |
1.24 (0.97, 1.57) |
||
Figura 4. Riesgo relativo en la tasa de eventos anualizados de exacerbaciones graves en el recuento basal de eosinófilos en sangre (células/μL) en el estudio AS 2.
El tiempo hasta la primera exacerbación fue más prolongado para los sujetos que recibieron DUPIXENT® en comparación con el placebo en el estudio AS 2 (Figura 5).
Figura 5. Curva de incidencia de Kaplan Meier para el tiempo hasta la primera exacerbación grave en sujetos con eosinófilos basales de ≥ 300 células/μL (estudio AS 2)a
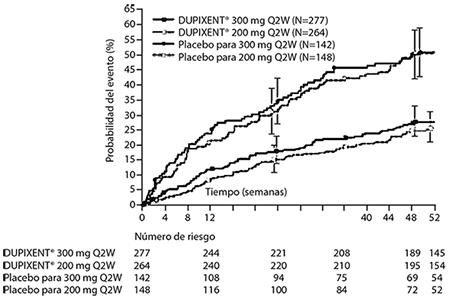
a En el momento del cierre de la base de datos, no todos los pacientes habían completado la Semana 52
Función pulmonar:
Se observaron aumentos significativos en el FEV1 prebroncodilatador en la Semana 12 para el estudio AS 1 y 2 en el análisis primario de población (sujetos con los recuentos basales de eosinófilos en sangre de ≥ 300 células/μL en el estudio AS 1 y en la población total del estudio AS 2).
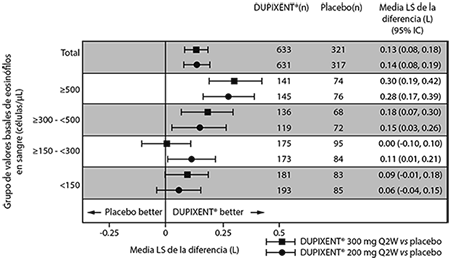
En la población total del estudio AS 2, el cambio medio de LS FEV1 desde los valores iniciales fue de 0.32 L (21%) y 0.34 L (23%) para DUPIXENT® 200 mg cada 2 semanas y 300 mg cada 2 semanas, respectivamente, en comparación con las medias de placebo correspondiente de 0.18 L (12%) y 0.21 L (14%).
La diferencia de tratamiento promedio respecto al placebo fue de 0.14 L (95% IC: 0.08, 0.19) y 0.13 L (95% IC: 0.08, 0.18) para DUPIXENT® 200 mg cada 2 semanas y 300 mg cada 2 semanas, respectivamente. Los resultados en sujetos con conteos basales de eosinófilos en sangre ≥ 300 células/μL en los Estudios AS 1 y 2 se muestran en la Tabla 8.
La mejoría en el FEV1 por los valores basales de eosinófilos en sangre para el estudio AS 2 se muestran en la Figura 6.
El análisis de subgrupos de los Estudios AS 1 y 2 demostró una mejoría mayor en los sujetos con valores basales mayores de eosinófilos en sangre.
Tabla 8. Media del cambio desde el valor basal respecto FEV1 pre-broncodilatador en la semana 12 de los estudios AS 1 y 2
|
Estudio |
Tratamiento |
EOS sanguíneos basales ≥ 300 células/μL (análisis primario de población, estudio 1) |
||
|
N |
Media LS del cambio desde valores basal L (%) |
Media LS Diferencia frente a placebo (IC 95%) |
||
|
Estudio AS 1 |
DUPIXENT® 200 mg cada 2 semanas |
65 |
0.43 (25.9) |
0.26 (0.11, 0.40) |
|
DUPIXENT® 300 mg cada 2 semanas |
64 |
0.39 (25.8) |
0.21 (0.06, 0.36) |
|
|
Placebo |
68 |
0.18 (10.2) |
||
|
Estudio AS 2 |
DUPIXENT® 200 mg cada 2 semanas |
264 |
0.43 (29.0) |
0.21 (0.13, 0.29) |
|
Placebo |
148 |
0.21 (15.6) |
||
|
DUPIXENT® 300 mg cada 2 semanas |
277 |
0.47 (32.5) |
0.24 (0.16, 0.32) |
|
|
Placebo |
142 |
0.22 (14.4) |
||
Figura 6. Media LS de la diferencia del cambio desde el valor basal respecto al placebo en la semana 12 en FEV1 pre-broncodilatador a través del conteo de eosinófilos basales (células/μL) en el estudio AS 2
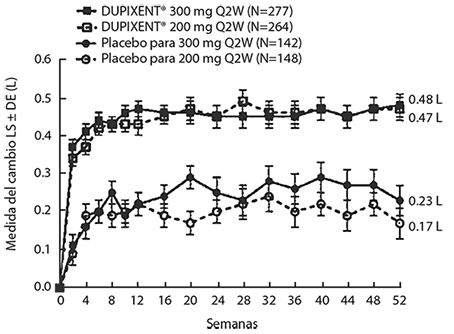
Los cambios medios en FEV1 a lo largo del tiempo en el Estudio AS 2 se muestran en la Figura 7.
Figura 7. Media del cambio desde el valor basal en el FEV1 pre-broncodilatador (L) a lo largo del tiempo en sujetos con eosinófilos basales ≥ 300 células/μL (Estudio AS 2)
Criterios de valoración secundarios adicionales: ACQ-5 y AQLQ (S) fueron analizados en el estudio AS 2 en la semana 52. La tasa de respuesta fue definida como una mejora en la puntuación de 0.5 o más [rango de la escala de 0-6 para ACQ-5 y 1-7 para AQLQ (S)].
• La tasa de respuesta ACQ-5 para DUPIXENT® 200 mg y 300 mg cada 2 semanas en la población total fue 69% respecto al 62% de grupo placebo (proporción de probabilidad 1.37; 95% IC: 1.01, 1.86) y 69% respecto al 63% de grupo placebo (proporción de probabilidad 1.28; 95% IC: 0.94, 1.73), respectivamente, y la tasa de respuesta de AQLQ(S) fue de 62% respecto al 54% de grupo placebo (proporción de probabilidad 1.61; 95% lC: 1.17, 2.21) y 62% respecto al 57% de grupo placebo (proporción de probabilidad 1.33; 95% lC: 0.98, 1.81), respectivamente.
• La tasa de respuesta ACQ-5 para DUPIXENT® 200 mg y 300 mg cada 2 semanas en sujetos con valores basales de eosinófilos en sangre de ≥ 300 células/μL fue de 75% respecto al 67% de grupo placebo (proporción de probabilidad 1.46; 95% lC: 0.90, 2.35) y 71% respecto al 64% de grupo placebo (proporción de probabilidad 1.39; 95% lC: 0.88, 2.19), respectivamente, y la tasa de respuesta de AQLQ (S) fue de 71% respecto al 55% de grupo placebo (proporción de probabilidad 2.02; 95% lC: 1.24, 3.32) y 65% respecto al 55 % de grupo placebo (proporción de probabilidad 1.79; 95% IC: 1.13, 2.85), respectivamente.
Reducción de corticosteroides orales (Ensayo AS 3): El estudio AS 3 evaluó el efecto de DUPIXENT® en la reducción del uso de corticosteroides orales de mantenimiento. La media basal del uso de corticosteroides orales fue de 12 mg en el grupo que recibió placebo y 11 mg en el grupo que recibió DUPIXENT®. Los resultados de los criterios de valoración primarios fueron la reducción del porcentaje desde el valor inicial de la dosis final de corticosteroides orales en la semana 24 mientras se mantenía el control del asma.
En comparación con el placebo, los sujetos que recibieron DUPIXENT® lograron mayores reducciones en el mantenimiento diario de la dosis de corticoesteroides orales, mientras mantenían el control del asma. El porcentaje medio de reducción en la dosis diaria de OCS desde el inicio del estudio fue del 70% (mediana del 100%) en sujetos que recibieron DUPIXENT® (95% lC: 60%, 80%) en comparación con el 42% (mediana del 50%) en sujetos que recibieron placebo (95% lC: 33%, 51%). Se observaron reducciones del 50% o más en la dosis de OCS en 82 (80%) sujetos que recibieron DUPIXENT® en comparación con 57 (53%) en los que recibieron placebo. La proporción de sujetos con una dosis final media inferior a 5 mg en la semana 24 fue del 72% para DUPIXENT® y del 37% para placebo (proporción de probabilidad 4.48; 95% lC: 2.39, 8.39).Un total de 54 (52%) sujetos que recibieron DUPIXENT® contra 31 (29%) sujetos en el grupo de placebo tuvieron una reducción del 100% en su dosis de OCS.
En este ensayo de 24 semanas, las exacerbaciones del asma (definidas como un aumento temporal en la dosis de corticosteroides orales durante al menos 3 días) fueron menores en los sujetos que recibieron DUPIXENT® en comparación con los que recibieron placebo (tasa anualizada de 0.65 y 1.60 para el grupo DUPIXENT® y placebo, respectivamente; el cociente de tasas 0.41 [95% IC 0.26, 0.63]) y la mejoría en el FEV1 pre broncodilatador desde el inicio hasta la Semana 24 fue mayor en los sujetos que recibieron DUPIXENT® en comparación con los que recibieron placebo (diferencia de medias de LS para DUPIXENT® respecto al placebo de 0.22 L (95% IC: 0.09 a 0.34 L]). Los efectos en la función pulmonar y en la reducción de esteroides orales y exacerbación fueron similares, independientemente de los niveles basales de eosinófilos en la sangre. El ACQ-5 y AQLQ (S) también se evaluaron en el Estudio AS 3 y mostraron mejoras similares a las del Estudio AS2.
Ensayo de extensión a largo plazo (TRAVERSE): La eficacia a largo plazo de DUPIXENT® en 2282 adultos y adolescentes con asma de moderada a grave, y los adultos con asma dependiente de corticoesteroides orales, que habían participado en ensayos clínicos anteriores de DUPIXENT®, se evaluó en el estudio abierto (TRAVERSE). En este estudio, el beneficio clínico de DUPIXENT®, incluyendo la reducción de las exacerbaciones y la mejora de la función pulmonar, se mantuvo hasta las 96 semanas. En la población con asma dependiente de los corticosteroides orales, hubo una reducción sostenida de las exacerbaciones y se mantuvo una mejoría en la función pulmonar, a pesar de la continua disminución o interrupción de la dosis oral de corticoesteroides hasta las 96 semanas. También se observó un mantenimiento similar del efecto para ACQ-5 y AQLQ(S) en la semana 48 (ver Tabla 9). También se observaron resultados consistentes en el subgrupo de pacientes con dosis altas de ICS.
Tabla 9. Tasa de exacerbaciones graves, cambio medio desde el valor basal en FEV1, ACQ-5 y AQLQ (s) Tasas de respuesta en TRAVERSEa (Niveles basales de Eosinófilos en Sangre 150 y 300 células/μL y FeNO a 25 ppb).
|
Tratamiento |
EOS ≥ 150 células/μL |
EOS ≥ 300 células/μL |
EOS ≥ 25 ppb |
|||
|
Tasa de exacerbaciones graves no ajustadas durante la semana 96 |
||||||
|
N |
Tasa |
N |
Tasa |
N |
Tasa |
|
|
DUPIXENT® 300 mg cada 2 semanas |
1496 |
0.30 |
905 |
0.27 |
1050 |
0.26 |
|
Cambio medio desde el valor basal en el FEV1 en la semana 96 |
||||||
|
N |
Media desde la línea basal L (%) |
N |
Media desde la línea basal L (%) |
N |
Media desde la línea basal L (%) |
|
|
DUPIXENT® 300 mg cada 2 semanas |
865 |
0.33 (21.1) |
511 |
0.42 (27.3) |
596 |
0.39 (24.6) |
|
ACQ-5 en la semana 48b |
||||||
|
N |
Tasa de respuesta (%) |
N |
Tasa de respuesta (%) |
N |
Tasa de respuesta (%) |
|
|
DUPIXENT® 300 mg cada 2 semanas |
1412 |
87.3 |
855 |
88.8 |
998 |
88.7 |
|
AQLQ(S) en la semana 48b |
||||||
|
N |
Tasa de respuesta (%) |
N |
Tasa de respuesta (%) |
N |
Tasa de respuesta (%) |
|
|
DUPIXENT® 300 mg cada 2 semanas |
1366 |
77.8 |
829 |
81.7 |
967 |
79.1 |
a . En los estudios de estudio de TRAVERSE, los pacientes pasaron por los estudios pivotales de asma DRI12544 y QUEST.
b ACQ-5 y AQLQ(S) no se recopilaron después de la semana 48.
Rinosinusitis crónica con poliposis nasal: El programa de desarrollo de rinosinusitis crónica con poliposis nasal (RSCcPN) incluyó dos estudios aleatorizados, doble ciego, de grupos paralelos, multicéntricos, controlados con placebo (Estudio CSNP 1 y Estudio CSNP 2) en 724 sujetos de 18 años o más con corticosteroides intranasales de base (INCS). Estos estudios incluyeron sujetos con RSCcPN a pesar de una cirugía o tratamiento sinonasal previo, o que no eran elegibles para recibir o eran intolerantes a corticosteroides sistémicos en los últimos 2 años. Los pacientes con rinosinusitis crónica sin poliposis nasal no se incluyeron en estos estudios. Se permitió el rescate con corticosteroides sistémicos o cirugía durante los estudios a discreción del investigador. En el Estudio CSNP 1, un total de 276 sujetos fueron aleatorizados para recibir 300 mg de DUPIXENT® (N = 143) o placebo (N = 133) cada dos semanas durante 24 semanas. En el Estudio CSNP 2, 448 sujetos fueron aleatorizados para recibir 300 mg de DUPIXENT® (N = 150) en semanas alternas durante 52 semanas, 300 mg de DUPIXENT® (N = 145) en semanas alternas hasta la semana 24 seguido de 300 mg de DUPIXENT® cada 4 semanas hasta semana 52, o placebo (N = 153). Todos los sujetos tenían evidencia de opacificación de los senos nasales en la clasificación tomográfica computarizada sinusal según Lund-Mackay (LMK) y del 73% al 90% de los sujetos presentaban opacificación de todos los senos nasales. Los sujetos fueron estratificados con base ensus antecedentes de cirugía previa y asma comórbida/enfermedad respiratoria agravada por fármacos antiinflamatorios no esteroideos (EREA-AINES). Un total del 63% de los sujetos informaron cirugía previa de los senos nasales, con un número medio de 2,0 cirugías previas, el 74% utilizó corticosteroides sistémicos en los 2 años anteriores con un número medio de 1.6 ciclos de corticosteroides sistémicos en los 2 años anteriores, el 59% tuvo asma mórbida, y el 28% tenía EREA-AINES.
Los criterios de valoración co-primarios de eficacia fueron el cambio desde el inicio hasta la semana 24 en la puntuación de pólipos nasales endoscópicos bilaterales (NPS; escala 0-8) según los lectores centrales cegados, y el cambio desde el inicio hasta la semana 24 en la puntuación de congestión/obstrucción nasal promediado sobre 28 días (NC; escala 0-3), según lo determinado por los sujetos que utilizan un registro diario. Para NPS, los pólipos a cada lado de la nariz se clasificaron en una escala categórica (0 = sin pólipos; 1 = pólipos pequeños en el meato medio que no llegan por debajo del borde inferior del cornete medio; 2 = pólipos que llegan por debajo del borde inferior del cornete medio; 3 = pólipos grandes que alcanzan el borde inferior del cornete inferior o pólipos medial al cornete medio; 4 = pólipos grandes que causan obstrucción completa de la cavidad nasal inferior). La puntuación total fue la suma de las puntuaciones derecha e izquierda. Los sujetos calificaron la congestión nasal diariamente en una escala de gravedad categórica de 0 a 3 (0 = sin síntomas; 1 = síntomas leves; 2 = síntomas moderados; 3 = síntomas graves).
En ambos estudios, los puntos finales secundarios clave en la semana 24 incluyeron cambios desde el valor inicial en: puntuación de la clasificación tomográfica computarizada sinusal según Lund-Mackay (LMK), pérdida diaria del olfato y prueba de resultado sino-nasal de 22 ítems (SNOT-22). La puntuación de la clasificación tomográfica computarizada sinusal según Lund-Mackay (LMK) evaluó la opacificación de cada seno utilizando una escala de 0 a 2 (0 normal; 1 = opacificación parcial; 2 = opacificación total) obteniendo una puntuación máxima de 12 por lado y una puntuación máxima total de 24 (las puntuaciones mayores indican más opacificación). El paciente puntuó reflexivamente la pérdida del olfato todas las mañanas en una escala de 0 a 3 (0 = sin síntomas, 1 = síntomas leves, 2 = síntomas moderados, 3 = síntomas graves). SNOT-22 incluye 22 ítems que evalúan los síntomas y el impacto de los síntomas asociados con RSCcPN con cada ítem puntuado de 0 (sin problema) a 5 (problema tan malo como puede ser) con una puntuación global que va de 0 a 110. SNOT-22 tuvo un periodo de recordatorio de 2 semanas. En los resultados de eficacia combinados, se evaluó la reducción en la proporción de sujetos rescatados con corticosteroides sistémicos y/o cirugía sino-nasal (hasta la semana 52).
Las características demográficas e iniciales de estos 2 ensayos se proporcionan en la Tabla 10 a continuación.
Tabla 10. Características demográficas y basales de los Estudios de RSCcPN
|
Parámetro |
Estudio CSNP 1 (N = 276) |
Estudio CSNP 2 (N = 448) |
|
Edad media (años) (DE) |
50 (13) |
52 (12) |
|
% hombres |
57 |
62 |
|
Duración media de RSCcPN (años) (DE) |
11 (9) |
11 (10) |
|
Pacientes con ≥ 1 cirugía previa (%) |
72 |
58 |
|
Pacientes con uso de corticosteroides sistémicos en los 2 años anteriores (%) |
65 |
80 |
|
NPS endoscópica bilateral mediaa (DE), rango 0-8 |
5.8 (1.3) |
6.1 (1.2) |
|
Puntuación media de congestión nasal (NC)a (SD), rango 0-3 |
2.4 (0.6) |
2.4 (0.6) |
|
Puntuación total media de la clsificación TC de LMK (DE)a, rango 0-24 |
19 (4.4) |
18 (3.8) |
|
Puntuación media de la pérdida del olfato (AM)a, (DE) rango 0-3 |
2.7 (0.5) |
2.8 (0.5) |
|
Puntuación total media (DE) de SNOT-22a, rango 0-110 |
49.4 (20.2) |
51.9 (20.9) |
|
Media de eosinófilos en sangre (células/μL) (DE) |
440 (330) |
430 (350) |
|
Medida total de IgE UI/mL (DE) |
212 (276) |
240 (342) |
|
Historia médica atópica En general |
75% |
82% |
|
Asma |
58 |
60 |
|
EREA (%) |
30 |
27 |
a Las puntuaciones más altas indican una mayor gravedad de la enfermedad.
DE = desviación estándar; AM = mañana; NPS = puntuación de pólipos nasales; SNOT-22 = prueba de resultado sino-nasal de 22 ítems; NSAID-ERD = asma/enfermedad respiratoria agravada por fármacos antiinflamatorios no esteroideos.
Respuesta clínica (Estudio CSNP 1 y Estudio CSNP 2): Los resultados de los criterios de valoración primarios en los Estudios RSCcPN se presentan en la Tabla 11.
Tabla 11. Características demográficas y basales de los Estudios de RSCcPN.
|
Placebo ( = 133) |
DUPIXENT® 300 mg cada 2 semanas (n=143) |
Diferencia de la media LS contra PLacbo (95% IC) |
Placebo ( n=133) |
DUPIXENT® 300 mg cada 2 semanas (n=295) |
Diferencia de la media LS contra PLacbo (95% IC) |
|||||
|
Criterios de valoración primarios en la semana 24 |
||||||||||
|
Puntuación |
Media basal |
Cambio de la media LS |
Media basal |
Cambio de la media LS |
Media basal |
Cambio de la media LS |
Media basal |
Cambio de la media LS |
||
|
NPS |
5.86 |
0.17 |
5.64 |
-1.89 |
-2.06 (-2.43, -1.69) |
5.96 |
0.10 |
6.18 |
-1.71 |
-1.80 (-2.10, 1.51) |
|
NC |
2.45 |
-0.45 |
2.26 |
-1.34 |
-0.89 (-1.07, -0.71) |
2.38 |
-0.38 |
2.46 |
-1.25 |
-0.87 (-1.03,-0.71) |
Una reducción en la puntuación indica una mejora.
NPS=puntuación de pólipos nasales; NC=congestión/obstrucción nasal.
Se observó una eficacia estadísticamente significativa en el Estudio CSNP 2 con respecto a la mejora en la puntuación NPS endoscópica bilateral en la semana 24 y la semana 52 (ver Figura 8).
Figura 8. Cambio en la puntuación media de LS de pólipos nasales bilaterales (NPS) desde el inicio hasta la semana 52 en el ensayo 2 de CSNP: población ITT.
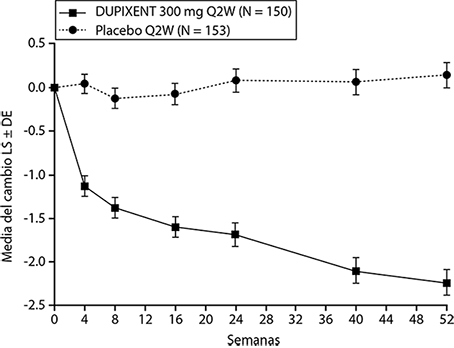
Se observaron resultados similares en el Estudio CSNP 1 en la semana 24. En el periodo posterior al tratamiento, cuando los sujetos estaban sin DUPIXENT®, el efecto del tratamiento disminuyó con el tiempo (ver Figura 9).
Figura 9. Cambio en la puntuación media de LS de pólipos nasales bilaterales (NPS) desde el inicio hasta la semana 48 en el ensayo 2 de CSNP: población ITT.
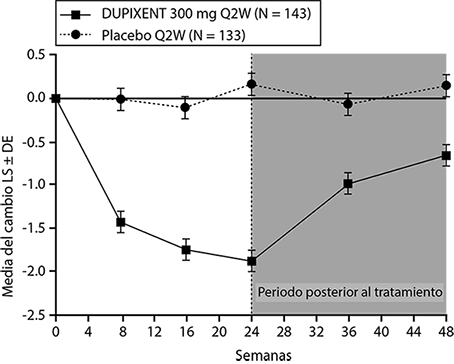
En la semana 52, la diferencia media de LS para la congestión nasal en el grupo DUPIXENT® con respecto al placebo fue de -0.98 (IC del 95%: -1.17, -0.79). En ambos estudios, se observaron mejoras significativas en la congestión nasal desde la primera evaluación en la semana 4. La diferencia media de LS para la congestión nasal en la semana 4 en el grupo DUPIXENT® con respecto al placebo fue -0,41 (IC del 95%: -0.52, -0.30) en el Estudio CSNP 1 y -0.37 (IC del 95%: -0.46, -0.27) en el Estudio CSNP 2.
Se observó una disminución significativa en la puntuación de la clasificación tomográfica computarizada sinusal según Lund-Mackay (LMK). La diferencia media LS para la puntuación de la tomografía computarizada del seno LMK en la semana 24 en el grupo DUPIXENT® con respecto al placebo fue -7.44 (IC del 95%: -8.35, -6.53) en el Estudio CSNP 1 y -5.13 (IC del 95%: -5.80, -4.46) en el Estudio CSNP 2. En la semana 52, en el ensayo 2 de CSNP, la diferencia media LS para la puntuación de la tomografía computarizada del seno LMK en el grupo DUPIXENT® con respecto al placebo fue -6.94 (IC del 95%: -7.87, -6.01).
Dupilumab mejoró significativamente la pérdida del olfato en comparación con el placebo. La diferencia media de MC para la pérdida del olfato en la semana 24 en el grupo DUPIXENT® con respecto al placebo fue -1.12 (IC del 95%: -1.31, -0.93) en el Estudio CSNP 1 y -0.98 (IC del 95%: -1.15, -0.81) en el Estudio CSNP 2. En la semana 52, la diferencia media de LS para la pérdida del olfato en el grupo DUPIXENT® con respecto al placebo fue -1.10 (IC del 95%: -1.31; -0.89). En ambos estudios, se observaron mejorías significativas en la severidad de la pérdida diaria del olfato desde la primera evaluación en la Semana 4. DUPIXENT® redujo significativamente los síntomas sino-nasales medidos por SNOT-22 en comparación con placebo. La diferencia media de MC para SNOT-22 en la semana 24 en el grupo DUPIXENT® con respecto al placebo fue -21.12 (IC del 95%: -25.17, -17.06) en el Estudio CSNP 1 y -17.36 (IC del 95%: -20.87, - 13.85) en Estudio CSNP 2. En la semana 52, la diferencia media de LS en el grupo DUPIXENT® con respecto al placebo fue -20.96 (IC del 95%: -25.03, -16.89).
En el análisis agrupado preespecificado y ajustado por multiplicidad de dos estudios, el tratamiento con DUPIXENT® dio como resultado una reducción significativa del uso de corticosteroides sistémicos y la necesidad de cirugía sino-nasal con respecto al placebo (HR de 0.24; IC del 95%: 0.17, 0.35) (ver Figura 10). La proporción de sujetos que requirieron corticosteroides sistémicos se redujo en un 74% (HR de 0,26; IC del 95%: 0.18, 0.38). El número total de ciclos de corticosteroides sistémicos por año se redujo en un 75% (RR de 0,25; IC del 95%: 0.17, 0.37). La proporción de sujetos que requirieron cirugía se redujo en un 83% (HR de 0.17; IC del 95%: 0.07, 0.46).
Figura 10. Cambio en la puntuación media de LS de pólipos nasales bilaterales (NPS) desde el inicio hasta la semana 52 en el ensayo 2 de CSNP: población ITT.
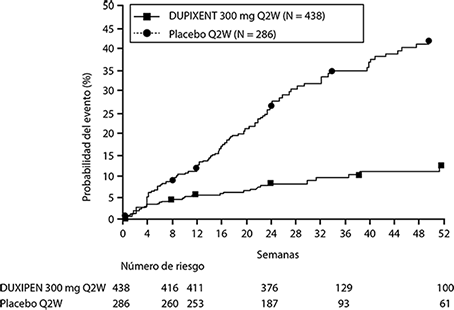
Los efectos de DUPIXENT® en los criterios de valoración primarios de NPS y congestión nasal, y los criterios secundarios de la puntuación de la tomografía computarizada sinusal de Lund-Mackay (LMK) fue consistente en pacientes con o sin cirugía previa.
En los sujetos con asma comórbida, la mejoría en el FEV1 pre-broncodilatadores fue similar a la de los pacientes del programa con asma.
CONTRAINDICACIONES:
DUPIXENT® está contraindicado en pacientes con hipersensibilidad conocida a dupilumab o a cualquiera de sus excipientes (ver Precauciones generales).
DUPIXENT® no debe administrarse durante el embarazo, la lactancia, ni en menores de 12 años con asma moderada a grave, en niños menres de 6 años con dermatitis atópica, en menores de 18 años con rinosinusitis crónica con poliposis nasal o en infecciones parasitarias activas.
RESTRICCIONES DE USO DURANTE EL EMBARAZO Y LA LACTANCIA:
Embarazo:
Resumen de riesgo: Los datos disponibles de informes de casos y series de casos con el uso de DUPIXENT® en mujeres embarazadas no han identificado un riesgo asociado con el fármaco y defectos de nacimiento mayores, aborto espontáneo o efectos adversos sobre la respuesta materna o fetales. Se sabe que los anticuerpos de lgG humanos cruzan la barrera placentaria; por lo tanto, DUPIXENT® puede ser transmitido de la madre al feto en desarrollo. Se observan efectos adversos sobre la respuesta materna y fetal asociados con el asma durante el embarazo (ver Consideraciones clínicas). En un estudio intensificado de desarrollo pre- y post-natal, no se observaron efectos adversos sobre el desarrollo en los recién nacidos de monos embarazadas después de la administración subcutánea de un anticuerpo homólogo contra el receptor alfa de interleucina 4 (IL-4Rα) durante la organogénesis hasta el parto, en dosis hasta 10 veces la dosis máxima recomendada en el humano (MRHD) [ver DATOS). Se desconoce el riesgo estimado subyacente de defectos mayores del nacimiento y embarazos interrumpidos para las poblaciones indicadas. DUPIXENT® no debe emplearse durante el embarazo.
Todos los embarazos poseen un riesgo inherente de defectos de nacimiento o pérdidas u otras evoluciones adversas. En la población general de los Estados Unidos, el riesgo inherente estimado de defectos mayores del nacimiento y abortos en embarazos clínicamente reconocidos es del 2% a 4% y 15% a 20%, respectivamente.
Consideraciones clínicas:
Riesgo materno y/o embriofetal asociado con la enfermedad: En las mujeres con asma poco o moderadamente controlada, la evidencia demuestra que hay un mayor riesgo de preeclampsia en la madre y prematurez, bajo peso al nacer y una disminución de la edad gestacional en el neonato. El nivel de control del asma debe ser cuidadosamente monitoreado en las mujeres embarazadas y debe ajustarse el tratamiento según sea necesario para mantener un control óptimo.
Datos:
Datos en animales: En un estudio intensificado de toxicidad del desarrollo pre- y post-natal, las monas cynomolgus embarazadas recibieron semanalmente dosis subcutáneas de anticuerpos homólogos contra IL-4Rα hasta 10 veces la MRHD (en base a mg/kg de 100 mg/kg/semana), desde el inicio de la organogénesis hasta el parto. No se observaron efectos adversos relacionados al tratamiento sobre malformaciones o toxicidad embriofetal, ni sobre el desarrollo morfológico, funcional o inmunológico en las crías, desde el nacimiento hasta los 6 meses de edad.
Lactancia: No existen datos sobre la presencia de dupilumab en la leche humana, de los efectos sobre la lactancia ni efectos sobre la producción de leche. Se sabe que la lgG humana está presente en la leche humana. Se desconocen los efectos de la exposición gastrointestinal local y la limitada exposición sistémica a dupilumab sobre el lactante. DUPIXENT® no debe administrarse durante el periodo de lactancia.
REACCIONES SECUNDARIAS Y ADVERSAS:
Las siguientes reacciones adversas son comentadas en mayor detalle más adelante:
• Hipersensibilidad (ver sección Precauciones generales).
• Conjuntivitis y queratitis (ver sección Precauciones generales).
Experiencia de estudios clínicos:
Debido a que los estudios clínicos fueron realizados bajo una amplia variedad de condiciones, las tasas de reacciones adversas observadas en los estudios clínicos de un fármaco no pueden ser comparadas directamente con las tasas de reacciones adversas de los estudios clínicos de otros fármacos y no pueden reflejar las tasas observadas en la práctica.
Adultos con dermatitis atópica: Tres estudios multicéntricos, aleatorizados, doble ciego, controlados con placebo (Estudios 1, 2 y 3) y un estudio de rango de dosis (Estudio 4) evaluaron la seguridad de DUPIXENT® en sujetos con dermatitis atópica moderada a grave. La población de seguridad tuvo una edad promedio de 38 años; 41% de los sujetos eran mujeres, 67% eran blancos, 24% eran asiáticos y 6% eran negros; en términos de condiciones comórbidas, el 48% de los sujetos tenían asma, 49% tenían rinitis alérgica, 37% tenían alergia alimentaria y 27% tenían conjuntivitis alérgica. En estos 4 estudios, 1472 sujetos fueron tratados con inyecciones subcutáneas de DUPIXENT®, con o sin corticoesteroides tópicos concomitantes (TCS). Un total de 739 sujetos fueron tratados con DUPIXENT® durante por lo menos 1 año en el programa de desarrollo para la dermatitis atópica moderada a grave.
Los Estudios 1, 2 y 4 compararon la seguridad de la monoterapia de DUPIXENT® con placebo hasta la semana 16. El Estudio 3 comparó la seguridad de DUPIXENT® + TCS al placebo + TCS hasta la hasta la semana 52.
El estudio 9 (estudio OLE) es un estudio de extensión abierto de fase 3 y multicéntrico, que evaluó la seguridad a largo plazo de dosis repetidas de DUPIXENT® en adultos con dermatitis atópica de moderada a grave que habían participado previamente en estudios controlados de DUPIXENT® o habían sido evaluados para un estudio de fase 3 (estudio 1 o estudio 2). Los datos de seguridad del estudio 9 reflejan la exposición a DUPIXENT® en 2677 sujetos adultos con dermatitis atópica, incluidos 2254 que completaron al menos 52 semanas, 1192 que completaron al menos 100 semanas y 357 que completaron al menos 148 semanas del estudio. La mayoría de los sujetos del estudio 9 (99.7%) fueron expuestos a la dosis de DUPIXENT® 300 mg semanal (cada semana).
Semanas 0 a 16 (Estudios 1 a 4): En los estudios de monoterapia de DUPIXENT® (Estudios 1, 2 y 4) hasta la semana 16, la cantidad de sujetos que suspendieron el tratamiento debido a eventos adversos fue del 1.9% en ambos grupos de DUPIXENT® 300 mg cada 2 semanas y placebo.
La Tabla 12 resume las reacciones adversas que ocurrieron con una tasa de por lo menos 1% en los grupos de monoterapia de DUPIXENT® 300 mg cada 2 semanas, y en el grupo de DUPIXENT® + TCS, todos en una tasa más alta que en sus respectivos grupos de comparación durante las primeras 16 semanas de tratamiento.
Tabla 12. Reacciones adversas que se presentaron en ≥ 1% del grupo de monoterapia de DUPIXEN® o el grupo de DUPIXENT® + TCS en los estudios de dermatitis atópica hasta la semana 16
|
Reacción adversa |
Monoterapia con DUPIXENT®a |
DUPIXENT® + TCSb |
||
|
DUPIXENT® 300 mg cada 2 semanasc N=529 n (%) |
Placebo N=517 n (%) |
DUPIXENT® 300 mg cada 2 semanasc + TCS N=110 n (%) |
Placebo + TCS N=315 n (%) |
|
|
Reacciones en el sitio de inyección |
51 (10) |
28 (5) |
11 (10) |
18 (6) |
|
Conjuntivitisd |
51 (10) |
12 (2) |
10 (9) |
15 (5) |
|
Blefaritis |
2 (< 1) |
1 (< 1) |
5 (5) |
2 (1) |
|
Herpes oral |
20 (4) |
8 (2) |
3 (3) |
5 (2) |
|
Queratitise |
1 (< 1) |
0 |
4 (4) |
0 |
|
Prurito ocular |
3 (1) |
1 (< 1) |
2 (2) |
2 (1) |
|
Otras infecciones por virus de herpes simplef |
10 (2) |
6 (1) |
1 (1) |
1 (< 1) |
|
Ojo seco |
1 (< 1) |
0 |
2 (2) |
1 (< 1) |
a Análisis concentrado de los Estudios 1, 2 y 4.
b El análisis de Estudio 3 incluyó a sujetos con antecedente de terapia con TCS.
c DUPIXENT® 600 mg en la semana 0, seguido por 300 mg cada dos semanas.
d Los racimos de conjuntivitis incluyeron a la conjuntivitis, conjuntivitis alérgica, conjuntivitis bacteriana, conjuntivitis viral, conjuntivitis papilar gigante, irritación ocular e inflamación ocular.
e Los racimos de queratitis incluyeron a la queratitis, queratitis ulcerativa, queratitis alérgica, queratoconjuntivitis atópica y herpes simple oftalmológico.
f Otras infecciones por el virus herpes simple incluyeron herpes simple, herpes genital, otitis externa por herpes simple e infección por virus herpes, pero se excluye el eccema herpeticum.
Seguridad hasta la semana 52 (Estudio 3): En el estudio de DUPIXENT® con TCS concomitantes (Estudio 3) hasta la Semana 52, la proporción de sujetos que suspendieron el tratamiento debido a eventos adversos fue del 1.8% en el grupo de DUPIXENT® 300 mg cada 2 semanas+ TCS y 7.6% en el grupo de placebo + TCS. Dos sujetos descontinuaron DUPIXENT® debido a reacciones adversas: dermatitis atópica (1 sujeto) y dermatitis exfoliativa (1 sujeto).
El perfil de seguridad de DUPIXENT® + TCS hasta la semana 52 fue generalmente consistente con el perfil de seguridad observado en la semana 16.
Seguridad a los 3 años (estudio 9): El perfil de seguridad a largo plazo observado en este estudio hasta los 3 años fue generalmente consistente con el perfil de seguridad de DUPIXENT® observado en estudios controlados.
Adolescentes con dermatitis atópica (de 12 a 17 años de edad): La seguridad de DUPIXENT®se evaluó en un estudio de 250 sujetos de 12 a 17 años de edad con dermatitis atópica de moderada a grave (estudio 6). El perfil de seguridad de DUPIXENT® en estos sujetos hasta la semana 16 fue similar al perfil de seguridad del estudio en adultos con dermatitis atópica.
La seguridad a largo plazo de DUPIXENT® se evaluó en un estudio de extensión de etiqueta abierta en sujetos de 12 a 17 años de edad con dermatitis atópica de moderada a grave (Estudio 7). El perfil de seguridad de DUPIXENT® en los sujetos seguidos durante la semana 52 fue similar al perfil de seguridad observado en la semana 16 en el estudio 6. El perfil de seguridad a largo plazo de DUPIXENT®observado en adolescentes fue consistente con el observado en adultos con dermatitis atópica.
Niños con dermatitis atópica (6 a 11 años de edad): La seguridad de DUPIXENT® con TCS concomitante se evaluó en un estudio clínico con 367 sujetos de 6 a 11 de edad con dermatitis atópica grave (Estudio 8). El perfil de seguridad de DUPIXENT® + TCS en estos sujetos hasta la semana 16 fue similar al perfil de seguridad de los estudios clínicos en adultos y adolescentes con dermatitis atópica.
La seguridad a largo plazo de DUPIXENT® + TCS se evaluó en un estudio de extensión de etiqueta abierta de 368 sujetos de 6 a 11 años de edad con dermatitis atópica (Estudio 7). Entre los sujetos que ingresaron al estudio, 110 (30%) tenían dermatitis atópica moderada y 72 (20%) tenían dermatitis atópica grave al momento de ingresar al estudio 7. El perfil de seguridad de DUPIXENT® + TCS en sujetos de seguimiento hasta la semana 52 fue similar al perfil de seguridad observado hasta la semana 16 en el Estudio 8. El perfil de seguridad de DUPIXENT® + TCS observado en sujetos pediátricos fue consistente con el observado en adultos y pacientes con dermatitis atópica (ver sección Uso en poblaciones especiales).
Asma:
Un total de 2888 sujetos adultos y adolescentes con asma moderada a grave (AS) fueron evaluados en 3 estudios aleatorizados, controlados por placebo, multicéntricos de 24 a 52 semanas de duración (Estudios AS 1, 2 y 3). De éstos, 2678 tuvieron antecedentes de 1 o más exacerbaciones graves en el año previo al reclutamiento a pesar del uso regular de corticosteroides inhalados a dosis media a alta más uno o más controladores adicionales (Estudios AS 1 y 2). Se reclutaron un total de 210 sujetos con asma dependiente de corticosteroides orales que recibieron altas dosis de corticosteroides inhalados y más de dos controladores adicionales (Estudio AS 3). La población de seguridad (Estudios AS 1 y 2) tenía una edad entre 12 y 87 años; 63% fueron mujeres; 82% fueron blancos. Se administró DUPIXENT® 200 mg o 300 mg por vía subcutánea cada 2 semanas, después de una dosis inicial de 400 mg o 600 mg, respectivamente.
En los Estudios AS 1 y 2, la proporción de sujetos que interrumpieron el tratamiento debido a eventos adversos fue de 4% del grupo con placebo, 3% del grupo con DUPIXENT® 200 mg cada 2 semanas y 6% del grupo con DUPIXENT® 300 mg cada 2 semanas.
La Tabla 13 resume las reacciones adversas que ocurrieron a una tasa de al menos 1% en sujetos tratados con DUPIXENT® y a una tasa mayor que sus respectivos grupos de comparación en los Estudios de Asma 1 y 2.
Tabla 13. Reacciones adversas que ocurren en ≥ 1% de los grupos con DUPIXENT® en los Ensayos de asma 1 y 2 y mayores que con placebo (mezcla de seguridad a los 6 meses)
|
Reacción adversa |
Estudios AS 1 y 2 |
||
|
DUPIXENT® 200 mg cada 2 semanas N=779 n (%) |
DUPIXENT® 300 mg cada 2 semanas N=788 n (%) |
Placebo N=792 n (%) |
|
|
Reacciones en el sitio de inyeccióna |
111 (14%) |
144 (18%) |
50 (6%) |
|
Dolor orofaríngeo |
13 (2%) |
19 (2%) |
7 (1%) |
|
Eosinofiliab |
17 (2%) |
16 (2%) |
2 (< 1%) |
a El grupo de reacciones en el lugar de inyección incluye eritema, edema, prurito, dolor e inflamación.
b Eosinofilia=eosinófilos en sangre ≥ 3,000 células/μL, o considerado por el investigador como un evento adverso. Ninguno cumplió con los criterios para condiciones eosinofílicas serias (ver sección Precauciones generales).
Las reacciones en el lugar de inyección fueron más comunes con la dosis de carga (inicial).
El perfil de seguridad de DUPIXENT® hasta la semana 52 fue generalmente consistente con el perfil de seguridad observado en la semana 24.
La seguridad a largo plazo de DUPIXENT® se evaluó en un estudio abierto en 2282 pacientes de 12 años o más, con asma de moderada a grave (estudio TRAVERSE). En este estudio, se siguió a los pacientes durante un máximo de 96 semanas, lo que dio lugar a una exposición acumulativa de 3169 pacientes-años a DUPIXENT®. El perfil de seguridad de DUPIXENT® en el estudio TRAVERSE fue consistente con el perfil de seguridad observado en estudios pivotales de asma durante un máximo de 52 semanas de tratamiento. No se identificaron reacciones adversas adicionales.
Rinosinusitis crónica con poliposis nasal: Un total de 722 sujetos adultos con Rinosinusitis crónica con poliposis nasal (RSCcPN) fueron evaluados en dos estudios multicéntricos, aleatorizados, controlados con placebo, de 24 a 52 semanas de duración (CSNP Estudio 1 y 2). Los datos de seguridad consistieron en las primeras semanas 24 de tratamiento para ambos estudios.
En la seguridad, la proporción de sujetos que descontinuaron el tratamiento debido a eventos adversos fue del 5% para el grupo placebo y del 2% para el grupo de DUPIXENT® 300 mg cada 2 semanas.
En la tabla 14 se resumen las reacciones adversas que ocurrieron a una tasa de al menos 1% en los sujetos tratados con DUPIXENT® y a una tasa más en comparación con sus respectivos grupos en los Estudios 1 y 2 de CSNP.
Tabla 14. Reacciones adversas que ocurren en ≥1% de los grupos con DUPIXENT® en los Ensayos de 1 y 2 de RSCcPN y mayores que con placebo (24 semanas).
|
Reacción adversa |
CSNP Estudios 1 y 2 |
|
|
DUPIXENT® 300 mg cada dos semanas N = 440 n (%) |
Placebo N = 282 n (%) |
|
|
Reacción en el sitio de inyecciónª |
28 (6%) |
12 (%) |
|
Conjuntivitisb |
7 (2%) |
2 (1%) |
|
Artralgia |
14 (3%) |
5 (2%) |
|
Gastritis |
7 (2%) |
2 (1%) |
|
Insomnio |
6 (1%) |
0 (< 1%) |
|
Eosinofilia |
5 (1%) |
1 (< 1%) |
|
Dolor dental |
5 (1%) |
1 (v 1%) |
a El grupo de las reacciones en el lugar de inyección incluyen reacción en el sitio de inyección, dolor, hematomas e inflamación.
b En el grupo de conjuntivitis incluyen conjuntivitis, conjuntivitis alérgica, conjuntivitis bacteriana, conjuntivitis viral, conjuntivitis papilar gigante, irritación ocular e inflamación ocular.
El perfil de seguridad de DUPIXENT® hasta la semana 52 fue generalmente consistente con el perfil de seguridad observado en la semana 24.
Reacciones adversas específicas:
Eventos relacionados con conjuntivitis y queratitis: En sujetos adultos con dermatitis atópica, durante el periodo de tratamiento de 16 semanas de los ensayos de monoterapia (estudio 1, 2 y 4), se reportó conjuntivitis en el 10% (34 por 100 sujetos-año) del grupo con dosis de 300 mg cada dos semanas y el 2% del grupo de placebo (8 por 100 sujetos-año). Durante el periodo de tratamiento de 52 semanas del estudio de dermatitis atópica con terapia concomitante (estudio 3), se reportó conjuntivitis en el 16% del grupo de DUPIXENT® 300 mg cada dos semanas + TCS (20 por 100 sujetos-año) y el 9% del grupo de placebo + TCS (10 por 100 sujetos-año). Durante el estudio de estudio OLE a largo plazo con datos de hasta 3 años, se reportó conjuntivitis en el 20% del grupo de DUPIXENT® (12 por cada 100 sujetos-año), la mayoría de estos fueron expuestos a DUPIXENT® 300 mg cada semana (estudio 9).
En los estudios de monoterapia de dermatitis atópica (Estudios 1, 2 y 4) con DUPIXENT® hasta la semana 16, se reportó queratitis en < 1% del grupo de DUPIXENT® (1 por cada 100 sujetos-año) y 0% en el grupo placebo (0 por cada 100 sujetos-año). En el estudio de dermatitis atópica de 52 semanas con DUPIXENT® + corticosteroides (TCS) (Estudio 3), se reportó 4% de queratitis en el grupo de DUPIXENT® + TCS (4 por cada 100 sujetos-año) y 2% en el grupo placebo + TCS (2 por cada 100 sujetos-año). Durante el estudio de OLE a largo plazo con datos de hasta 3 años (estudio 9), se reportó queratitis en el 3% del grupo de DUPIXENT® (2 por cada 100 sujetos-año). En sujetos con dermatitis atópica que recibieron DUPIXENT® ocurrieron con mayor frecuencia conjuntivitis y queratitis. La conjuntivitis fue el trastorno ocular notificado con más frecuencia. La mayoría de los sujetos con queratitis o conjuntivitis, se recuperaron o se estaban recuperando durante el periodo de tratamiento.
Entre los sujetos con asma, la frecuencia de conjuntivitis y queratitis fue similar entre DUPIXENT® y el placebo. En sujetos con RSCcPN la frecuencia de conjuntivitis fue de 2% en el grupo tratado con DUPIXENT® en comparación con el 1% en el grupo placebo en el grupo de seguridad de 24 semana, todos los sujetos se recuperaron. En el estudio RSCcPN de 52 semanas (ensayo CSNP 2), la frecuencia de conjuntivitis fue del 3% en los sujetos DUPIXENT® y el 1% en los sujetos con placebo; todos estos sujetos se recuperaron. No se notificaron casos de queratitis en el programa de desarrollo de RSCcPN (ver sección Precauciones generales).
Eccema herpeticum y herpes zoster: La incidencia de eccema herpeticum fue similar en los grupos de placebo y DUPIXENT® en los ensayos de dermatitis atópica. Las tasas se mantuvieron estables en los 3 años del estudio OLE a largo plazo (estudio 9).
El herpes zoster fue reportado en < 1% de los grupos de DUPIXENT® (1 por 100 sujetos-año) y en < 1% del grupo placebo (1 por 100 sujetos-año) en los estudios de monoterapia para dermatitis atópica de 16 semanas. En el estudio de 52 semanas de DUPIXENT® + TCS, el herpes zoster fue reportado en 1% del grupo de DUPIXENT® + TCS (1 por 100 sujetos-año) y en 2% del grupo de placebo + TCS (2 por 100 sujetos-año). Durante el estudio de OLE a largo plazo con datos de hasta 3 años (estudio 9), el 1.9% de los sujetos tratados con DUPIXENT® notificaron herpes zoster (0.99 por cada 100 sujetos-año de seguimiento). Entre los sujetos con asma, la frecuencia de herpes zoster fue similar entre DUPIXENT® y placebo. Entre los sujetos RSCcPN no se informaron casos de herpes zóster o eccema herpético.
Reacciones de hipersensibilidad: Las reacciones de hipersensibilidad fueron reportadas en < 1% de los sujetos tratados con DUPIXENT®. Estos incluyeron la reacción de enfermedad del suero, reacciones similares a enfermedad del suero, urticaria generalizada, erupción cutánea, eritema nodoso y anafilaxia (ver sección Contraindicaciones, Precauciones generales y Reacciones secundarias y adversas). Se han reportado reacciones de hipersensibilidad, incluyendo anafilaxia, enfermedad del suero o reacciones similares a la enfermedad del suero y angioedema.
Eosinófilos: Los sujetos tratados con DUPIXENT® tuvieron una media más grande inicial de incremento a partir del registro basal en la cuenta de eosinófilos en sangre, en comparación con los sujetos tratados con placebo. En sujetos con dermatitis atópica, la media y la mediana de los aumentos de eosinófilos en sangre desde el comienzo hasta la semana 4 fueron de 100 y 0 células/μL, respectivamente. En sujetos con asma, la media y la mediana de los aumentos de los eosinófilos en sangre desde el comienzo hasta la semana 4 fueron 130 y 10 células/μL respectivamente. La incidencia de eosinofilia surgida durante el tratamiento (≥ 500 células/μL), fue similar en los grupos de DUPIXENT® y placebo. En sujetos con RSCcPN, la media y la mediana de los aumentos de eosinófilos en sangre desde el comienzo hasta la semana 16 fue de 150 y 50 células/μL, respectivamente.
En todas las indicaciones, la incidencia de eosinofilia emergente del tratamiento (≥ 500 células/μL) fue similar en los grupos de DUPIXENT® y placebo. La eosinofilia surgida durante el tratamiento (≥ 5,000 células/μL) fue reportada en < 2% de los pacientes tratados con DUPIXENT® y en < 0.5% de los pacientes tratados con placebo. La cuenta de eosinófilos en sangre declinó hasta cerca de niveles basales durante el tratamiento del estudio. El recuento de eosinófilos continuó disminuyendo por debajo del nivel basal durante el estudio abierto en pacientes con asma.
Cardiovascular: En el estudio controlado con placebo de 1 año en sujetos con asma (Estudio AS 2), eventos tromboembólicos cardiovasculares (muertes cardiovasculares, infartos de miocardio no fatales y accidentes cerebrovasculares no fatales) fueron reportados en 1 (0.2%) del grupo DUPIXENT® 200 mg cada 2 semanas, 4 (0.6%) del grupo DUPIXENT® 300 mg cada 2 semanas y 2 (0.3%) del grupo placebo.
En el estudio controlado con placebo de 1 año en sujetos con dermatitis atópica (Estudio 3), se reportaron eventos tromboembólicos cardiovasculares (muertes cardiovasculares, infartos de miocardio no fatales y accidentes cerebrovasculares no fatales) en 1 (0.9%) del grupo DUPIXENT® + TCS 300 mg cada 2 semanas, 0 (0.0%) del grupo DUPIXENT® + TCS 300 mg cada semana y 1 (0.3%)del grupo placebo+ TCS.
En el estudio controlado con placebo de 24 semanas en sujetos con RSCcPN (ensayo CSNP 1), se reportaron eventos tromboembólicos cardiovasculares (muertes cardiovasculares, infartos de miocardio no fatales y accidentes cerebrovasculares no fatales) en 1 (0.7%) del grupo DUPIXENT® y 0 (0.0%) del grupo placebo. En el estudio controlado con placebo de 1 año en sujetos con RSCcPN (ensayo CSNP 2), no se informaron casos de eventos tromboembólicos cardiovasculares (muertes cardiovasculares, infartos de miocardio no fatales y accidentes cerebrovasculares no fatales) en ningún grupo de tratamiento.
lnmunogenicidad: Como sucede con todas las proteínas terapéuticas, existe el potencial de inmunogenicidad. La detección de formación de anticuerpos es altamente dependiente de la sensibilidad y especificidad del ensayo.
Adicionalmente, la incidencia observada de anticuerpos (incluyendo anticuerpos neutralizantes) positiva en un ensayo puede estar influenciada por diversos factores, incluyendo la metodología del ensayo, el manejo de muestras, el momento de recolección de muestras, medicamentos concomitantes y enfermedades subyacentes.
Por estos motivos, la comparación de la incidencia de anticuerpos hacia dupilumab en los estudios descritos abajo con la incidencia de anticuerpos en otros estudios u otros productos puede ser errónea.
Aproximadamente el 5% de los sujetos con dermatitis atópica, asma o RSCcPN que recibieron DUPIXENT® 300 mg cada 2 semanas durante 52 semanas desarrollaron anticuerpos contra dupilumab; aproximadamente el 2% exhibió respuestas ADA persistentes y aproximadamente el 2% tenía anticuerpos neutralizantes. Se observaron resultados similares en sujetos pediátricos (de 6 a 11 años de edad) con dermatitis atópica que recibieron DUPIXENT® 200 mg cada 2 semanas o 300 mg cada 4 semanas durante 16 semanas.
Aproximadamente el 16% de los sujetos adolescentes con dermatitis atópica que recibieron DUPIXENT® 300 mg o 200 mg cada dos semanas durante 16 semanas desarrollaron anticuerpos contra dupilumab; aproximadamente el 3% exhibió respuestas persistentes a ADA y aproximadamente el 5% tenía anticuerpos neutralizantes.
Aproximadamente 9% de los sujetos con asma que recibieron DUPIXENT® 200 mg cada 2 semanas durante 52 semanas desarrollaron anticuerpos contra dupilumab; de este porcentaje, aproximadamente 4% exhibió respuestas persistentes a ADA y aproximadamente 4% tuvo anticuerpos neutralizantes.
Independientemente de la edad o la población, aproximadamente del 2% al 4% de los sujetos en los grupos de placebo fueron positivos para anticuerpos contra DUPIXENT®; aproximadamente el 2% exhibió respuestas persistentes a ADA y aproximadamente el 1% tenía anticuerpos neutralizantes.
Los títulos de anticuerpos detectados tanto en los sujetos tratados con DUPIXENT® como con placebo fueron generalmente bajos. En los sujetos quienes recibieron DUPIXENT®, el desarrollo de un alto título de anticuerpos hacia dupilumab fue asociado a una menor concentración sérica de dupilumab (ver sección Farmacocinética y farmacodinamia).
Dos sujetos que experimentaron respuestas de títulos altos de anticuerpos desarrollaron enfermedad del suero o reacciones similares a enfermedad del suero, durante la terapia con DUPIXENT® (ver sección Precauciones generales).
Experiencia Posterior a la comercialización: Se han identificado las siguientes reacciones adversas adicionales durante el uso posterior a la comercialización de DUPIXENT®. Debido a que estas reacciones se notifican voluntariamente a partir de una población de tamaño incierto, no siempre es posible estimar de forma fiable su frecuencia o establecer una relación causal con la exposición a los medicamentos.
Trastornos de la piel y del tejido subcutáneo:
Erupción facial.
Trastornos del sistema inmunitario:
Angioedema.
Trastornos musculoesqueléticos y del tejido conectivo:
Artralgia.
PRECAUCIONES EN RELACIÓN CON EFECTOS DE CARCINOGÉNESIS, MUTAGÉNESIS, TERATOGÉNESIS Y SOBRE LA FERTILIDAD:
No se han realizado estudios en animales para evaluar el potencial carcinogénico o mutagénico de dupilumab.
No se observaron efectos sobre los parámetros de fertilidad, tales como órganos reproductivos, duración del ciclo menstrual o análisis de esperma, en ratones sexualmente maduros que recibieron por vía subcutánea un anticuerpo homólogo contra IL-4Rα en dosis hasta de 200 mg/kg/semana.
INTERACCIONES MEDICAMENTOSAS Y DE OTRO GÉNERO:
Vacunas de microorganismos vivos: Evitar el uso de vacunas de microorganismos vivos en pacientes tratados con DUPIXENT®.
Vacunas de microorganismos no vivos: Las respuestas inmunes a la vacunación fueron evaluadas en un estudio en el cual los sujetos con dermatitis atópica fueron tratados una vez a la semana durante 16 semanas, con 300 mg de dupilumab (dos veces la frecuencia recomendada de dosificación). Después de 12 semanas de administración de DUPIXENT®, los sujetos fueron vacunados con una vacuna Tdap (Adacel®) y una vacuna de polisacáridos de meningococos (Menomune®). Las respuestas de los anticuerpos al toxoide tetánico y a los polisacáridos de meningococos del serogrupo C fueron evaluadas 4 semanas después. Las respuestas de los anticuerpos a ambas vacunas de tétanos y a la vacuna de polisacáridos meningocócicos fueron similares en los sujetos tratados con dupilumab y con placebo. Las respuestas inmunes a otros componentes activos de las vacunas de Adacel® y Menomune® no fueron evaluadas.
ALTERACIONES EN LOS RESULTADOS DE PRUEBAS DE LABORATORIO: En estudios clínicos realizados con DUPIXENT® se ha reportado eosinofilia (ver sección Reacciones secundarias y adversas).
PRECAUCIONES GENERALES:
Hipersensibilidad: En menos del 1% de los sujetos que recibieron DUPIXENT® en los estudios clínicos, se reportaron reacciones de hipersensibilidad, incluyendo urticaria generalizada, erupción cutánea, eritema nodoso, anafilaxia, enfermedad del suero o reacciones similares a enfermedad del suero, y angioedema (ver Reacciones adversas y secundarias). Dos sujetos en el programa de desarrollo de dermatitis atópica experimentaron enfermedad del suero o reacciones similares a la enfermedad del suero que fueron asociados con títulos altos de anticuerpos contra dupilumab. Si ocurre una reacción de hipersensibilidad clínicamente significativa, establezca el tratamiento adecuado e interrumpa la administración de DUPIXENT® (ver Reacciones adversas y secundariaS).
Conjuntivitis y eventos relacionados con la queratitis: Se han reportado eventos relacionados de conjuntivitis y queratitis con DUPIXENT®, durante los ensayos clínicos y posterior a la comercialización, predominantemente en pacientes con dermatitis atópica. Algunos pacientes reportaron alteraciones visuales (por ejemplo, visión borrosa) asociadas con conjuntivitis o queratitis (ver sección Reacciones secundarias y adversas).
Indicar a los pacientes que reporten nuevos síntomas oculares o empeoramiento de síntomas oculares, a su profesional de salud. Considere la posibilidad de realizar un examen oftalmológico a pacientes que desarrollen conjuntivitis y que persista después de un tratamiento estándar o signos y síntomas que sugieran queratitis, según corresponda (ver sección Reacciones secundarias y adversas).
Condiciones eosinofílicas: Los pacientes que fueron tratados para el asma pueden presentar eosinofilia sistémica grave que a veces presenta características clínicas de neumonía eosinofílica o vasculitis compatible con granulomatosis eosinofílica con poliangeitis granulomatosa eosinofílica, con poliangitis, condiciones que a menudo son tratadas con terapia sistémica con corticosteroides. Estos eventos pueden estar asociados con la reducción del tratamiento con corticosteroides orales. Los médicos deben estar atentos a la erupción vasculítica, el empeoramiento de los síntomas pulmonares, las complicaciones cardiacas y/o neuropatía que se presenta en sus pacientes con eosinofilia. Se han reportado casos de neumonía eosinofílica y casos de vasculitis compatibles con granulomatosis eosinofílica con poliangitis con DUPIXENT® en pacientes adultos que participaron en el programa de desarrollo de asma, así como en pacientes adultos con asma comórbida en el programa de desarrollo de RSCcPN. No se ha establecido una asociación causal entre DUPIXENT® y estas condiciones.
Síntomas agudos de asma o exacerbación asmática: DUPIXENT® no debe ser utilizado para tratar los síntomas agudos de asma o las exacerbaciones agudas. No use DUPIXENT® para tratar broncoespasmo agudo o el estado asmático. Los pacientes deben consultar a su médico si su asma no se controla o empeora después de iniciar el tratamiento con DUPIXENT®.
Reducción de dosificación de corticosteroides: No interrumpa bruscamente los corticosteroides sistémicos, tópicos o inhalados al inicio del tratamiento con DUPIXENT®. Las reducciones en la dosis de corticosteroides, si son apropiadas, deben ser graduales y realizarse bajo la supervisión directa de un médico. La reducción en la dosis de corticosteroides puede estar asociada con síntomas de abstinencia sistémica y/o desenmascarar afecciones previamente suprimidas por el tratamiento sistémico con corticosteroides.
Pacientes con dermatitis atópica o con RSCcNP o con asma comórbida: Aconseje a los pacientes con dermatitis atópica o RSCcPN que tienen o asma comórbida, que no ajusten o suspendan sus tratamientos para el asma, sin consultar con su médico.
Infecciones parasitarias (helmintos): Los pacientes con infecciones helmínticas conocidas fueron excluidos de su participación en los estudios clínicos. Se desconoce si DUPIXENT® influye en la respuesta inmune contra las infecciones helmínticas.
Trate a los pacientes con infecciones helmínticas preexistentes antes de iniciar DUPIXENT®. Si los pacientes se infectan mientras reciben DUPIXENT® y no responden al tratamiento antihelminto, suspenda el tratamiento con DUPIXENT® hasta que la infección se resuelva.
DOSIS Y VÍA DE ADMINISTRACIÓN:
Vía de administración: DUPIXENT® se administra por inyección subcutánea.
Dosis:
Dermatitis atópica:
Dosis en adultos: La dosis recomendada de DUPIXENT® para pacientes adultos consiste en una dosis inicial de 600 mg (dos inyecciones de 300 mg), seguido por 300 mg administrados cada dos semanas.
Dosis en pacientes pediátricos (6 a 17 años de edad): La dosis recomendada de DUPIXENT® para pacientes adolescentes de 6 a 17 años de edad está especificada en la Tabla 15.
Tabla 15. Dosis de DUPIXENT® administrada por vía subcutánea en pacientes adolescentes de 6 a 17 años de edad con dermatitis atópica
|
Peso corporal del paciente |
Dosis inicial |
Dosis subsecuente |
|
15 Kg a menos de 30 Kg |
600 mg (dos inyecciones de 300 mg) |
300 mg cada 4 semanas |
|
30 Kg a menos de 60 Kg |
400 mg (dos inyecciones de 200 mg) |
200 mg cada 2 semanas |
|
60 Kg o más |
600 mg (dos inyecciones de 300 mg) |
300 mg cada 2 semanas |
Terapias concomitantes tópicas: DUPIXENT® puede ser usado con o sin corticoesteroides tópicos. Pueden ser usados los inhibidores tópicos de la calcineurina, pero deben ser reservados para áreas problemáticas únicamente, tales como la cara, cuello, genitales y áreas intertriginosas.
Se debe considerar la interrupción del tratamiento en pacientes que no han demostrado respuesta después de 16 semanas de tratamiento. Algunos pacientes con respuesta parcial inicial pueden mejorar posteriormente con el tratamiento continuado después de 16 semanas.
Asma: La dosis recomendada de DUPIXENT® para adultos y adolescentes (de 12 años en adelante) es:
• Para el resto de los pacientes, una dosis inicial de 400 mg (dos inyecciones de 200 mg) seguida de 200 mg administrada cada dos semanas.
• Para pacientes con asma dependiente de corticosteroides orales o con dermatitis atópica de moderada a grave comórbida para la cual DUPIXENT® está indicado, empezar el tratamiento con una dosis inicial de 600 mg (dos inyecciones de 300 mg) seguida de 300 mg administrados cada dos semanas.
Rinosinusitis crónica con poliposis nasal: La dosis recomendada de DUPIXENT® para pacientes adultos es de 300 mg administrados en semanas alternas.
Instrucciones importantes de administración: Se pretende que DUPIXENT® sea usado bajo la asesoría de un profesional de la salud. El paciente puede auto- inyectarse DUPIXENT® después de recibir entrenamiento en técnicas de inyección subcutánea, usando la jeringa prellenada. En adolescentes de 12 años de edad o mayores, se recomienda que DUPIXENT® sea administrado por un adulto o bajo la supervisión de éste. La jeringa precargada de DUPIXENT® debe ser administrada por un médico o cuidador entrenado en niños de 6 a 11 años. Proporcionar entrenamiento adecuado a los pacientes y/o cuidadores acerca de la preparación y administración de DUPIXENT® previo al uso, de acuerdo a las “Instrucciones de uso”.
Para pacientes con asma y dermatitis atópica la dosis inicial de 600 mg, administrar cada una de las dos inyecciones de DUPIXENT® 300 mg en diferentes sitios de inyección.
Para pacientes con asma y dermatitis atópica la dosis inicial de 400 mg, administrar cada una de las dos inyecciones de DUPIXENT® 200 mg en diferentes sitios de inyección.
Administrar la inyección subcutánea en el muslo o abdomen, excepto en los 5 centímetros (2 pulgadas) alrededor del ombligo. También puede ser usada la parte superior del brazo, si un cuidador administra la inyección. Alternar el sitio de inyección en cada aplicación. NO inyectar DUPIXENT® en la piel que está adolorida, inflamada, con moretones o cicatrizada.
Si se olvida una dosis cada dos semanas, indique al paciente que se administre la inyección dentro de los 7 días posteriores a la dosis olvidada y luego reanude el programa original del paciente. Si la dosis omitida no se administra dentro de los 7 días, indique al paciente que espere hasta la siguiente dosis en el horario original.
Si se omite una dosis cada 4 semanas, indique al paciente que se administre la inyección dentro de los 7 días posteriores a la dosis omitida y luego reanude el horario original del paciente. Si la dosis omitida no se administra dentro de los 7 días, indique al paciente que se administre la dosis, comenzando un nuevo horario basado en esta fecha.
Existen dos presentaciones de DUPIXENT® (es decir, jeringa prellenada con o sin protector de aguja). Las “Instrucciones de uso” de DUPIXENT® de cada presentación, contiene instrucciones más detalladas sobre la preparación y administración de DUPIXENT® (ver sección Instrucciones de uso).
Preparación para el uso de la jeringa prellenada de DUPIXENT®: Antes de la inyección, retirar DUPIXENT® del refrigerador y permitir que DUPIXENT® alcance la temperatura ambiente (45 minutos para la jeringa prellenada de 300 mg/2 mL y 30 minutos para la jeringa prellenada de 200 mg/1.14 mL) sin retirar la tapa de la aguja.
Inspeccionar visualmente DUPIXENT® en busca de partículas suspendidas o decoloración, previo a la administración. DUPIXENT® es una solución clara a ligeramente opalescente, incolora a amarillo pálida. No administrar si el líquido contiene partículas suspendidas, está decolorado o nuboso (diferente de claro a ligeramente opalescente, incolora a amarillo pálida). DUPIXENT® no contiene conservadores; por lo tanto, desechar todo producto no usado o sobrante en la jeringa prellenada.
Poblaciones especiales:
Uso en pediatría:
Dermatitis atópica: Se ha establecido la seguridad y eficacia de DUPIXENT® en pacientes pediátricos a partir de los 6 años de edad con dermatitis atópica de moderada a grave.
Un total de 251 adolescentes de 12 a 17 años de edad con dermatitis atópica de moderada a grave fueron reclutados en el Estudio 6 y el Estudio 8, que incluyó a 367 niños de 6 a 11 años con dermatitis atópica grave. La seguridad y eficacia fueron generalmente consistentes entre adolescentes y adultos (ver sección Reacciones secundarias y adversas y Farmacocinética y farmacodinamia: “Estudios clínicos”). DUPIXENT® está contraindicado en menores de 6 años de edad.
El uso de DUPIXENT® también está respaldado por el Estudio 7, el cual es un estudio de extensión de etiqueta abierta que inscribió a sujetos que completaron los Estudios 6 y 8. El Estudio 7 incluyó a 136 adolescentes del Estudio 6 y 110 niños del Estudio 8 con dermatitis atópica moderada en el momento de la inscripción en el estudio de extensión. El Estudio 7 incluyó a 64 adolescentes del Estudio 6 y 72 niños del Estudio 8 con dermatitis atópica grave en el momento de la inscripción. No se identificaron nuevas señales de seguridad en el Estudio 7 (ver sección Reacciones secundarias y adversas).
No se ha establecido la seguridad y eficacia en pacientes pediátricos < 6 años con dermatitis atópica.
Asma: Un total de 107 adolescentes de 12 a 17 años con asma de moderada a grave fueron reclutados en el Estudio AS 2 y recibieron 200 mg (N=21) o 300 mg (N=18) de DUPIXENT® (o el placebo correspondiente de 200 mg [N=34] o 300 mg [N=34]) cada 2 semanas. Las exacerbaciones del asma y la función pulmonar fueron evaluadas tanto en los adolescentes como en los adultos. Para las dosis de 200 mg y 300 mg cada 2 semanas, se observaron mejorías en el FEV1 (Media LS de cambio desde el valor basal en la semana 12) (0.36 L y 0.27 L, respectivamente). Para la dosis de 200 mg cada 2 semanas, los sujetos tuvieron una reducción en la tasa de exacerbaciones graves que fue consistente con los adultos. No se ha establecido la seguridad y eficacia en pacientes pediátricos (< 12 años de edad) con asma. La exposición a dupilumab fue mayor en los pacientes adolescentes que en los adultos al nivel de dosis respectivo, lo que se debió principalmente a la diferencia en el peso corporal (ver sección Farmacocinética y farmacodinamia).
El perfil de eventos adversos en adolescentes fue generalmente similar a los adultos (ver sección Reacciones secundarias y adversas).
Rinosinusitis crónica con poliposis nasal: RSCcPN normalmente no ocurre en niños. No se ha establecido la seguridad y eficacia en pacientes pediátricos (<18 años) con RSCcPN.
Uso en geriatría: De los 1472 sujetos con dermatitis atópica expuestos a DUPIXENT® en un estudio de rango de dosis y en estudios controlados con placebo, 67 sujetos tenían 65 años o más. Aun cuando no se observaron diferencias en la seguridad o eficacia entre los sujetos mayores y los más jóvenes, el número de sujetos de 65 años y más no fue suficiente para determinar si respondieron de manera diferente a los sujetos más jóvenes (ver sección Farmacocinética y farmacodinamia). De los 1977 sujetos con asma expuestos a DUPIXENT®, un total de 240 sujetos tenían 65 años o más. La eficacia y la seguridad en este grupo de edad fue similar a la población total del estudio. De los 440 sujetos con RSCcPN expuestos a DUPIXENT®, un total de 79 sujetos tenían 65 años o más. La eficacia y seguridad en este grupo de edad fueron similares a la de la población general del estudio.
MANIFESTACIONES Y MANEJO DE LA SOBREDOSIFICACIÓN O INGESTA ACCIDENTAL:
No existe un tratamiento específico para la sobredosis de DUPIXENT®. En caso de sobredosis, monitorear al paciente en busca de cualquier signo o síntoma de reacción adversa e instituir el tratamiento sintomático apropiado inmediatamente.
PRESENTACIONES:
DUPIXENT® 300 mg/2 mL:
• Caja con 1 ó 2 jeringa(s) prellenada(s) con protector de aguja e instructivo anexo.
• Caja con 1 ó 2 jeringa(s) prellenada(s) e instructivo anexo.
DUPIXENT® 200 mg/1.14 mL:
• Caja con 1 ó 2 jeringa(s) prellenada(s) con protector de aguja e instructivo anexo.
RECOMENDACIONES SOBRE ALMACENAMIENTO:
DUPIXENT® es una solución estéril, libre de conservadores.
Desechar cualquier cantidad no usada. Consérvese en refrigeración entre 2ºC a 8ºC en el empaque original para protegerla de la luz.
Si es necesario, las jeringas prellenadas pueden ser mantenidas a temperatura ambiente hasta 25ºC durante un máximo de 14 días. Después de retirar el medicamento del refrigerador, DUPlXENT® debe ser usado dentro de los 14 días siguientes, una vez transcurrido este periodo, el producto deberá desecharse.
No almacenar a temperaturas superiores a 25ºC. No exponer la jeringa al calor o a luz solar directa.
Todo el medicamento no usado o material de desperdicio debe ser desechado de acuerdo con los requerimientos locales.
NO congelar. NO exponer al calor. NO agitar.
LEYENDAS DE PROTECCIÓN:
Literatura exclusiva para médicos. Su venta requiere receta médica. No se deje al alcance de los niños. No se administre en niños menores de 6 años con dermatitis atópica. No se administre en niños menores de 12 años con asma moderada a grave. No se administre en menores de 18 años con rinosinusitis crónica con poliposis nasal. No administrar durante el embarazo y la lactancia.
Reporte las sospechas de reacción adversa al correo: farmacovigilancia@cofepris.gob.mx
SANOFI-AVENTIS DE MÉXICO, S.A. de C.V.
Acueducto del Alto Lerma No. 2,
Zona Industrial Ocoyoacac,
C.P. 52740, Ocoyoacac, México, México
Reg. Núm. 177M2018, SSA IV


