
COBAS HIV-1/HIV-2 QUALITATIVE
Para visualizar el contenido completo de la IPPA (información para prescribir amplia), deberá iniciar sesión.
RESULTADOS:
El cobas® 5800 System y los cobas® 6800/8800 Systems detectan y diferencian de forma automática y simultánea el HIV-1 y el HIV-2 de muestras y controles, además de mostrar la validez de la prueba, los resultados generales y los resultados individuales de cada diana.
Control de calidad y validez de los resultados en el cobas® 5800 System:
• Se procesan un control negativo para plasma humano normal [(–) C] y dos controles positivos [HIV- 1M/HIV-2 (+)C y HIV-1O (+)C] al menos cada 72 horas o con cada lote de kit nuevo. Los controles positivo y/o negativo pueden programarse con mayor frecuencia en función de los procedimientos de laboratorio y/o la reglamentación local.
• Compruebe los avisos y los resultados asociados tanto en el software cobas® 5800 System como en el informe para garantizar la validez de la serie.
El software cobas® 5800 invalida automáticamente los resultados cuando los controles positivos o negativos fallan.
Nota: El cobas® 5800 System se suministra con la configuración estándar para el análisis de un conjunto de controles (positivo y negativo) con cada serie, pero se puede modificar por un programa menos frecuente de hasta cada 72 horas según los procedimientos de laboratorio y la reglamentación local. Póngase en contacto con su ingeniero técnico de Roche y/o con el represente del servicio técnico de Roche para obtener más información.
Resultados del control en el cobas® 5800 System:
Los resultados de los controles se muestran en el software cobas® 5800, en la aplicación de controles.
• Los controles se marcan como “Válido” en la columna “Resultados de control” cuando todas las dianas del control se han notificado como válidas. Los controles se marcan como “No válido” en la columna “Resultados de control” cuando todas o alguna de las dianas del control se han notificado como no válidas.
• Los controles marcados como “No válido” muestran un aviso en la columna “Aviso”. En la vista de detalles podrá encontrar más información sobre el motivo por el que el control se ha notificado como no válido, además de información sobre el aviso.
• Si uno de los controles positivos no válido, repita el análisis de todos los controles positivos y de todas las muestras asociadas. Si el control negativo no es válido, repita el análisis de todos los controles y de todas las muestras asociadas.
Control de calidad y validez de los resultados en los cobas® 6800/8800 Systems:
• Con cada serie se procesan un control negativo para plasma humano normal [(–) C] y dos controles positivos [HIV-1M/HIV-2 (+)C y HIV-1O (+)C].
• Compruebe los avisos y los resultados asociados tanto en el software cobas® 6800/8800 como en el informe para garantizar la validez de la serie.
• La serie se considera válida cuando no hay avisos para ninguno de los tres controles.
El software cobas® 6800/8800 invalida automáticamente los resultados cuando los controles positivos y negativos fallan.
Avisos de controles en los cobas® 6800/8800 Systems:
Tabla 15. Avisos de controles para los controles negativo y positivo
|
Control negativo |
Aviso |
Resultado |
Interpretación |
|
(–) C |
Q02 (Serie de control errónea) |
No válida |
Toda la serie se considera inválida si el resultado del (–) C no es válido. |
|
Control positivo |
Aviso |
Resultado |
Interpretación |
|
HIV-1M/HIV-2 (+)C |
Q02 (Serie de control errónea) |
No válida |
Toda la serie se considera no válida si el resultado del HIV-1M/HIV-2 (+)C no es válido. |
|
HIV-1O (+)C |
Q02 (Serie de control errónea) |
No válida |
Toda la serie se considera no válida si el resultado del HIV-1O (+)C no es válido. |
Si la serie no es válida, repita las pruebas para toda la serie, incluyendo muestras y controles.
(-) C hace referencia al control negativo para NHP, HIV-1M/HIV-2 (+)C es el control positivo de cobas® HIV-1M/HIV-2 mientras que HIV-1O (+)C hace referencia al control positivo para cobas® HIV-1O en el software cobas® 6800/8800.
Interpretación de los resultados en el cobas® 5800 System:
Los resultados de las muestras se muestran en el software cobas® 5800, en la aplicación de controles.
En las series de control válidas, compruebe cada muestra para detectar avisos en el software cobas® 5800 y/o en el informe. La interpretación de resultados se debe realizar del siguiente modo:
• Una serie de control válida puede incluir resultados de muestras tanto válidos como no válidos.
• Las muestras asociadas a una serie de control válida se muestran como “Válido” en la columna “Resultados de control” cuando todos los resultados de la diana de control se han notificado como válidos. Las muestras asociadas a una serie de control errónea se muestran como “No Válido” en la columna “Resultados de control” cuando todos los resultados de la diana de control se han notificado como no válidos.
• Si los controles asociados al resultado de una muestra no son válidos, se añade un aviso específico al resultado de la muestra de la siguiente manera:
° Q05D: fallo de validación del resultado por un control positivo no válido.
° Q06D: fallo de validación del resultado por un control negativo no válido.
• Los valores de la columna “Resultado” para el resultado individual de la diana de la muestra deben interpretarse tal como se muestra en la Tabla 16.
• Si una o más dianas de la muestra se marcan como “No válido”, el software cobas® 5800 muestra un aviso en la columna “Aviso”. En la vista de detalles podrá encontrar más información sobre el motivo por el que las dianas de la muestra se han notificado como no válidas, además de información sobre el aviso.
Los resultados y sus interpretaciones correspondientes para la detección del HIV-1 y el HIV-2 se muestran en la Tabla 16.
Tabla 16. Resultados para la interpretación de los resultados individuales de cada diana en el cobas® 5800 System
|
Resultado |
Interpretación |
|
|
HIV-1 Reactive |
HIV-2 Reactive |
Todos los resultados solicitados son válidos. Se ha detectado una señal de diana para HIV-1 e HIV-2. |
|
HIV-1 Reactive |
HIV-2 Non-Reactive |
Todos los resultados solicitados son válidos. Se ha detectado una señal de diana para HIV-1. No se ha detectado ninguna señal de diana para HIV-2. |
|
HIV-1 Non-Reactive |
HIV-2 Reactive |
Todos los resultados solicitados son válidos. No se ha detectado ninguna señal de diana para HIV-1. Se ha detectado una señal de diana para HIV-2. |
|
HIV-1 Non-Reactive |
HIV-2 Non-Reactive |
Todos los resultados solicitados son válidos. No se ha detectado ninguna señal de diana para HIV-1 o HIV-2. |
|
Invalid |
Invalid |
Ni el resultado de HIV-1 ni el de HIV-2 son válidos. La muestra original debe volver a analizarse para obtener resultados válidos de HIV-1 e HIV-2. Si los resultados siguen siendo no válidos, deberá obtenerse una nueva muestra. |
Interpretación de los resultados en los cobas® 6800/8800 Systems:
En las series válidas, compruebe cada muestra para detectar avisos en el software cobas® 6800/8800 Systems y/o en el informe. La interpretación de resultados se debe realizar del siguiente modo:
• Una serie válida puede incluir resultados de muestras tanto válidos como no válidos.
• Las muestras presentan un “Yes” en la columna “Válida” si todos los resultados de las dianas solicitadas muestran resultados válidos. Las muestras que presentan un “No” en la columna “Válida” pueden requerir alguna interpretación o acción adicional.
• Los valores de la columna “Resultado global” para muestras individuales deberían interpretarse del modo siguiente:
° Reactive: todos los resultados solicitados son reactivos, o bien uno de los resultados solicitados es reactivo y, el otro, no reactivo.
° Non-Reactive: todos los resultados solicitados son no reactivos.
° Invalid: al menos uno de los resultados solicitados es inválido.
• Los resultados de la diana obtenidos para las muestras individuales son válidos, salvo que se especifique lo contrario.
Los resultados y sus interpretaciones correspondientes para la detección del HIV-1 y el HIV-2 se muestran en la Tabla 17.
Tabla 17. Resultados de la diana para la interpretación de los resultados individuales de cada diana en los cobas® 6800/8800 Systems
|
Válido |
Resultado general |
Diana 1 |
Diana 2 |
Interpretación |
|
Yes |
Reactive |
HIV-1 Reactive |
HIV-2 Reactive |
Todos los resultados solicitados son válidos. Se ha detectado una señal de diana para HIV-1 e HIV-2. |
|
Yes |
Reactive |
HIV-1 Reactive |
HIV-2 Non-Reactive |
Todos los resultados solicitados son válidos. Se ha detectado una señal de diana para HIV-1. No se ha detectado ninguna señal de diana para HIV-2. |
|
Yes |
Reactive |
HIV-1 Non-Reactive |
HIV-2 Reactive |
Todos los resultados solicitados son válidos. No se ha detectado ninguna señal de diana para HIV-1. Se ha detectado una señal de diana para HIV-2. |
|
Yes |
Non-Reactive |
HIV-1 Non-Reactive |
HIV-2 Non-Reactive |
Todos los resultados solicitados son válidos. No se ha detectado ninguna señal de diana para HIV-1 o HIV-2. |
|
No |
Invalid |
Invalid |
Invalid |
Ni el resultado de HIV-1 ni el de HIV-2 son válidos. La muestra original debe volver a analizarse para obtener resultados válidos de HIV-1 e HIV-2. Si los resultados siguen siendo no válidos, deberá obtenerse una nueva muestra. |
Limitaciones del procedimiento:
• La prueba COBAS® HIV-1/HIV-2 QUALITATIVE se ha evaluado para ser utilizada únicamente con el cobas® HIV- 1/HIV-2 Qualitative Control Kit, el cobas® NHP Negative Control Kit, el cobas omni MGP Reagent, el cobas omni Lysis Reagent, el cobas omni Specimen Diluent, el cobas omni Wash Reagent y el cobas® Specimen Pre-Extraction Reagent (utilizado en el flujo de trabajo con muestras de sangre seca) exclusivamente en los cobas® 5800/6800/8800 Systems.
• La fiabilidad de los resultados depende de que el tipo de muestra sea el adecuado (plasma conservado en EDTA o suero) y de la obtención de la muestra, así como de los procedimientos de almacenamiento y manipulación. La utilización del ensayo con otros tipos de muestras no garantiza la exactitud de los resultados.
• La detección de ácidos nucleicos del HIV-1 y el HIV-2 depende del número de partículas víricas presentes en la muestra y se puede ver afectada por los métodos de obtención de las mismas, el almacenamiento y la manipulación, factores propios del paciente (como la edad o la presencia de síntomas) y/o la fase de infección.
• Aunque es poco probable, las mutaciones en las regiones muy conservadas del genoma vírico cubiertas por la prueba COBAS® HIV-1/HIV-2 QUALITATIVE pueden afectar a la unión de cebadores y/o sondas e impedir la detección del virus.
• Debido a las diferencias específicas entre tecnologías, se recomienda a los usuarios que realicen estudios de correlación en el laboratorio para determinar las diferencias tecnológicas antes de cambiar de una a otra. Los usuarios deberán adherirse a las políticas y los procedimientos específicos.
• La prueba COBAS® HIV-1/HIV-2 QUALITATIVE no se ha concebido como prueba de cribado para la presencia de HIV-1/HIV-2 en donaciones de sangre o productos sanguíneos.
OBTENCIÓN, TRANSPORTE Y ALMACENAMIENTO DE LAS MUESTRAS:
Nota: Manipule todas las muestras y los controles como si pudieran transmitir agentes infecciosos.
Almacene todas las muestras a las temperaturas especificadas.
La estabilidad de las muestras se ve afectada por las temperaturas elevadas.
Si se utilizan muestras congeladas en tubos secundarios, deje que se descongelen a temperatura ambiente (15-30 °C) por completo y, a continuación, agítelas unos instantes (entre 3 y 5 segundos) y centrifúguelas para que todo el volumen de la muestra se deposite en la parte inferior del tubo.
Muestras:
Muestras de suero y plasma conservado en EDTA:
• La sangre entera debería recogerse en tubos de separación de suero SST™, en tubos para preparación de plasma BD Vacutainer® PPT™ para métodos de pruebas de diagnóstico molecular o en tubos estériles y utilizar EDTA como anticoagulante. Siga las instrucciones del fabricante de los tubos de recogida de muestras.
• La sangre entera recogida en tubos de separación de suero SST™, tubos para preparación de plasma BD Vacutainer® PPT™ para métodos de pruebas de diagnóstico molecular o tubos estériles con EDTA como anticoagulante pueden almacenarse y/o transportarse durante un máximo de 24 horas a una temperatura comprendida entre 2 °C y 25 °C antes de la preparación del plasma o el suero. La centrifugación debe realizarse conforme a las instrucciones del fabricante.
• Después de la separación, las muestras de suero o plasma conservado en EDTA pueden almacenarse en tubos secundarios durante un máximo de 24 horas a 30 °C seguido de hasta 5 días de almacenamiento a una temperatura comprendida entre 2 °C y 8 °C o un máximo de 6 semanas a ≤ –20 °C. Para un almacenamiento a más largo plazo se recomiendan temperaturas ≤ –60 °C.
• Las muestras de plasma y suero se mantienen estables hasta un máximo de tres ciclos de congelación/descongelación si se congelan a ≤ –20 °C.
• Consulte la Ilustración 1 para conocer las condiciones de almacenamiento.
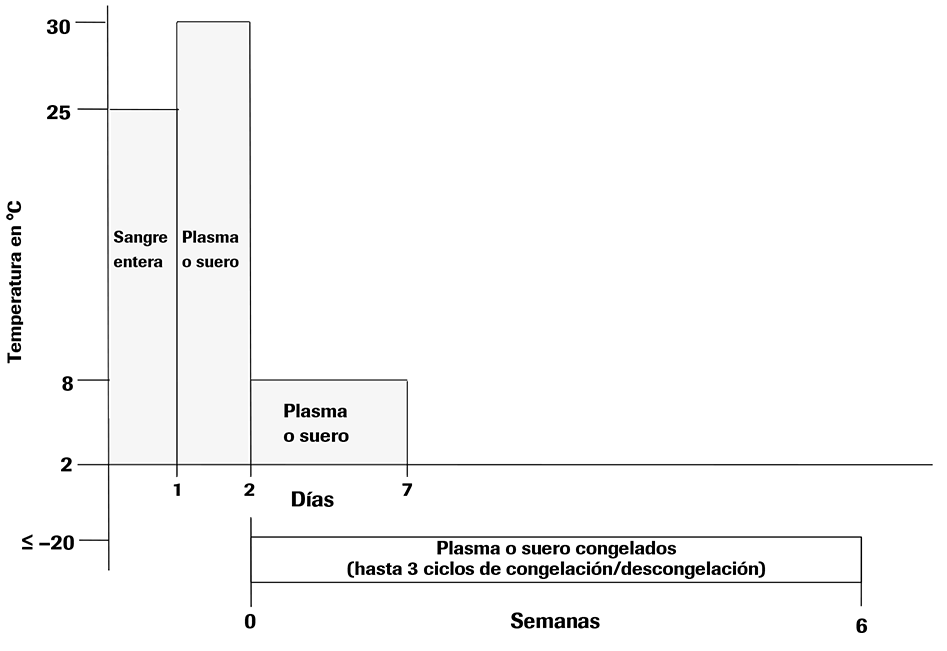
Ilustración 1. Condiciones de almacenamiento de muestras de suero, plasma y sangre entera
• Si las muestras se van a transportar, es recomendable empaquetarlas y etiquetarlas de acuerdo con la reglamentación estatal y/o internacional relativa al transporte de muestras y agentes etiológicos.
Muestras de sangre seca:
• Obtenga las muestras DBS mediante los procedimientos clínicos correspondientes.
• Se recomienda aplicar un mínimo de 70 μL de sangre capilar dentro de cada círculo delineado en la tarjeta para muestras DBS.
• Asegúrese de que AMBAS caras del papel están saturadas y de que el círculo delineado está completamente cubierto.
• Deje secar la muestra de sangre seca a temperatura ambiente (18-25 °C) durante 3 horas como mínimo, protegiendo la tarjeta DBS de la luz solar directa.
• Para conocer más información, consulte el boletín técnico de las tarjetas de papel de filtro que utilice.
• Se recomienda preparar un mínimo de 3 discos de papel por cada muestra de paciente.
• Almacene las DBS en bolsas resellables individuales con una bolsita desecante en cada bolsa.
• Las DBS se pueden transportar o almacenar a 15-30 °C hasta un máximo de tres meses.
• Si las muestras se van a transportar, es recomendable empaquetarlas y etiquetarlas de acuerdo con la reglamentación estatal y/o internacional relativa al transporte de muestras y agentes etiológicos.
PRECAUCIONES Y REQUISITOS DE MANIPULACIÓN:
Advertencias y precauciones:
Como sucede con cualquier procedimiento analítico, resulta esencial seguir las buenas prácticas de laboratorio recomendadas para obtener un rendimiento correcto del ensayo. Debido a la elevada sensibilidad de esta prueba, deben extremarse las precauciones para evitar cualquier tipo de contaminación de los reactivos y las mezclas de amplificación.
• Para diagnóstico in vitro exclusivamente.
• Esta prueba no está diseñada para utilizarse en el cribado de donantes de sangre o plasma.
• Todas las muestras de paciente deben tratarse como si fueran infecciosas, utilizando los procedimientos de laboratorio recomendados tal como se describe en la publicación Biosafety in Microbiological and Biomedical Laboratories y en el documento M29-A4 del CLSI.18,19 Solamente el personal competente en la manipulación de material biopeligroso y en el uso de la prueba COBAS® HIV-1/HIV-2 QUALITATIVE y los cobas® 5800/6800/8800 Systems deberían llevar a cabo este procedimiento.
• Todos los materiales de origen humano deben considerarse potencialmente infecciosos y manipularse teniendo en cuenta las precauciones generales. En caso de que se produzca algún derrame, lleve a cabo una desinfección inmediata siguiendo los procedimientos apropiados del laboratorio.
° Si se produce un derrame de muestras DBS en el cobas® Specimen Pre-Extraction Reagent (que contiene tiocianato de guanidina), no permita que entre en contacto con soluciones de hipoclorito de sodio que contengan desinfectantes como la lejía. Tales mezclas pueden producir gases de alta toxicidad.
• El COBAS® HIV-1/HIV-2 Qualitative Control Kit y el COBAS® NHP Negative Control Kit contienen plasma obtenido de sangre humana. Se ha analizado el material original mediante las pruebas para anticuerpos autorizadas y no se ha considerado reactivo para la presencia de anticuerpos del HIV-1/2. En el análisis del plasma humano normal con métodos de PCR no se ha detectado ARN de HIV-1 (grupos M y O) ni ARN de HIV-2. Ningún método de prueba conocido puede garantizar totalmente que un producto derivado de la sangre humana no transmita agentes infecciosos.
• No congele la sangre entera ni las muestras almacenadas en tubos primarios.
• El cobas® Specimen Pre-Extraction Reagent es sensible a la luz y se suministra en botellas con protección lumínica.
• Utilice solo el material fungible suministrado o que se requiera expresamente para garantizar el óptimo rendimiento de la prueba.
• Puede solicitar Hojas de datos de seguridad (Safety Data Sheets, SDS) al representante local de Roche.
• Siga al pie de la letra los procedimientos y las directrices que se suministran para garantizar la correcta realización de la prueba. Cualquier variación de dichos procedimientos y directrices podría afectar al rendimiento óptimo de la prueba.
• Podrían producirse resultados falsos positivos si no se evita la contaminación por arrastre de las muestras durante la manipulación y el procesamiento de las mismas.
• Informe a la autoridad competente local de cualquier incidente grave que pueda tener lugar durante la realización del ensayo.
Manipulación de reactivos:
• Manipule todos los reactivos, controles y muestras de acuerdo con las prácticas de laboratorio recomendadas para evitar la contaminación por arrastre de las muestras o los controles.
• Antes de utilizarlos, revise cada casete de reactivo, diluyente, reactivo de lisis, reactivo de lavado y cobas® Specimen Pre-Extraction Reagent (requerido únicamente para el uso con DBS) para comprobar que no existen signos de fugas. No utilice el material si hay alguna evidencia de fuga.
• El cobas omni Lysis Reagent y el cobas® Specimen Pre-Extraction Reagent contienen tiocianato de guanidina, una sustancia química potencialmente peligrosa. Evite el contacto de reactivos con la piel, los ojos o las membranas mucosas. En caso de contacto, lave inmediatamente la zona afectada con abundante agua para evitar quemaduras.
• Los kits de la prueba COBAS® HIV-1/HIV-2 QUALITATIVE, el cobas omni MGP Reagent y el cobas omni Specimen Diluent contienen azida sódica como conservante. Evite el contacto de estos reactivos con la piel, los ojos o las membranas mucosas. En caso de contacto, lave inmediatamente la zona afectada con abundante agua para evitar quemaduras. Si se producen salpicaduras de reactivos, diluya las manchas con agua antes de secarlas con un paño.
• No permita que el cobas omni Lysis Reagent o el cobas® Specimen Pre-Extraction Reagent, que contienen tiocianato de guanidina, entren en contacto con la solución de hipoclorito de sodio (lejía). Tales mezclas pueden producir gases de alta toxicidad.
• Elimine todos los materiales que hayan estado en contacto con las muestras y los reactivos de acuerdo con la reglamentación nacional, estatal y local.
Buenas prácticas de laboratorio:
• No pipetee con la boca.
• No se debe comer, beber ni fumar en las áreas de trabajo.
• Utilice guantes, bata de laboratorio y protección ocular cuando manipule las muestras y los reactivos. Es necesario cambiarse los guantes entre la manipulación de las muestras y de los kits de la prueba COBAS® HIV-1/HIV-2 QUALITATIVE y los reactivos cobas omni para evitar la contaminación. Evite la contaminación de los guantes durante la manipulación de las muestras y de los controles.
• Lávese bien las manos después de manipular las muestras y los reactivos del kit, y cuando se saque los guantes.
• Limpie y desinfecte minuciosamente todas las superficies de trabajo del laboratorio usando una solución recién preparada de hipoclorito de sodio al 0,5% en agua destilada o desionizada (lejía doméstica diluida a 1:10).
A continuación, límpielas con un trapo impregnado en etanol al 70%.
• Si el derrame se produce sobre un instrumento cobas® 5800, siga las instrucciones descritas en la Asistencia al usuario y/o la Guía del usuario del cobas® 5800 System para limpiar y descontaminar correctamente la superficie del instrumento.
• Si el derrame se produce sobre un instrumento cobas® 6800/8800, siga las instrucciones descritas en la Asistencia al usuario o la Guía del usuario de los cobas® 6800/8800 Systems para limpiar y descontaminar correctamente la superficie de los instrumentos.
REACTIVOS Y MATERIALES:
Reactivos y controles para la prueba COBAS® HIV-1/HIV-2 QUALITATIVE:
Todos los reactivos y controles sin abrir deben almacenarse como se recomienda desde la Tabla 1 hasta la Tabla 5.
Tabla 1. COBAS® HIV-1/HIV-2 QUALITATIVE
|
COBAS® HIV-1/HIV-2 QUALITATIVE Almacenar a 2-8 °C Casete para 192 pruebas (P/N 09040528190) |
||
|
Componentes del kit |
Composición del reactivo |
Cantidad por kit 192 pruebas |
|
Solución de proteinasa (PASE) |
Buffer Tris, < 0,05% de EDTA, cloruro de calcio, acetato de calcio, 8% (p/v) de proteinasa, glicerol EUH210: Puede solicitarse la ficha de datos de seguridad. EUH208: Contiene subtilisina. Puede provocar una reacción alérgica. |
22,3 mL |
|
Control interno (IC) |
Buffer Tris, < 0,05% de EDTA, < 0,001% de constructo de Armored RNA como control interno (ARN no infeccioso encapsulado en bacteriófago MS2), < 0,002% de ARN Poli rA (sintético), < 0,1% de azida sódica |
21,2 mL |
|
Buffer de elución (EB) |
Buffer Tris, 0,2% de metil-4-hidroxibenzoato |
21,2 mL |
|
Reactivo 1 de Master Mix (MMX-R1) |
Acetato de manganeso, hidróxido potásico, < 0,1% de azida sódica |
7,5 mL |
|
Reactivo 2 de Master Mix para HIV-1/HIV-2 (HIV-1/HIV-2 MMX-R2) |
Buffer Tricina, acetato de potasio, 18% de sulfóxido de dimetilo, glicerol, Tween 20, EDTA, < 0,06% de dATP, dCTP, dGTP, < 0,14% de dUTP, < 0,01% de cebadores ascendente y descendente de HIV-1, HIV-2 y control interno, < 0,01% de sondas marcadas con fluorescente para HIV-1 y HIV-2, < 0,01% de sonda de control interno marcada con fluorescente, < 0,01% de aptámero oligonucleótido, < 0,01% de polimerasa de ADN Z05D, < 0,01% de enzima AmpErase (uracil-N-glicosilasa) (microbiana), < 0,1% de azida sódica |
9,7 mL |
Tabla 2. cobas® HIV-1/HIV-2 Qualitative Control Kit
|
COBAS® HIV-1/HIV-2 Qualitative Control Kit Almacenar a 2-8 °C Para uso en el sistema cobas® 5800 (P/N 09040536190) Para uso en sistemas cobas® 6800/8800 (P/N 07862091190 y P/N 09040536190) |
|||
|
Componentes del kit |
Composición del reactivo |
Cantidad por kit |
Símbolo de seguridad y advertencia* |
|
Control positivo para HIV-1M/HIV-2 (HIV-1M/HIV-2 (+)C) |
< 0,001% de Armored RNA sintético de HIV-1 grupo M encapsulado en proteína recubierta de bacteriófago MS2, < 0,001% de Armored RNA sintético de HIV-2 encapsulado en proteína recubierta de bacteriófago MS2, plasma humano normal, no reactivo según las pruebas autorizadas para anticuerpos del HIV-1/2; ARN de HIV-1 y ARN de HIV-2 no detectables mediante métodos de PCR 0,1% de conservante ProClin® 300** |
5,2 mL (8 × 0,65 mL) |
ADVERTENCIA H317: Puede provocar una reacción alérgica en la piel. H412: Nocivo para los organismos acuáticos, con efectos nocivos duraderos. P261: Evitar respirar el polvo/el humo/el gas/la niebla/los vapores/el aerosol. P273: Evítese su liberación al medio ambiente. P280: Utilice guantes protectores. P333 + P313: En caso de irritación o erupción cutánea: consultar a un médico. P362 + P364: Quitarse las prendas contaminadas y lavarlas antes de volver a usarlas. P501: Eliminar el contenido/el recipiente en una planta de eliminación de residuos autorizada. 55965-84-9 masa reactiva de: 5-cloro-2-metil-4-isotiazolin-3-ona [n.º CE 247-500-7] y 2-metil-2H- isotiazol-3-ona [n.º CE 220-239-6] (3:1). |
|
Control positivo para HIV-1O (HIV-1O (+)C) |
< 0,001% de Armored RNA sintético de HIV-1 grupo O encapsulado en proteína recubierta de bacteriófago MS2, plasma humano normal, no reactivo según las pruebas autorizadas para anticuerpos del HIV-1/2; ARN de HIV-1 y ARN de HIV-2 no detectables mediante métodos de PCR 0,1% de conservante ProClin® 300** |
5,2 mL (8 × 0,65 mL) |
ADVERTENCIA H317: Puede provocar una reacción alérgica en la piel. H412: Nocivo para los organismos acuáticos, con efectos nocivos duraderos. P261: Evitar respirar el polvo/el humo/el gas/la niebla/los vapores/el aerosol. P273: Evítese su liberación al medio ambiente. P280: Utilice guantes protectores. P333 + P313: En caso de irritación o erupción cutánea: consultar a un médico. P362 + P364: Quitarse las prendas contaminadas y lavarlas antes de volver a usarlas. P501: Eliminar el contenido/el recipiente en una planta de eliminación de residuos autorizada. 55965-84-9 masa reactiva de: 5-cloro-2-metil-4-isotiazolin-3-ona [n.º CE 247-500-7] y 2-metil-2H- isotiazol-3-ona [n.º CE 220-239-6] (3:1). |
* Las etiquetas de seguridad del producto se basan fundamentalmente en la regulación GHS de la UE.
** Sustancia peligrosa.
Tabla 3. cobas® NHP Negative Control Kit
|
cobas® NHP Negative Control Kit Almacenar a 2-8 °C Para uso en el sistema cobas® 5800 (P/N 09051554190) Para uso en sistemas cobas® 6800/8800 (P/N 07002220190 y P/N 09051554190) |
|||
|
Componentes del kit |
Composición del reactivo |
Cantidad por kit |
Símbolo de seguridad y advertencia* |
|
Control negativo para plasma humano normal (NHP-NC) |
Plasma humano normal, no reactivo según pruebas autorizadas para anticuerpos del VIH-1/2; ARN de VIH-1 y ARN de VIH-2 no detectables mediante métodos de PCR < 0,1% de conservante ProClin® 300** |
16 mL (16 × 1 mL) |
ADVERTENCIA H317: Puede provocar una reacción alérgica en la piel. P261: Evitar respirar el polvo/el humo/el gas/la niebla/los vapores/el aerosol. P272: Las prendas de trabajo contaminadas no podrán sacarse del lugar de trabajo. P280: Utilice guantes protectores. P333 + P313: En caso de irritación o erupción cutánea: consultar a un médico. P362 + P364: Quitarse las prendas contaminadas y lavarlas antes de volver a usarlas. P501: Eliminar el contenido/el recipiente en una planta de eliminación de residuos autorizada. 55965-84-9 masa reactiva de: 5-cloro-2-metil-4-isotiazolin-3-ona [n.º CE 247-500-7] y 2-metil- 2H-isotiazol-3-ona [n.º CE 220-239-6] (3:1). |
* Las etiquetas de seguridad del producto se basan fundamentalmente en la regulación GHS de la UE.
** Sustancia peligrosa.
Reactivos cobas omni para la preparación de muestras:
Tabla 4. Reactivos cobas omni para la preparación de las muestras*
|
Reactivos |
Composición del reactivo |
Cantidad por kit |
Símbolo de seguridad y advertencia** |
|
cobas omni MGP Reagent (MGP) Almacenar a 2-8 °C (P/N: 06997546190) |
Partículas de vidrio magnéticas, buffer Tris, 0,1% de metil-4 hidroxibenzoato, < 0,1% de azida sódica |
480 pruebas |
No aplicable |
|
cobas omni Specimen Diluent (SPEC DIL) Almacenar a 2-8 °C (P/N: 06997511190) |
Buffer Tris, 0,1% de metil-4 hidroxibenzoato, < 0,1% de azida sódica |
4 × 875 mL |
No aplicable |
|
cobas omni Lysis Reagent (LYS) Almacenar a 2-8 °C (P/N: 06997538190) |
43% (p/p) de tiocianato de guanidina***, 5% (p/v) de polidocanol***, 2% (p/v) de ditiotreitol, citrato de sodio dihidratado |
4 × 875 mL |
PELIGRO H302 + H332: Nocivo en caso de ingestión o inhalación. H314: Provoca quemaduras graves en la piel y lesiones oculares graves. H412: Nocivo para los organismos acuáticos, con efectos nocivos duraderos. EUH032: En contacto con ácidos libera gases muy tóxicos. P261: Evitar respirar el polvo/el humo/el gas/la niebla/los vapores/el aerosol. P273: Evítese su liberación al medio ambiente. P280: Llevar gafas/máscara de protección. P303 + P361 + P353 EN CASO DE CONTACTO CON LA PIEL (o el pelo): quitar inmediatamente todas las prendas contaminadas. Enjuagar la piel con agua. P304 + P340 + P310 EN CASO DE INHALACIÓN: transportar a la persona al aire libre y mantenerla en una posición que le facilite la respiración. Llamar a un CENTRO DE TOXICOLOGÍA/médico en caso de malestar. P305 + P351 + P338 + P310 EN CASO DE CONTACTO CON LOS OJOS: aclarar cuidadosamente con agua durante varios minutos. Quitar las lentes de contacto, si lleva y resulta fácil. Seguir aclarando. Llamar inmediatamente a un CENTRO DE TOXICOLOGÍA/médico. 593-84-0 Tiocianato de guanidina 9002-92-0 Polidocanol 3483-12-3 (R*,R*)-1,4-dimercaptobutano-2,3-diol |
|
cobas omni Wash Reagent (WASH) Almacenar a 15-30 °C (P/N: 06997503190) |
Citrato de sodio dihidratado, 0,1% de metil-4-hidroxibenzoato |
4,2 l |
No aplicable |
* Estos reactivos no están incluidos en el kit de la prueba COBAS® HIV-1/HIV-2 QUALITATIVE. Consulte el listado de material adicional necesario (Tabla 11 y Tabla 12).
** Las etiquetas de seguridad del producto se basan fundamentalmente en la regulación GHS de la UE.
*** Sustancia peligrosa.
cobas® Specimen Pre-Extraction Reagent:
Tabla 5. cobas® Specimen Pre-Extraction Reagent*
|
cobas® Specimen Pre-Extraction Reagent Almacenar a 2-8 °C (P/N 08064695190) |
|||
|
Reactivo |
Composición del reactivo |
Cantidad por kit |
Símbolo de seguridad y advertencia** |
|
cobas® Specimen Pre-Extraction Reagent (SPER) |
28% (p/p) de tiocianato de guanidina, 6% (p/v) de polidocanol, 1% (p/v) de ditiotreitol, citrato de sodio dihidratado |
600 mL (15 × 40 mL) |
PELIGRO H302: Nocivo por ingestión. H314 Provoca quemaduras graves en la piel y lesiones oculares graves. H412: Nocivo para los organismos acuáticos, con efectos nocivos duraderos. EUH032: En contacto con ácidos libera gases muy tóxicos. P273: Evítese su liberación al medio ambiente. P280: Llevar gafas/máscara de protección. P301 + P330 + P331 EN CASO DE INGESTIÓN: Enjuagarse la boca. NO provocar el vómito. P303 + P361 + P353 EN CASO DE CONTACTO CON LA PIEL (o el pelo): quitar inmediatamente todas las prendas contaminadas. Enjuagar la piel con agua. P304 + P340 + P310 EN CASO DE INHALACIÓN: transportar a la persona al aire libre y mantenerla en una posición que le facilite la respiración. Llamar inmediatamente a un CENTRO DE TOXICOLOGÍA/médico. P305 + P351 + P338 + P310 EN CASO DE CONTACTO CON LOS OJOS: aclarar cuidadosamente con agua durante varios minutos. Quitar las lentes de contacto, si lleva y resulta fácil. Seguir aclarando. Llamar inmediatamente a un CENTRO DE TOXICOLOGÍA/médico. 593-84-0 Tiocianato de guanidina 9002-92-0 Polidocanol |
* Este reactivo no está incluidos en el kit de la prueba COBAS® HIV-1/HIV-2 QUALITATIVE. Consulte el listado de material adicional necesario (Tabla 11 y Tabla 12).
** Las etiquetas de seguridad del producto se basan fundamentalmente en la regulación GHS de la UE.
Requisitos de almacenamiento de los reactivos:
Los reactivos deben almacenarse y manipularse según las indicaciones de la Tabla 6, la Tabla 7 y la Tabla 8. El cobas® Specimen Pre-Extraction Reagent (SPER), utilizado en el flujo de trabajo de DBS, debe almacenarse y manipularse tal como se especifica en la Tabla 9 y la Tabla 10.
Cuando los reactivos no están cargados en el cobas® 5800 System o los cobas® 6800/8800 Systems, almacénelos a la temperatura correspondiente especificada en la Tabla 6.
Tabla 6. Almacenamiento de reactivos (cuando el reactivo no está cargado en el sistema)
|
Reactivo |
Temperatura de almacenamiento |
|
COBAS® HIV-1/HIV-2 QUALITATIVE |
2-8 °C |
|
COBAS® HIV-1/HIV-2 Qualitative Control Kit |
2-8 °C |
|
cobas® NHP Negative Control Kit |
2-8 °C |
|
cobas omni Lysis Reagent |
2-8 °C |
|
cobas omni MGP Reagent |
2-8 °C |
|
cobas omni Specimen Diluent |
2-8 °C |
|
cobas omni Wash Reagent |
15-30 °C |
Requisitos para la manipulación de reactivos en el cobas® 5800 System:
Los reactivos cargados en el cobas® 5800 System se almacenan a la temperatura correspondiente adecuada y el sistema controla su fecha de caducidad. El sistema solamente permite utilizar los reactivos cuando se cumplen todas las condiciones indicadas en la Tabla 7. El sistema evita automáticamente el uso de reactivos caducados. La Tabla 7 ayuda al usuario a entender las condiciones de manipulación de los reactivos del cobas® 5800 System.
Tabla 7. Condiciones de caducidad de los reactivos del cobas® 5800 System
|
Reactivo |
Fecha de caducidad del kit |
Estabilidad del kit abierto |
Series en las que se puede utilizar el kit |
Periodo de estabilidad |
|
COBAS® HIV-1/HIV-2 QUALITATIVE |
No caducado |
90 días desde el primer uso |
Máx. 40 series |
Máx. 36 días* |
|
COBAS® HIV-1/HIV-2 Qualitative Control Kit |
No caducado |
No aplicable** |
No aplicable |
Máx. 36 días* |
|
COBAS® NHP Negative Control Kit |
No caducado |
No aplicable** |
No aplicable |
Máx. 36 días* |
|
cobas omni Lysis Reagent |
No caducado |
30 días desde la carga* |
No aplicable |
No aplicable |
|
cobas omni MGP Reagent |
No caducado |
30 días desde la carga* |
No aplicable |
No aplicable |
|
cobas omni Specimen Diluent |
No caducado |
30 días desde la carga* |
No aplicable |
No aplicable |
|
cobas omni Wash Reagent |
No caducado |
30 días desde la carga* |
No aplicable |
No aplicable |
* El tiempo se calcula desde la primera vez que se carga el reactivo en el cobas® 5800 System.
** Reactivo de un solo uso.
Requisitos para la manipulación de reactivos en los cobas® 6800/8800 Systems:
Los reactivos cargados en los cobas® 6800/8800 Systems se almacenan a la temperatura correspondiente adecuada y el sistema controla su fecha de caducidad. Los cobas® 6800/8800 Systems solamente permiten utilizar los reactivos cuando se cumplen todas las condiciones indicadas en la Tabla 8. El sistema evita automáticamente el uso de reactivos caducados. Tabla 8 ayuda al usuario a entender las condiciones de manipulación de los reactivos de los cobas® 6800/8800 Systems.
Tabla 8. Condiciones de caducidad de los reactivos de los cobas® 6800/8800 Systems
|
Reactivo |
Fecha de caducidad del kit |
Estabilidad del kit abierto |
Series en las que se puede utilizar el kit |
Periodo de estabilidad (horas acumuladas de carga fuera de la nevera) |
|
COBAS® HIV-1/HIV-2 QUALITATIVE |
No caducado |
90 días desde el primer uso |
Máx. 40 series |
Máx. 40 horas |
|
COBAS® HIV-1/HIV-2 Qualitative Control Kit |
No caducado |
No aplicablea |
No aplicable |
Máx. 8 horas |
|
COBAS® NHP Negative Control Kit |
No caducado |
No aplicablea |
No aplicable |
Máx. 10 horas |
|
cobas omni Lysis Reagent |
No caducado |
30 días desde la carga* |
No aplicable |
No aplicable |
|
cobas omni MGP Reagent |
No caducado |
30 días desde la carga* |
No aplicable |
No aplicable |
|
cobas omni Specimen Diluent |
No caducado |
30 días desde la carga* |
No aplicable |
No aplicable |
|
cobas omni Wash Reagent |
No caducado |
30 días desde la carga* |
No aplicable |
No aplicable |
a Reactivo de un solo uso.
* El tiempo se calcula desde la primera vez que se carga el reactivo en los cobas® 6800/8800 Systems.
Almacene el cobas® Specimen Pre-Extraction Reagent (utilizado en el flujo de trabajo de DBS) a la temperatura correspondiente especificada en la Tabla 9.
Tabla 9. Almacenamiento del cobas® Specimen Pre-Extraction Reagent
|
Reactivo |
Temperatura de almacenamiento |
|
cobas® Specimen Pre-Extraction Reagent |
2-8 °C |
El cobas® Specimen Pre-Extraction Reagent permanece estable hasta la fecha de caducidad indicada. Una vez abierto, este reactivo se mantiene estable durante 30 días cuando se almacena a una temperatura comprendida entre 2 y 8 °C, incluidas 13 horas acumuladas a 30 °C, o bien hasta la fecha de caducidad, lo que se produzca primero, según se especifica en la Tabla 10.
Tabla 10. Condiciones de caducidad del cobas® Specimen Pre-Extraction Reagent
|
Reactivo |
Fecha de caducidad del kit |
Estabilidad del kit abierto |
Series en las que se puede utilizar el kit |
Estabilidad a 30 °C fuera de la nevera (tiempo acumulado) |
|
cobas® Specimen Pre-Extraction Reagent |
No caducado |
30 días desde el primer uso |
No aplicable |
Máx. 13 horas |
Material adicional necesario para el cobas® 5800 System:
Tabla 11. Material y fungibles para el uso en el cobas® 5800 System
|
Material |
P/N |
|
cobas omni Processing Plate 24 |
08413975001 |
|
cobas omni Amplification Plate 24 |
08499853001 |
|
cobas omni Liquid Waste Plate 24 |
08413983001 |
|
Punta CORE TIPS con filtro, 1 mL |
04639642001 |
|
Punta CORE TIPS con filtro, 300 μL |
07345607001 |
|
cobas omni Liquid Waste Container |
07094388001 |
|
cobas omni Lysis Reagent |
06997538190 |
|
cobas omni MGP Reagent |
06997546190 |
|
cobas omni Specimen Diluent |
06997511190 |
|
cobas omni Wash Reagent |
06997503190 |
|
Bolsa para residuos sólidos o bolsa para residuos sólidos con complemento |
07435967001 o 08030073001 |
Material adicional necesario para los cobas® 6800/8800 Systems:
Tabla 12. Materiales y fungibles para el uso en los cobas® 6800/8800 Systems
|
Material |
P/N |
|
cobas omni Processing Plate |
05534917001 |
|
cobas omni Amplification Plate |
05534941001 |
|
cobas omni Pipette Tips |
05534925001 |
|
cobas omni Liquid Waste Container |
07094388001 |
|
cobas omni Lysis Reagent |
06997538190 |
|
cobas omni MGP Reagent |
06997546190 |
|
cobas omni Specimen Diluent |
06997511190 |
|
cobas omni Wash Reagent |
06997503190 |
|
Bolsa para residuos sólidos y recipiente de residuos sólidos o Bolsa para residuos sólidos con complemento y kit del cajón |
07435967001 y 07094361001 o 08030073001 y 08387281001 |
Tabla 13. Otros materiales y consumibles necesarios únicamente para el uso con DBS
|
Materiales |
|
Tarjeta de papel de filtro Whatman 903®, tarjeta para la obtención de muestras Munktell TFN o equivalente (diámetro de mancha: 12-13 mm) |
|
Tubos de polipropileno de 5 mL y 12,5 mm de diámetro con rosca interior (p. ej., Cryo.s™) y con tapones |
|
Eppendorf Thermomixer (p. ej., el modelo R 5355 o equivalente) con termobloque para 24 criotubos |
|
Fórceps o pinzas estériles o desechables |
|
Bolsas resellables y bolsitas de desecante (para el almacenamiento de las DBS) |
Instrumentos y software necesarios:
Es preciso instalar el software cobas® 5800 y el paquete de análisis para la prueba COBAS® HIV-1/HIV-2 QUALITATIVE para el cobas® 5800 System, el software cobas® HIV-1/2 Qual-Serum/Plasma ASAP y/o el software cobas® 5800 HIV-1/2 Qual- DBS ASAP en el instrumento cobas® 5800. El software Data Manager y el PC para el cobas® 5800 System se suministran con el sistema.
Es preciso instalar en los instrumentos el software cobas® 6800/8800 y los paquetes de análisis para la prueba COBAS® HIV-1/HIV-2 QUALITATIVE para los cobas® 6800/8800 Systems, el software COBAS® HIV-1/2 Qual-Serum/Plasma ASAP y/o el software COBAS® HIV-1/2 Qual-DBS ASAP. El IG (Instrument Gateway) se suministra con el sistema.
Tabla 14. Instrumentos
|
Equipo |
P/N |
|
cobas® 5800 System |
08707464001 |
|
cobas® 6800 System (opción móvil) |
05524245001 y 06379672001 |
|
cobas® 6800 System (fijo) |
05524245001 y 06379664001 |
|
cobas® 8800 System |
05412722001 |
|
Módulo de suministro de muestras |
06301037001 |
Consulte la Asistencia al usuario y/o las Guías del usuario del cobas® 5800 System o los cobas® 6800/8800 Systems para obtener información adicional.
Nota: póngase en contacto con su representante local de Roche para obtener una lista de pedido detallada para racks de muestras, racks para puntas obstruidas y bandejas de racks compatibles con cada instrumento.
EVALUACIÓN CLÍNICA DEL RENDIMIENTO:
Reproducibilidad:
La reproducibilidad de la prueba COBAS® HIV-1/HIV-2 QUALITATIVE se evaluó en plasma conservado en EDTA entre lotes de reactivo, centro de pruebas/sistema de instrumento, operador, días, series e intraserie. El análisis de la reproducibilidad se llevó a cabo en tres centros utilizando tres lotes de reactivo, dos cobas® 6800 Systems y un cobas® 8800 System, dos operadores durante 6 días; en cada serie se analizaron tres réplicas de cada miembro del panel. Cada panel estaba formado por un miembro de panel negativo y seis miembros de panel positivos. El porcentaje de concordancia de negativos se estimó en un 100%, con un IC exacto del 95% de (98,9%, 100,0%), mientras que el porcentaje de concordancia de positivos fue del 100% para cada miembro del panel tanto para HIV-1 como para HIV-2.
Para los miembros del panel positivos para HIV-1, el coeficiente de variación (CV (%)) de todos los miembros de panel fue ≤ 1,9%, lo que demuestra una variabilidad muy baja de los resultados de la prueba COBAS® HIV-1/HIV-2 QUALITATIVE entre lotes de reactivo, centros/instrumentos, días, operadores y series.
Tabla 36. Porcentaje atribuible de la variación total, desviación estándar de la precisión total y CV (%) de los valores de umbral de ciclo de los resultados reactivos para HIV-1 con la prueba COBAS® HIV-1/HIV-2 QUALITATIVE por miembro del panel positivo para HIV-1
|
Miembro del panel |
Na |
Loteb |
Centrob |
Operadorb |
Díab |
Serieb |
Intra-serieb |
Desviación estándar de la precisión totalc |
Precisión total CV (%)d |
|
~3 x LoD (3,78E1) HIV-1, Negativo para HIV-2 |
324 |
0,0% (0,0%) |
0,0% (0,0%) |
2,2% (0,3%) |
6,6% (0,5%) |
0,0% (0,0%) |
91,1% (1,8%) |
0,69 |
1,9% |
|
> 3 x LoD (1,00E5) HIV-1, Negativo para HIV-2 |
322 |
0,0% (0,0%) |
15,5% (0,4%) |
0,0% (0,0%) |
33,1% (0,5%) |
4,5% (0,2%) |
46,9% (0,6%) |
0,24 |
0,9% |
|
> 3 x LoD (1,00E5) HIV-1, ~3 x LoD (8,37E1) HIV-2 |
323 |
0,0% (0,0%) |
7,8% (0,3%) |
0,0% (0,0%) |
45,0% (0,6%) |
10,9% (0,3%) |
36,3% (0,6%) |
0,25 |
1,0% |
|
~3 x LoD (3,78E1) HIV-1, > 3 x LoD (1,00E5) HIV-2 |
323 |
1,5% (0,2%) |
0,0% (0,0%) |
0,0% (0,0%) |
0,0% (0,0%) |
0,0% (0,0%) |
98,5% (1,8%) |
0,67 |
1,8% |
Nota: la tabla únicamente incluye resultados con analito detectable.
a Número de pruebas válidas con analito detectable.
b Las celdas de las columnas para los componentes de lote, centro, operador, día, serie e intraserie muestran el porcentaje de la variación total (%) y, entre paréntesis, el porcentaje del coeficiente de variación (CV%) del componente.
c Cálculo realizado utilizando la variabilidad total del procedimiento SAS MIXED.
d CV (%) = (desviación estándar ÷ media) × 100%.
Para los miembros del panel positivos para HIV-2, el coeficiente de variación (CV (%)) de todos los miembros de panel fue ≤ 1,7%, lo que demuestra una variabilidad muy baja de los resultados de la prueba COBAS® HIV-1/HIV-2 QUALITATIVE entre lotes de reactivo, centros/instrumentos, días, operadores y series.
Tabla 37. Porcentaje atribuible de la variación total, desviación estándar de la precisión total y CV (%) de los valores de umbral de ciclo de los resultados reactivos para HIV-2 con la prueba COBAS® HIV-1/HIV-2 QUALITATIVE por miembro del panel positivo para HIV-2
|
Miembro del panel |
Na |
Loteb |
Centrob |
Operadorb |
Díab |
Serieb |
Intra-serieb |
Desviación estándar de la precisión totalc |
Precisión total CV (%)d |
|
Negativo para HIV-1, ~3 x LoD (8,37E1) HIV-2 |
324 |
60,0% (1,3%) |
3,3% (0,3%) |
0,0% (0,0%) |
14,6% (0,7%) |
4,5% (0,4%) |
17,6% (0,7%) |
0,59 |
1,7% |
|
Negativo para HIV-1, > 3 x LoD (1,00E5) HIV-2 |
324 |
31,9% (0,9%) |
10,0% (0,5%) |
0,0% (0,0%) |
32,9% (1,0%) |
0,0% (0,0%) |
25,1% (0,8%) |
0,42 |
1,7% |
|
> 3 x LoD (1,00E5) HIV-1, ~3 x LoD (8,37E1) HIV-2 |
323 |
26,0% (0,7%) |
4,0% (0,3%) |
0,0% (0,0%) |
9,9% (0,4%) |
4,2% (0,3%) |
55,9% (1,0%) |
0,46 |
1,3% |
|
~3 x LoD (3,78E1) HIV-1, > 3 x LoD (1,00E5) HIV-2 |
323 |
38,2% (0,9%) |
8,2% (0,4%) |
0,0% (0,0%) |
24,7% (0,8%) |
0,0% (0,0%) |
28,9% (0,8%) |
0,39 |
1,5% |
Nota: la tabla únicamente incluye resultados con analito detectable.
a Número de pruebas válidas con analito detectable.
b Las celdas de las columnas para los componentes de lote, centro, operador, día, serie e intraserie muestran el porcentaje de la variación total (%) y, entre paréntesis, el porcentaje del coeficiente de variación (CV%) del componente.
c Cálculo realizado utilizando la variabilidad total del procedimiento SAS MIXED.
d CV (%) = (desviación estándar ÷ media) × 100%.
Comparación de métodos clínicos:
Sensibilidad clínica del HIV-1 y HIV-2:
Se comparó el rendimiento de la prueba COBAS® HIV-1/HIV-2 QUALITATIVE en los cobas® 6800/8800 Systems con el de una prueba NAT cualitativa alternativa para el HIV-1 o una prueba NAT para el HIV-2 en sujetos infectados con el virus de inmunodeficiencia humana tipo 1 (HIV-1) o el virus de inmunodeficiencia humana tipo 2 (HIV-2).
Análisis de muestras de sujetos infectados con el HIV-1:
Se analizó un total de 1.030 muestras de sujetos positivos conocidos para HIV-1 con cargas virales de HIV-1 ≥ 100 copias/ml. De las 1.030 muestras evaluables para el análisis estadístico, 537 (52,1%) pertenecían a mujeres, 752 (73,0%) se obtuvieron de sujetos africanos/afroamericanos y la media de edad de los sujetos era de 37 años (rango: 18-81 años). 736 muestras HIV-1 eran del subtipo B y 294 eran muestras HIV-1 diferentes del subtipo B.
La sensibilidad para el HIV-1 de la prueba COBAS® HIV-1/HIV-2 QUALITATIVE fue del 100% (1030/1030, IC del 95%: entre 99,6% y 100%). La sensibilidad para las muestras con concentraciones de ARN viral es igual o mayor que 100 copias/ml (Tabla 38). Se observó un rendimiento similar entre las muestras de plasma y de suero.
Tabla 38. Sensibilidad para el HIV-1 de la prueba COBAS® HIV-1/HIV-2 QUALITATIVE para la población positiva conocida para HIV-1
|
Población, tipo de muestra |
Total de muestras positivas conocidas |
Número de reactivos por prueba |
Estimación de sensibilidad del HIV-1 |
IC exacto del 95% |
|
HIV-1, global |
1030 |
1030 |
100% |
(99,6%, 100%) |
|
HIV-1, plasma |
712 |
712 |
100% |
(99,5%, 100%) |
|
HIV-1, suero |
318 |
318 |
100% |
(98,8%, 100%) |
Análisis de muestras de sujetos infectados con el HIV-2:
Se analizó un total de 183 muestras de sujetos positivos conocidos para HIV-2 con cargas virales de HIV-2 de ≥ 100 copias/mL según el ensayo cuantitativo de ARN de HIV-2 en plasma. De las 183 muestras evaluables, 92 (50,3%) pertenecían a hombres, todos ellos africanos, con una media de edad de 50 años (rango: 23-77 años).
La sensibilidad para el HIV-2 de la prueba COBAS® HIV-1/HIV-2 QUALITATIVE fue del 99,5% (182/183, IC del 95%: entre 97,0% y 99,99%) (Tabla 39). La sensibilidad para las muestras con concentraciones de ARN viral es igual o mayor que 100 copias/mL. Se observó un rendimiento similar entre las muestras de plasma y de suero.
Tabla 39. Sensibilidad para el HIV-2 de la prueba COBAS® HIV-1/HIV-2 QUALITATIVE para la población positiva conocida para HIV-2
|
Población, tipo de muestra |
Total de muestras positivas conocidas |
Número de reactivos por prueba |
Estimación de sensibilidad del HIV-2 |
IC exacto del 95% |
|
HIV-2, global |
183 |
182 |
99,5% |
(97,0%, 99,99%) |
|
HIV-2, plasma |
115 |
115 |
100,0% |
(96,8%, 100%) |
|
HIV-2, suero |
68 |
67 |
98,5% |
(92,1%, 99,96%) |
Especificidad clínica:
La especificidad clínica de la prueba COBAS® HIV-1/HIV-2 QUALITATIVE en los cobas® 6800/8800 Systems se determinó mediante comparación con un algoritmo clínico utilizando dos inmunoensayos seguidos de un análisis NAT para resolver resultados indeterminados en una población con bajo riesgo para HIV (algoritmo para análisis clínicos del HIV de los Centros para el control y la prevención de enfermedades de los EE. UU.). La población de riesgo bajo para el HIV estaba formada por 1.988 donantes de sangre sanos y 3.929 sujetos de bajo riesgo de un área con una prevalencia de HIV inferior al 1%. De las 5.917 muestras evaluables para el análisis estadístico, 3.301 (55,8%) pertenecían a mujeres, 3.942 (66,6%) se obtuvieron de sujetos blancos/caucásicos y la media de edad de los mismos era de 36 años (rango: 17-92 años).
La especificidad global para el HIV-1 y la especificidad para el HIV-2 de la prueba COBAS® HIV-1/HIV-2 QUALITATIVE fue del 100%, sin diferencia entre las muestras de plasma y suero (Tabla 40).
Tabla 40. Especificidad de la prueba COBAS® HIV-1/HIV-2 QUALITATIVE por analito analizado en la población con riesgo reducido para el HIV
|
Analito analizado, tipo de muestra |
Total de sujetos no reactivos para la prueba COBAS® HIV-1/HIV-2 QUALITATIVE |
Sujetos con estado negativo según el algoritmo de análisis del HIV de los CDC |
Estimación de especificidad |
IC exacto del 95% |
|
HIV-1, globala |
5902 |
5902 |
100% |
(99,94%, 100%) |
|
HIV-1, plasmaa |
3608 |
3608 |
100% |
(99,90%, 100%) |
|
HIV-1, sueroa |
2294 |
2294 |
100% |
(99,84%, 100%) |
|
HIV-2, globalb |
5914 |
5914 |
100% |
(99,94%, 100%) |
|
HIV-2, plasmab |
3618 |
3618 |
100% |
(99,90%, 100%) |
|
HIV-2, suerob |
2296 |
2296 |
100% |
(99,84%, 100%) |
a En el análisis de especificidad para la diana del HIV-1 no se incluyeron quince muestras positivas para HIV-1 según el algoritmo de análisis del HIV de los CDC (10 muestras de plasma y 5 muestras de suero).
b No se incluyeron tres muestras de suero porque no se pudo obtener el resultado del algoritmo de análisis del HIV de los CDC debido a un volumen de muestra insuficiente para el análisis NAT del HIV-2.
Estudio prospectivo de individuales con riesgo elevado de infección por el HIV-1:
Se comparó el rendimiento de la prueba COBAS® HIV-1/HIV-2 QUALITATIVE con el de un análisis NAT alternativo cualitativo para HIV-1 en sujetos con un riesgo elevado de infección por HIV-1. Las 519 muestras de sujetos con un riesgo elevado de infección por HIV-1 eran de sujetos con 1 factor de riesgo como mínimo para la infección por HIV: usuario de drogas inyectables (actualmente o en alguna ocasión), sexo sin protección con una persona infectada por HIV (actualmente o en alguna ocasión), diagnóstico de enfermedad de transmisión sexual durante el último año, múltiples parejas sexuales (más de 1 pareja en los últimos 12 meses), HSH (actualmente o en algún momento), sexo sin protección con una persona diagnosticada con una enfermedad de transmisión sexual durante el transcurso del último año. De las 519 muestras evaluables, 345 (66,5%) pertenecían a hombres, 289 (55,7%) eran de sujetos africanos/afroamericanos, con una media de edad de 39 años (rango: 18-80 años).
El PCP para el HIV-1 de la prueba COBAS® HIV-1/HIV-2 QUALITATIVE fue del 100% (5/5) y el PCN para el HIV-1 fue del 100% (514/514, IC del 95%: entre 99,3% y 100%). Se observó un rendimiento similar entre las muestras de plasma y de suero.
Estudio prospectivo de individuos con riesgo elevado de infección por el HIV-2:
Se comparó el rendimiento de la prueba COBAS® HIV-1/HIV-2 QUALITATIVE con un algoritmo clínico utilizando dos inmunoensayos seguidos de una prueba NAT para resolver los resultados indeterminados en sujetos con riesgo elevado de infección por HIV-2 (algoritmo para análisis clínicos del HIV de los Centros para el control y la prevención de enfermedades de los EE. UU.).
Todas las muestras de sujetos con un riesgo elevado de infección por HIV-2 procedían de un área de África occidental donde el HIV-2 es endémico (es decir, Guinea Bissau, Camerún y Costa de Marfil). De las 499 muestras evaluables, 366 (73,3%) pertenecían a hombres, todos ellos africanos, con una media de edad de 28 años (rango: 19-66 años).
El PCP de la prueba COBAS® HIV-1/HIV-2 QUALITATIVE para HIV-1 en la población de alto riesgo para el HIV-2 fue del 79,0% (79/100; IC del 95%: 69,7-86,5%), mientras que el PCN fue del 99,5% (395/397; IC del 95%: 98,2-99,9%). El PCP de la prueba COBAS® HIV-1/HIV-2 QUALITATIVE para HIV-2 en la población de alto riesgo para el HIV-2 fue del 44,4% (4/9; IC del 95%: 13,7-78,8%), mientras que el PCN fue del 100% (490/490; IC del 95%: 99,2-100%). Se observó un rendimiento similar entre las muestras de plasma y de suero.
Equivalencia entre sistemas/comparación de sistemas:
La equivalencia entre los cobas® 5800, cobas® 6800 y los cobas® 8800 Systems se demostró a partir de estudios de rendimiento.
Los resultados incluidos en las Instrucciones de uso hacen patente la equivalencia de rendimiento entre todos los sistemas.
EVALUACIÓN NO CLÍNICA DEL RENDIMIENTO:
Características clave de rendimiento de los cobas® 6800/8800 Systems:
Límite de detección (LoD):
Estándares internacionales de la OMS/Estándares primarios de Roche:
El límite de detección de la prueba COBAS® HIV-1/HIV-2 QUALITATIVE se ha determinado a partir de los siguientes estándares:
• Tercer estándar internacional de la OMS para el ARN de HIV-1 grupo M (código NIBSC 10/152) para muestras de plasma conservado en EDTA, suero y DBS.
• Estándar internacional de la OMS para el ARN de HIV-2 (código NIBSC 08/150) para muestras de plasma y suero.
• Estándares primarios de Roche para el ARN de HIV-2 para muestras DBS.
• Estándares primarios de Roche para el ARN de HIV-1 grupo O para muestras de plasma conservado en EDTA y suero.
Actualmente no hay ningún estándar internacional disponible para el ARN de HIV-1 grupo O. El estándar de Roche para el ARN de HIV-1 grupo O puede encontrarse en el Lote 01 del panel de referencia 1 del ARN para el subtipo HIV-1 de CBER. Los estándares primarios de Roche para el ARN del HIV-1 grupo O son cultivos de virus conocidos comerciali- zados, P/N 2420 (n.º de catálogo 500493, SeraCare Life Sciences). El estándar del ARN de HIV-2 de Roche puede encontrarse en el estándar internacional de la OMS para el ARN de HIV-2 (código NIBSC 08/150). Los estándares primarios de Roche para el ARN de HIV-2 son cultivos de virus conocidos comercializados, P/N HIV-2 NIH-Z (n.º de catálogo 10-27-000, Applied Biotechnologies, Inc.). Una copia del ARN de HIV-1 equivale a 1,7 unidades internacionales (UI) y una copia del ARN del HIV-2 equivale a 0,2 UI.
Se prepararon diluciones en serie de los estándares en plasma humano conservado en EDTA, suero o sangre entera de muestras DBS negativas para el HIV. Se analizaron paneles con 5 o 6 niveles de concentración más una muestra negativa con 3 lotes de reactivo para la prueba COBAS® HIV-1/HIV-2 QUALITATIVE, con múltiples series analíticas, días, operadores e instrumentos.
Para estimar el LoD de cada virus se utilizó el análisis PROBIT del 95% con los datos combinados de series de dilución y de lotes de reactivos, junto con el límite inferior y superior del intervalo de confianza del 95% (Tabla 18). En la Tabla 19 a la Tabla 21 se resumen las tasas de reactividad observadas en los estudios de LoD para cada virus.
Tabla 18. Resultados del análisis PROBIT del 95% con datos del LoD obtenidos para los estándares víricos de plasma conservado en EDTA, suero y DBS
|
Matrices |
Analito |
Unidades de medición |
LoD |
Límite inferior del intervalo de confianza del 95% |
Límite superior del intervalo de confianza del 95% |
|
Plasma conservado en EDTA |
HIV-1 grupo M |
Copias/mL |
12,6 |
10,9 |
15,2 |
|
HIV-1 grupo O |
Copias/mL |
14,8 |
12,8 |
17,7 |
|
|
HIV-2 |
Copias/mL |
27,9 |
22,9 |
36,6 |
|
|
Suero |
HIV-1 grupo M |
Copias/mL |
12,1 |
10,5 |
14,5 |
|
HIV-1 grupo O |
Copias/mL |
12,6 |
10,9 |
15,2 |
|
|
HIV-2 |
Copias/mL |
23,4 |
19,6 |
29,7 |
|
|
DBS |
HIV-1 grupo M |
Copias/mL |
255 |
224 |
299 |
|
HIV-2 |
Copias/mL |
984 |
856 |
1169 |
Tabla 19. Resumen de las tasas de reactividad para HIV-1 grupo M en plasma conservado en EDTA, suero y DBS
|
Matrices |
Concentración de ARN de |
Número de reactivos |
Número de réplicas válidas |
% de reactividad |
|
Plasma conservado en EDTA |
40 |
189 |
189 |
100% |
|
30 |
189 |
189 |
100% |
|
|
20 |
187 |
189 |
99% |
|
|
10 |
174 |
189 |
92% |
|
|
5 |
124 |
189 |
66% |
|
|
2,5 |
91 |
189 |
48% |
|
|
0 |
0 |
189 |
0% |
|
|
Suero |
40 |
189 |
189 |
100% |
|
30 |
189 |
189 |
100% |
|
|
20 |
187 |
189 |
99% |
|
|
10 |
176 |
189 |
93% |
|
|
5 |
126 |
189 |
67% |
|
|
2,5 |
86 |
189 |
46% |
|
|
0 |
0 |
189 |
0% |
|
|
DBS |
750 |
252 |
252 |
100% |
|
600 |
252 |
252 |
100% |
|
|
360 |
246 |
250 |
98% |
|
|
180 |
220 |
249 |
88% |
|
|
90 |
163 |
252 |
65% |
|
|
45 |
109 |
250 |
44% |
|
|
0 |
0 |
107 |
0% |
Tabla 20. Resumen de las tasas de reactividad para HIV-1 grupo O en plasma conservado en EDTA y suero
|
Matrices |
Concentración de ARN de HIV-1 grupo O (cp/mL) |
Número de reactivos |
Número de réplicas válidas |
% de reactividad |
|
Plasma conservado en EDTA |
40 |
189 |
189 |
100% |
|
30 |
189 |
189 |
100% |
|
|
20 |
185 |
188 |
98% |
|
|
10 |
163 |
189 |
86% |
|
|
5 |
117 |
189 |
62% |
|
|
2,5 |
78 |
189 |
41% |
|
|
0 |
0 |
189 |
0% |
|
|
Suero |
40 |
189 |
189 |
100% |
|
30 |
189 |
189 |
100% |
|
|
20 |
186 |
189 |
98% |
|
|
10 |
173 |
189 |
92% |
|
|
5 |
132 |
189 |
70% |
|
|
2,5 |
91 |
189 |
48% |
|
|
0 |
0 |
189 |
0% |
Tabla 21. Resumen de las tasas de reactividad para HIV-2 en muestras de plasma conservado en EDTA, suero y DBS
|
Matrices |
Concentración de ARN de HIV-2 (cp/ml) |
Número de reactivos |
Número de réplicas válidas |
% de reactividad |
|
Plasma conservado en EDTA |
80 |
126 |
126 |
100% |
|
40 |
124 |
126 |
98% |
|
|
20 |
115 |
126 |
91% |
|
|
10 |
81 |
126 |
64% |
|
|
5 |
61 |
126 |
48% |
|
|
0 |
0 |
189 |
0% |
|
|
Suero |
80 |
126 |
126 |
100% |
|
40 |
125 |
126 |
99% |
|
|
20 |
114 |
126 |
90% |
|
|
10 |
96 |
126 |
76% |
|
|
5 |
49 |
126 |
39% |
|
|
0 |
0 |
189 |
0% |
|
|
DBS |
3000 |
252 |
252 |
100% |
|
1450 |
241 |
247 |
98% |
|
|
725 |
226 |
246 |
92% |
|
|
362 |
167 |
248 |
67% |
|
|
181 |
103 |
250 |
41% |
|
|
0 |
0 |
108 |
0% |
Precisión (intralaboratorio):
La precisión de la prueba COBAS® HIV-1/HIV-2 QUALITATIVE se ha determinado a partir de los siguientes estándares:
• Estándar Secundario de Roche para HIV-1 grupo M
• Estándar Primario de Roche para HIV-2
En el estudio se analizaron 2 paneles de dianas formuladas individualmente para el HIV-1 grupo M y HIV-2, cada una de ellas formada por 3 miembros de panel con concentraciones de aproximadamente 0,6 ×, 1 × y 3 × el LoD de la prueba COBAS® HIV-1/HIV-2 QUALITATIVE. Las pruebas se han realizado para determinar los siguientes componentes de variabilidad:
• Variabilidad entre días durante 4 días.
• Variabilidad entre lotes con 3 lotes de reactivos diferentes de la prueba COBAS® HIV-1/HIV-2 QUALITATIVE.
• Variabilidad entre instrumentos con 3 cobas® 6800/8800 Systems diferentes.
Se analizaron aproximadamente 84 réplicas con cada uno de los 3 miembros de panel para cada lote de reactivos para un total de 252 réplicas en todos los lotes de reactivos por diana. Los resultados de precisión se evaluaron calculando el porcentaje de resultados de las pruebas reactivas en cada nivel de concentración para cada uno de los componentes de variabilidad analizados.
Se calcularon los límites de los intervalos de confianza del 95% bilaterales de cada tasa de reactividad para cada uno de los tres niveles de HIV-1 grupo M y HIV-2 analizados durante 4 días, con 3 lotes de reactivos y 3 cobas® 6800/8800 Systems distintos. La prueba COBAS® HIV-1/HIV-2 QUALITATIVE garantiza la reproducibilidad durante varios días, con varios lotes de reactivos y en diferentes equipos. En la Tabla 22 y la Tabla 23 se resumen los resultados de la variabilidad entre lotes.
Tabla 22. Resumen de la precisión entre lotes de los reactivos de la prueba COBAS® HIV-1/HIV-2 QUALITATIVE (plasma conservado en EDTA)
|
Analito |
Concentración |
Lote de reactivos |
% de reactividad (réplicas reactivas/válidas) |
Límite inferior del intervalo de confianza del 95% |
Límite superior del intervalo de confianza del 95% |
|
HIV-1 grupo M |
~3 × LoD |
1 |
100% (84/84) |
95,7% |
100% |
|
2 |
100% (84/84) |
95,7% |
100% |
||
|
3 |
100% (84/84) |
95,7% |
100% |
||
|
~1 × LoD |
1 |
98,8% (83/84) |
93,5% |
100% |
|
|
2 |
98,8% (83/84) |
93,5% |
100% |
||
|
3 |
100% (84/84) |
95,7% |
100% |
||
|
~0,6 × LoD |
1 |
77,4% (65/84) |
67,0% |
85,8% |
|
|
2 |
76,2% (64/84) |
65,7% |
84,8% |
||
|
3 |
82,1% (69/84) |
72,3% |
89,6% |
||
|
HIV-2 |
~3 × LoD |
1 |
98,8% (83/84) |
93,5% |
100% |
|
2 |
96,4% (81/84) |
89,9% |
99,3% |
||
|
3 |
100% (84/84) |
95,7% |
100% |
||
|
~1 × LoD |
1 |
98,8% (83/84) |
93,5% |
100% |
|
|
2 |
98,8% (83/84) |
93,5% |
100% |
||
|
3 |
97,6% (82/84) |
91,7% |
99,7% |
||
|
~0,6 × LoD |
1 |
66,7% (56/84) |
55,5% |
76,6% |
|
|
2 |
69,0% (58/84) |
58,0% |
78,7% |
||
|
3 |
69,0% (58/84) |
58,0% |
78,7% |
Tabla 23. Resumen de la precisión entre lotes del reactivo de la prueba COBAS® HIV-1/HIV-2 QUALITATIVE (DBS)
|
Analito |
Concentración |
Lote de reactivos |
% de reactividad (réplicas reactivas/válidas) |
Límite inferior del intervalo de confianza del 95% |
Límite superior del intervalo de confianza del 95% |
|
HIV-1 grupo M |
~3 × LoD |
1 |
100% (84/84) |
95,7% |
100% |
|
2 |
100% (83/83) |
95,7% |
100% |
||
|
3 |
97,6% (82/84) |
91,7% |
99,7% |
||
|
~1 × LoD |
1 |
96,4% (81/84) |
89,9% |
99,3% |
|
|
2 |
96,4% (81/84) |
89,9% |
99,3% |
||
|
3 |
95,2% (80/84) |
88,3% |
98,7% |
||
|
~0,6 × LoD |
1 |
88,0% (73/83) |
79,0% |
94,1% |
|
|
2 |
83,3% (70/84) |
73,6% |
90,6% |
||
|
3 |
88,1% (74/84) |
79,2% |
94,1% |
||
|
HIV-2 |
~3 × LoD |
1 |
100% (83/83) |
95,7% |
100% |
|
2 |
100% (84/84) |
95,7% |
100% |
||
|
3 |
100% (83/83) |
95,7% |
100% |
||
|
~1 × LoD |
1 |
97,6% (82/84) |
91,7% |
99,7% |
|
|
2 |
97,6% (82/84) |
91,7% |
99,7% |
||
|
3 |
98,8% (83/84) |
93,5% |
100% |
||
|
~0,6 × LoD |
1 |
88,1% (74/84) |
79,2% |
94,1% |
|
|
2 |
91,7% (77/84) |
83,6% |
96,6% |
||
|
3 |
85,7% (72/84) |
76,4% |
92,4% |
Verificación del grupo/subtipo e inclusividad:
El rendimiento de la prueba COBAS® HIV-1/HIV-2 QUALITATIVE para los subtipos de HIV-1 grupo M, grupo O y grupo N y del HIV-2 grupo B se evaluó mediante:
• La verificación del límite de detección para los subtipos de HIV-1 grupo M, grupo O (verificado mediante dilución en sangre entera de DBS) y grupo N y de HIV-2 grupo B.
• La verificación de la inclusividad para los subtipos de HIV-1 grupo M, grupo O y grupo N y de HIV-2 grupos A y B.
Verificación del límite de detección para los subtipos del grupo M de HIV-1, grupo O y grupo N y de HIV-2 grupo B:
Se diluyeron muestras clínicas o cultivos de muestras del HIV para el HIV-1 grupo M (A, C, D, F, G, H) y formas recombinantes circulantes (CRF01_AE, CRF02_AG), el HIV-1 grupo N y el HIV-2 grupo B en plasma conservado en EDTA, suero o sangre entera de DBS y, de forma adicional, el HIV-1 grupo O en sangre entera de DBS, en la concentración LoD del grupo/subtipo predominante (HIV-1 grupo M subtipo B o HIV-2 grupo A) basado en el LoD determinado con el análisis PROBIT del 95% en todos los lotes combinados. La determinación de la tasa de reactividad se llevó a cabo con 42 réplicas. El análisis se realizó con 1 lote de reactivos para la prueba COBAS® HIV-1/HIV-2 QUALITATIVE. Los resultados para HIV-1 se muestran en la Tabla 24 y los resultados para HIV-2, en la Tabla 25. Estos resultados verifican que la prueba COBAS® HIV-1/HIV-2 QUALITATIVE es capaz de detectar el HIV del grupo M de HIV-1 (A, C, D, F, G, H, CRF01_AE, CRF02_AG), el HIV-1 grupo O, el HIV-1 grupo N y el HIV-2 grupo B en la concentración esperada para cada matriz o en una concentración inferior con un intervalo de confianza superior del 95% igual o mayor que la tasa de reactividad esperada del 95%.
Tabla 24. Verificación del LoD para los subtipos del HIV-1 grupo M, grupo O y grupo N en plasma conservado en EDTA, suero y sangre entera para DBS
|
Grupo |
Subtipo |
Plasma: 12,6 cp/mL |
Suero: 12,1 cp/mL |
DBS: 255 cp/mL |
||||||
|
Número de réplicas válidas |
Número de reactivos |
% de reactivos (IC* del 95%) |
Número de réplicas válidas |
Número de reactivos |
% de reactivos (IC* del 95%) |
Número de réplicas válidas |
Número de reactivos |
% de reactivos (IC* del 95%) |
||
|
M |
A |
42 |
40 |
95% (99%) |
42 |
40 |
95% (99%) |
41 |
37 |
90% (97%) |
|
C |
42 |
41 |
98% (100%) |
42 |
42 |
100% (99,4%) |
42 |
42 |
100% (100%) |
|
|
D |
42 |
37 |
88% (96%) |
42 |
37 |
88% (96%) |
42 |
39 |
93% (99%) |
|
|
F |
42 |
38 |
90% (97%) |
42 |
38 |
90% (97%) |
42 |
40 |
95% (99%) |
|
|
G |
42 |
40 |
95% (99%) |
42 |
39 |
93% (99%) |
42 |
42 |
100% (100%) |
|
|
H |
42 |
38 |
90% (97%) |
42 |
41 |
98% (100%) |
42 |
41 |
98% (100%) |
|
|
CRF01_AE |
42 |
38 |
90% (97%) |
42 |
38 |
90% (97%) |
42 |
41 |
98% (100%) |
|
|
CRF02_AG |
42 |
36 |
86% (95%) |
42 |
39 |
93% (99%) |
42 |
42 |
100% (100%) |
|
|
O |
N/D |
N/D |
N/D |
N/D |
N/D |
N/D |
41 |
39 |
95% (99%) |
|
|
N |
42 |
39 |
93% (99%) |
42 |
37 |
88% (96%) |
41 |
40 |
98% (100%) |
|
* Intervalo de confianza superior del 95%
Tabla 25. Verificación del LoD para HIV-2 grupo B en plasma conservado en EDTA, suero o sangre entera para DBS
|
Grupo |
Plasma: 27,9 cp/mL |
Suero: 23,4 cp/mL |
DBS: 984 cp/mL |
||||||
|
Número de réplicas válidas |
Número de reactivos |
% de reactivos (IC* del 95%) |
Número de réplicas válidas |
Número de reactivos |
% de reactivos (IC* del 95%) |
Número de réplicas válidas |
Número de reactivos |
% de reactivos (IC* del 95%) |
|
|
B |
42 |
42 |
100% (100%) |
42 |
42 |
100% (100%) |
42 |
42 |
100% (100%) |
* Intervalo de confianza superior del 95%
La verificación de la inclusividad para los subtipos de HIV-1 grupo M, grupo O y grupo N y de HIV-2 grupos A y B:
El rendimiento de la prueba COBAS® HIV-1/HIV-2 QUALITATIVE para la detección de los subtipos de HIV-1 grupo M (A, C, D, F, G, H, J, K) y formas recombinantes circulantes (CRF01_AE, CRF02_AG, CRF12_BF, CRF14_BG), HIV-1 grupo O, HIV-1 grupo N, HIV-2 grupo A y HIV-2 grupo B se determinó analizando muestras clínicas únicas y/o cultivos de muestras aislados para cada grupo o subtipo en suero o plasma conservado en EDTA.
HIV-1 grupo M:
Se analizó un total de 105 muestras clínicas únicas de HIV-1 grupo M con subtipo de HIV-1 conocido tanto sin diluir como diluidas con una concentración ~5 × LoD de la prueba COBAS® HIV-1/HIV-2 QUALITATIVE. Las 105 muestras clínicas con subtipos conocidos se detectaron sin diluir y con una concentración de ~5 × LoD (Tabla 26).
También se analizaron 4 muestras de HIV-1 grupo M subtipo CRF12_BF y 1 muestra clínica de HIV-1 grupo M subtipo CRF14_BG tras la preparación de las series de dilución. Se analizó 1 réplica de cada una de las muestras sin diluir y 1 de cada dilución de 1:1,0E+01 a 1:5,0E+02 (de 2 a 4 diluciones por muestra) para HIV-1 grupo M subtipo CRF12_BF y de 1:2,0E+01 a 1:1,2E+02 (4 diluciones) para HIV-1 grupo M subtipo CRF14_BG. Todos los resultados obtenidos fueron reactivos. Todas las muestras clínicas analizadas se detectaron con una concentración ≤ 5 × LoD.
Tabla 26. Muestras clínicas de HIV-1 grupo M
|
Subtipo/Formas recombinantes circulantes |
% de reactividad (muestras reactivas/analizadas) sin diluir |
% de reactividad (muestras reactivas/analizadas) diluidas a ~5 × LoD |
|
A |
100% (10/10) |
100% (10/10) |
|
C |
100% (10/10) |
100% (10/10) |
|
D |
100% (10/10) |
100% (10/10) |
|
F |
100% (10/10) |
100% (10/10) |
|
G |
100% (10/10) |
100% (10/10) |
|
H |
100% (10/10) |
100% (10/10) |
|
J |
100% (5/5) |
100% (5/5) |
|
K |
100% (9/9) |
100% (9/9) |
|
CRF01_AE |
100% (10/10) |
100% (10/10) |
|
CRF02_AG |
100% (10/10) |
100% (10/10) |
|
CRF12_BF |
100% (2/2) |
100% (2/2) |
|
CRF14_BG |
100% (9/9) |
100% (9/9) |
HIV-1 grupo O y HIV-1 grupo N:
Se analizó un total de 10 muestras clínicas o cultivos de muestras de HIV-1 grupo O y 1 muestra clínica o cultivo de muestra de HIV-1 grupo N tras la preparación de la series de dilución. Se analizaron 2 réplicas de cada una de las muestras sin diluir y 4 de cada dilución de 1:1,0E+01 a 1:4,8E+05 (de 3 a 5 diluciones por muestra) para HIV-1 grupo O. Todos los resultados obtenidos fueron reactivos. Se analizaron 2 réplicas de muestra sin diluir y 4 de cada dilución de 1:1,0E+04 a 1:1,4E+05 (5 diluciones) para HIV-1 grupo N. La muestra sin diluir y las diluciones de 1:1,0E+04 a 1:4,5E+04 obtuvieron resultados 100% reactivos, mientras que la dilución 1:1,4E+05 obtuvo un resultado reactivo al 50%. Todas las muestras analizadas se detectaron con una concentración ≤ 3 × LoD.
HIV-2:
Se analizó un total de 16 muestras clínicas únicas o cultivos de muestras de HIV-2 grupo A y grupo B tanto sin diluir como diluidas con una concentración ~5 × LoD de la prueba COBAS® HIV-1/HIV-2 QUALITATIVE. Las 16 muestras de HIV-2 se detectaron sin diluir y con una concentración de ~5 × LoD (Tabla 27).
También se analizaron 6 muestras clínicas de HIV-2 grupo A y 4 de HIV-2 grupo B tras la preparación de las series de dilución. Se analizó 1 réplica de cada una de las muestras sin diluir y 1 de cada dilución de 1:1,0E+01 a 1:9,0E+02 (de 2 a 5 diluciones por muestra) para HIV-2 grupo A y de 1:2,0E+01 a 1:6,0E+01 (2-4 diluciones) para HIV-2 grupo B. Todos los resultados obtenidos fueron reactivos. Todas las muestras clínicas analizadas se detectaron con una concentración ≤ 3 × LoD.
Tabla 27. Muestras clínicas o cultivos de muestras del HIV-2
|
Subtipo |
% de reactividad (muestras reactivas/analizadas) sin diluir |
% de reactividad (muestras reactivas/analizadas) diluidas a ~5 × LoD |
|
A |
100% (4/4) |
100% (4/4) |
|
B |
100% (6/6) |
100% (6/6) |
Especificidad:
La especificidad de la prueba COBAS® HIV-1/HIV-2 QUALITATIVE se determinó mediante el análisis de muestras de plasma conservado en EDTA negativas para HIV, suero negativo para HIV o DBS negativas para HIV obtenidas de donantes de sangre individuales. Se analizó un total de 613 muestras individuales de plasma conservado en EDTA y 607 muestras individuales de suero con 2 lotes de reactivos para la prueba COBAS® HIV-1/HIV-2 QUALITATIVE. También se analizaron 604 muestras DBS individuales con 3 lotes de reactivos para la prueba COBAS® HIV-1/HIV-2 QUALITATIVE. Todas las muestras analizadas obtuvieron resultados no reactivos para HIV-1 y HIV-2. En todas las muestras de plasma conservado en EDTA, suero o DBS, la especificidad de la prueba COBAS® HIV-1/HIV-2 QUALITATIVE fue del 100% (límite de confianza del 95%: ≥ 99,5%).
Paneles de seroconversión:
El rendimiento de la prueba COBAS® HIV-1/HIV-2 QUALITATIVE se evaluó mediante paneles de seroconversión comercializados para HIV-1 grupo M.
Paneles de seroconversión para HVI-1 grupo M:
Se utilizaron 25 paneles de seroconversión comercializados. Se analizó cada miembro del panel sin diluir con la prueba COBAS® HIV-1/HIV-2 QUALITATIVE y se compararon los resultados con los resultados obtenidos mediante una prueba serológica de HIV Ag/Ab de 4.a generación autorizada por la FDA y una prueba de ácidos nucleicos de comparación autorizada por la FDA realizadas con miembros del panel sin diluir. Los resultados de rendimiento generales se muestran en la Tabla 28.
Tabla 28. Rendimiento de la prueba COBAS® HIV-1/HIV-2 QUALITATIVE en los paneles de seroconversión de HIV
|
Panel de sero conversión de HIV |
Número de miembros del panel analizados |
Número de miembros del panel con resultado reactivo |
Días hasta el primer resultado reactivo |
Días de antelación en la detección con la prueba COBAS® HIV-1/HIV-2 QUALITATIVE |
|||||
|
COBAS® HIV-1/HIV-2 Qualitative |
NAT compa rativa |
Ensayo de HIV Ag/Ab |
COBAS® Qualitative |
NAT comparativa |
Ensayo de HIV Ag/Ab |
NAT comparativa |
Ensayo de HIV Ag/Ab |
||
|
HIV6243 |
10 |
6 |
6 |
4 |
18 |
18 |
25 |
0 |
7 |
|
HIV9011 |
11 |
3 |
3 |
2 |
30 |
30 |
38 |
0 |
8 |
|
HIV9012 |
8 |
5 |
5 |
3 |
9 |
7 |
16 |
-2 |
7 |
|
HIV9013 |
7 |
3 |
2 |
2 |
18 |
23 |
23 |
5 |
5 |
|
HIV9018 |
10 |
5 |
5 |
3 |
21 |
21 |
28 |
0 |
7 |
|
HIV9020 |
21 |
5 |
5 |
3 |
83 |
83 |
90 |
0 |
7 |
|
HIV9022 |
9 |
3 |
4 |
2 |
23 |
17 |
25 |
-6 |
2 |
|
HIV9030 |
16 |
6 |
5 |
3 |
40 |
40 |
47 |
0 |
7 |
|
HIV9031 |
19 |
8 |
6 |
4 |
120 |
131 |
146 |
11 |
26 |
|
HIV9034 |
13 |
4 |
4 |
3 |
41 |
41 |
46 |
0 |
5 |
|
HIV9076 |
9 |
3 |
3 |
3 |
66 |
66 |
66 |
0 |
0 |
|
HIV9089 |
6 |
5 |
5 |
3 |
7 |
7 |
16 |
0 |
9 |
|
HIV12008 |
13 |
7 |
7 |
5 |
21 |
21 |
28 |
0 |
7 |
|
PRB954 |
7 |
5 |
5 |
2 |
7 |
7 |
17 |
0 |
10 |
|
PRB956 |
5 |
4 |
4 |
2 |
40 |
40 |
47 |
0 |
7 |
|
PRB958 |
6 |
6 |
6 |
4 |
0 |
0 |
7 |
0 |
7 |
|
PRB961 |
9 |
4 |
4 |
2 |
19 |
19 |
27 |
0 |
8 |
|
PRB962 |
6 |
4 |
4 |
2 |
7 |
7 |
14 |
0 |
7 |
|
PRB963 |
7 |
4 |
5 |
2 |
9 |
7 |
17 |
-2 |
8 |
|
PRB967 |
6 |
5 |
5 |
3 |
3 |
3 |
17 |
0 |
14 |
|
PRB968 |
10 |
6 |
6 |
4 |
15 |
15 |
26 |
0 |
11 |
|
PRB969 |
10 |
7 |
6 |
3 |
53 |
53 |
70 |
0 |
17 |
|
PRB973 |
4 |
4 |
4 |
2 |
0 |
0 |
7 |
0 |
7 |
|
PRB976 |
4 |
4 |
4 |
2 |
0 |
0 |
7 |
0 |
7 |
|
PRB977 |
4 |
4 |
2 |
2 |
0 |
-* |
13 |
-* |
13 |
|
Total |
230 |
120 |
115 |
70 |
|||||
* Resultado no válido para la prueba NAT de comparación del primer miembro del panel PRB977, para el que se esperaba un resultado NAT reactivo. Por este motivo, el panel (PRB977) no se incluyó en la evaluación de los resultados de la prueba NAT de comparación.
Especificidad analítica:
Se evaluó la especificidad analítica de la prueba COBAS® HIV-1/HIV-2 QUALITATIVE para la reactividad cruzada con un panel de microorganismos con una concentración de 105 o 106 partículas, copias o UFP/ml de aislados víricos y cepas bacterianas/aislados de levadura, respectivamente (Tabla 29). Los microorganismos se añadieron a plasma humano conservado en EDTA negativo para el HIV y se analizaron con y sin virus del HIV-1 y el HIV-2 añadido a una concentración de aproximadamente 3 × LoD de la prueba COBAS® HIV-1/HIV-2 QUALITATIVE para cada virus. La prueba COBAS® HIV-1/HIV-2 QUALITATIVE generó resultados no reactivos para todas las muestras de microorganismos sin diana de HIV-1 y HIV-2 y resultados reactivos para todas las muestras de microorganismos con dianas del HIV-1 y el HIV-2. Los microorganismos analizados no presentan reactividad cruzada ni interfieren con la prueba COBAS® HIV-1/HIV-2 QUALITATIVE.
Tabla 29. Microorganismos analizados para reactividad cruzada
|
Virus |
Bacterias |
Levadura |
|
|
Adenovirus tipo 5 |
Virus de la varicela zóster |
Propionibacterium acnes |
Candida albicans |
|
Citomegalovirus |
Virus del Nilo Occidental |
Staphylococcus aureus |
|
|
Virus de Epstein-Barr |
Virus de la encefalitis de St. Louis |
||
|
Virus de la hepatitis A |
Virus de la encefalitis del Valle Murray |
||
|
Virus de la hepatitis B |
Virus del dengue tipos 1, 2, 3 y 4 |
||
|
Virus de la hepatitis C |
Virus TBE (cepa HYPR) |
||
|
Virus de la hepatitis D |
Virus de la gripe A |
||
|
Virus linfotrópico de células T humanas tipos 1 y 2 |
Virus Zika |
||
|
Virus del herpes humano tipo 6 |
Virus del papiloma humano |
||
|
Virus del herpes simple tipos 1 y 2 |
Virus de la fiebre amarilla |
||
Se analizaron muestras de plasma conservado en EDTA pertenecientes a cada uno de los estadios de la enfermedad (una del adenovirus tipo 5 y diez del resto de los estadios de la enfermedad) indicados en la Tabla 30 con y sin virus del HIV-1 y el HIV-2 añadido a una concentración de aproximadamente 3 × LoD de la prueba COBAS® HIV-1/HIV-2 QUALITATIVE para cada virus. Estas fases de la enfermedad no presentan reactividad cruzada ni interfieren con la prueba COBAS® HIV-1/HIV-2 QUALITATIVE.
Tabla 30. Muestras de cada estadio de la enfermedad analizadas para la especificidad analítica
|
Estadio de la enfermedad |
||
|
Adenovirus tipo 5 |
Virus de la hepatitis B |
Virus del herpes simple tipo 2 |
|
Citomegalovirus |
Virus de la hepatitis C |
Virus linfotrópico de células T humanas tipo I |
|
Virus del dengue |
Virus de la hepatitis E |
Virus linfotrópico de células T humanas tipo II |
|
Virus de Epstein-Barr |
Virus del herpes simple tipo 1 |
Virus del Nilo Occidental |
Especificidad analítica: sustancias interferentes:
Se analizaron niveles elevados de triglicéridos (33 g/l), bilirrubina conjugada (0,2 g/l), bilirrubina no conjugada (0,2 g/l), albúmina (60 g/l), hemoglobina (2 g/l) y ADN humano (2 mg/l) en muestras, así como la presencia de enfermedades autoinmunes como el lupus eritematoso sistémico (LES), la artritis reumatoide (AR) y anticuerpos antinucleares (ANA), en presencia y ausencia de ARN de HIV-1 y HIV-2.
También se analizaron los compuestos farmacológicos de la Tabla 31 con una concentración tres veces superior a la Cmax tanto en presencia como en ausencia de ARN de HIV-1 y HIV-2.
Ninguna de las posibles sustancias interferentes afecta al rendimiento de la prueba a excepción de los triglicéridos y el ADN humano. Con concentraciones mayores de 25 g/l de triglicéridos y de 1,5 mg/l de ADN humano, pueden obtenerse resultados inválidos a causa de la interferencia. La prueba COBAS® HIV-1/HIV-2 QUALITATIVE generó resultados no reactivos para todas las muestras sin diana del HIV y resultados reactivos para todas las muestras con dianas del HIV-1 y el HIV-2.
Tabla 31. Compuestos farmacológicos analizados para la interferencia mediante la prueba COBAS® HIV-1/HIV-2 QUALITATIVE
|
Clase de fármaco |
Nombre genérico del fármaco |
|
|
Moduladores del sistema inmunológico |
Peginterferón α-2a |
Ribavirina |
|
Peginterferón α-2b |
||
|
Inhibidores HCV |
Simeprevir |
Sofosbuvir |
|
Inhibidores de la transcriptasa inversa o de la polimerasa de ADN |
Emtricitabina |
Tenofovir |
|
Entecavir |
Adefovir dipivoxil |
|
|
Foscarnet |
Telbivudina |
|
|
Cidofovir |
Aciclovir |
|
|
Lamivudina |
Valganciclovir |
|
|
Ganciclovir |
||
|
Compuestos para el tratamiento de infecciones oportunistas |
Azitromicina. Claritromicina. Etambutol Fluconazol Isoniazida |
Pirazinamida. Rifabutina. Rifampicina. Sulfametoxazol. Trimetoprima |
|
Estatina |
Atorvastatina |
|
|
Inhibidor selectivo de la recaptación de serotonina |
Fluoxetina |
Paroxetina |
|
Sertralina |
||
|
Antihistamínico |
Loratadina |
|
|
Beta bloqueador |
Nadolol |
|
|
Descongestivo |
Fenilefrina HCI |
|
|
Fármaco antiinflamatorio no esteroideo |
Naproxeno |
Ibuprofeno |
|
Analgésico |
Acetaminofeno |
Ácido acetilsalicílico |
|
Vitaminas |
Ácido ascórbico |
|
Correlación de métodos:
Suero y plasma conservado en EDTA:
Se comparó el rendimiento de la prueba COBAS® HIV-1/HIV-2 QUALITATIVE con el de la prueba COBAS® AmpliPrep/COBAS® TaqMan® HIV-1 Qualitative (muestras de plasma conservado en EDTA del HIV-1) y con el de una prueba de diferenciación de anticuerpos de HIV-1/HIV-2 con marcado CE (muestras de suero y plasma de HIV-1 y HIV-2 conservado en EDTA).
Para la correlación con la prueba COBAS® AmpliPrep/COBAS® TaqMan® HIV-1 Qualitative v2.0, se analizaron muestras clínicas de plasma conservado en EDTA en un centro externo. Para las muestras clínicas de plasma conservado en EDTA positivas para el HIV-1 y negativas para el HIV-1, ambas pruebas mostraron un porcentaje de concordancia del 100%, lo que demuestra que el rendimiento de la prueba COBAS® HIV-1/HIV-2 QUALITATIVE y el de la prueba COBAS® AmpliPrep/COBAS® TaqMan® HIV-1 Qualitative v2.0 son equivalentes (Tabla 32).
Tabla 32. Resumen de resultados de la correlación de métodos para las muestras de plasma conservado en EDTA de HIV-1
|
Correlación de métodos Muestras de plasma conservadas en EDTA de HIV-1 |
Prueba COBAS® AmpliPrep/COBAS® TaqMan® HIV-1 Qualitative, v2.0 |
||
|
Reactivo |
No reactivo |
||
|
COBAS® HIV-1/HIV-2 QUALITATIVE |
Reactivo |
68 |
0 |
|
No reactivo |
0 |
80 |
|
Para la comparación con una prueba de diferenciación de anticuerpos de HIV-1/HIV-2 con marcado CE, se analizaron muestras clínicas de suero o plasma conservado en EDTA en un centro externo. Las muestras de plasma y suero conservados en EDTA positivas para HIV-1 y negativas para HIV-1 mostraron un porcentaje de concordancia total del 100% entre ambas pruebas. Las muestras de plasma y suero conservados en EDTA positivas para HIV-2 y negativas para HIV-2 mostraron un porcentaje de concordancia total del 99,7% entre ambas pruebas. Esto demuestra que el rendimiento de la prueba COBAS® HIV-1/HIV-2 QUALITATIVE y el de la prueba de diferenciación de anticuerpos de HIV-1 y HIV-2 con marcado CE es equivalente (Tabla 33 y Tabla 34).
Tabla 33. Resumen de los resultados de la correlación de métodos para muestras de suero y plasma conservados en EDTA de HIV-1
|
Correlación de métodos Muestras de suero y plasma conservado en EDTA de HIV-1 |
Prueba de diferenciación de anticuerpos HIV-1/HIV-2 con marcado CE |
|||
|
Reactivo |
No reactivo |
Indeterminado |
||
|
COBAS® HIV-1/HIV-2 QUALITATIVE |
Reactivo |
138 |
0 |
0 |
|
No reactivo |
0 |
164 |
1* |
|
* Se confirmó que la muestra con un resultado no reactivo con la prueba COBAS® HIV-1/HIV-2 QUALITATIVE y un resultado indeterminado con la prueba de diferenciación de anticuerpos de HIV-1/HIV-2 con marcado CE era negativa mediante una prueba serológica del HIV Ag/Ab de 4a generación con marcado CE.
Tabla 34. Resumen de los resultados de la correlación de métodos para muestras de suero y plasma conservados en EDTA de HIV-2
|
Correlación de métodos Muestras de suero y plasma conservado en EDTA de HIV-2 |
Prueba de diferenciación de anticuerpos HIV-1/HIV-2 con marcado CE |
|||
|
Reactivo |
No reactivo |
Indeterminado |
||
|
COBAS® HIV-1/HIV-2 QUALITATIVE |
Reactivo |
14 |
0 |
0 |
|
No reactivo |
1 |
287 |
1* |
|
* Se confirmó que la muestra con un resultado no reactivo con la prueba COBAS® HIV-1/HIV-2 QUALITATIVE y un resultado indeterminado con la prueba de diferenciación de anticuerpos de HIV-1/HIV-2 con marcado CE era negativa mediante una prueba serológica del HIV Ag/Ab de 4a generación con marcado CE.
DBS:
Se comparó el rendimiento de la prueba COBAS® HIV-1/HIV-2 QUALITATIVE con el de la prueba COBAS® AmpliPrep/COBAS® TaqMan® HIV-1 Qualitative v2.0 mediante el análisis de muestras clínicas DBS procedentes de bebés en un centro externo. Para las muestras DBS positivas para el HIV-1 y negativas para el HIV-1, ambas pruebas mostraron un porcentaje de concordancia del 99,6%, lo que demuestra que el rendimiento de la prueba COBAS® HIV-1/HIV-2 QUALITATIVE y el de la prueba COBAS® AmpliPrep/COBAS® TaqMan® HIV-1 Qualitative v2.0 es equivalente (Tabla 35).
Tabla 35. Resumen de los resultados de la correlación de métodos para las muestras DBS de HIV-1
|
Correlación de métodos Muestras DBS de HIV-1 |
Prueba COBAS® AmpliPrep/COBAS® TaqMan® HIV-1 Qualitative, v2.0 |
||
|
Reactivo |
No reactivo |
||
|
COBAS® HIV-1/HIV-2 QUALITATIVE |
Reactivo |
127 |
1* |
|
No reactivo |
0 |
151 |
|
* Se confirmó que la muestra con un resultado reactivo para la prueba COBAS® HIV-1/HIV-2 QUALITATIVE y un resultado no reactivo para la prueba COBAS® AmpliPrep/COBAS® TaqMan® HIV- 1 Qualitative v2.0 era positiva para HIV-1 mediante PCR anidada.
Fallo de todo el sistema:
La tasa de fallo de todo el sistema para la prueba COBAS® HIV-1/HIV-2 QUALITATIVE se determinó mediante el análisis de 100 réplicas de plasma conservado en EDTA y 100 réplicas de sangre entera de DBS a las que se añadió HIV-1 grupo M subtipo B y HIV-2. Estas muestras se analizaron con una concentración de la diana de aproximadamente 3 × LoD.
Los resultados del estudio indican que todas las réplicas fueron válidas y positivas para las dianas de HIV-1 y HIV-2, lo que representa una tasa de fallo de todo el sistema del 0% para las muestras de plasma conservado en EDTA y DBS. El intervalo de confianza bilateral exacto del 95% fue del 0% para el límite inferior y del 3,6% para el límite superior de cada diana y matriz.
Contaminación por arrastre:
La tasa de contaminación cruzada de la prueba COBAS® HIV-1/HIV-2 QUALITATIVE se determinó mediante el análisis de 240 réplicas de una muestra DBS negativa para HIV y 225 réplicas de una muestra DBS con un título elevado de HIV-1 con una concentración de 2,0E+07 cp/ml. El estudio se realizó siguiendo el flujo de trabajo de preparación de muestras DBS. En total, se realizaron cinco series con muestras positivas y negativas utilizando un método de ensayo con configuración de tablero de ajedrez.
Las 240 réplicas de la muestra negativa resultaron no reactivas, por lo que la tasa de contaminación cruzada es del 0%. El intervalo de confianza del 95% fue del 0% para el límite inferior y del 1,5% para el límite superior.
USO PREVISTO:
La prueba de ácidos nucleicos COBAS® HIV-1/HIV-2 QUALITATIVE para uso en los cobas® 5800/6800/8800 Systems es una prueba de amplificación de ácidos nucleicos in vitro para la detección cualitativa y diferenciación del virus de inmunodeficiencia humano (HIV) tipo 1 (HIV-1) y tipo 2 (HIV-2) en muestras de suero, plasma y sangre seca (DBS) humanas.
La prueba está concebida para su uso como ayuda en el diagnóstico del HIV-1/HIV-2. La detección de ácidos nucleicos de HIV-1 o HIV-2 es indicativa de infección por HIV-1 o HIV-2, respectivamente. La presencia de ácidos nucleicos HIV-1 o HIV-2 en el plasma o suero de personas sin anticuerpos frente al HIV-1 o HIV-2 es indicativo de una infección agua o primaria. En bebés de madres infectadas por HIV y que presentan anticuerpos maternos frente al HIV-1 o el HIV-2, la presencia de ácidos nucleicos del HIV indica una infección activa. La prueba COBAS® HIV-1/HIV-2 QUALITATIVE también puede utilizarse para confirmar una infección por HIV-1 o HIV-2 en un sujeto con muestras reactivas para anticuerpos o antígenos del HIV-1 o HIV-2.
INSTRUCCIONES DE USO:
Notas sobre el procedimiento:
• No utilice los reactivos para la prueba COBAS® HIV-1/HIV-2 QUALITATIVE, el cobas® HIV-1/HIV-2 Qualitative Control Kit, el cobas® NHP Negative Control Kit, los reactivos cobas omni o el cobas® Specimen Pre-Extraction Reagent después de la fecha de caducidad.
• No reutilice el material fungible. Son de un solo uso.
• Consulte la Asistencia al usuario o la Guía del usuario del cobas® 5800 System o los cobas® 6800/8800 Systems para obtener información sobre el correcto mantenimiento de los instrumentos.
Preparación de muestras de sangre seca:
• Deje que el cobas® Specimen Pre-Extraction Reagent (SPER) alcance la temperatura ambiente antes de utilizarlo.
• Extraiga una DBS de la tarjeta para muestras DBS.
• Transfiera la muestra a un tubo (5 mL, rosca interior, 12,5 mm de diámetro, polipropileno [Cryo.s™]) mediante fórceps o pinzas estériles o desechables.
• Asegúrese de que la DBS se deposita en la parte inferior del tubo tal como se muestra en la Ilustración 2.
• Pipetee 1.150 μL de SPER en el tubo que contiene la DBS y coloque el tapón.
• Asegúrese de que la DBS está totalmente cubierta con SPER.
• Coloque los tubos en las posiciones de la 1 a la 24 en un Eppendorf Thermomixer (p. ej., modelo R 5355 o equivalente) precalentado con termobloque para 24 criotubos e incube durante 10 minutos a 56 °C y 1.000 rpm para extraer el virus de la muestra de sangre seca.
• Quite el tapón de los tubos y asegúrese de que la DBS está adherida a la pared del tubo (Ilustración 3) para evitar coágulos en la muestra.
• Elimine cualquier película líquida que se haya podido crear por encima del nivel del líquido (Ilustración 4) mediante una punta de pipeta estéril para evitar la detección de nivel precoz.
• Transfiera los tubos al cobas® 5800 System o a los cobas® 6800/8800 Systems.

Ilustración 2 DBS en el tubo
Ilustración 3 Ubicación de la DBS
Ilustración 4 Película líquida
Realización de la prueba COBAS® HIV-1/HIV-2 QUALITATIVE en el cobas® 5800 System:
La prueba COBAS® HIV-1/HIV-2 QUALITATIVE puede ejecutarse con volúmenes de muestra obligatorios de 650 μL (para el flujo de trabajo de muestras de plasma o suero de 500 μL) o de 1.150 μL de cobas® Specimen Pre-Extraction Reagent (para el flujo de trabajo de muestras DBS de 850 μL). Tenga en cuenta que las muestras DBS pueden analizarse en el modo de serie combinada con flujos de trabajo de plasma, suero o COBAS® HIV-1 PSC. El procedimiento de la prueba se describe con detalle en la Asistencia al usuario o la Guía del usuario del cobas® 5800 System. La Ilustración 5 siguiente resume el procedimiento.
Ilustración 5. Procedimiento de la prueba COBAS® HIV-1/HIV-2 QUALITATIVE en el cobas® 5800 System
|
1 |
Inicie una sesión en el sistema. |
|
2 |
Cargue las muestras en el sistema: • Cargue los racks de muestras en el sistema. • El sistema se prepara automáticamente. • Solicite las pruebas. |
|
3 |
Cargue los reactivos y el material fungible según las indicaciones del sistema: • Cargue el casete de reactivo específico de la prueba. • Cargue los miniracks de control. • Cargue las puntas de procesamiento. • Cargue las puntas de elución. • Cargue las placas de procesamiento. • Cargue las placas de residuos líquidos. • Cargue las placas de amplificación. • Cargue el casete de MGP. • Cargue el diluyente de muestras. • Cargue el reactivo de lisis. • Cargue el reactivo de lavado. |
|
4 |
Seleccione el botón de inicio de procesamiento en la interfaz de usuario para iniciar la serie analítica. Las series siguientes se iniciarán de forma automática si no se posponen manualmente. |
|
5 |
Revise y exporte los resultados. |
|
6 |
Retire y tape todos los tubos de muestra que cumplan los requisitos de volumen mínimo para utilizarlos en el futuro, en caso necesario. Limpieza del instrumento: • Descargue los miniracks de control vacíos. • Descargue el casete de reactivo específico de la prueba vacío. • Vacíe el cajón de placas de amplificación. • Vacíe los residuos líquidos. • Vacíe los residuos sólidos. |
Realización de la prueba COBAS® HIV-1/HIV-2 QUALITATIVE en los cobas® 6800/8800 Systems:
La prueba COBAS® HIV-1/HIV-2 QUALITATIVE puede ejecutarse con un volumen de muestra mínimo obligatorio de 650 μL (para el flujo de trabajo de muestras de plasma o suero de 500 μL) o de 1.150 μL de cobas® Specimen Pre-Extraction Reagent (para el flujo de trabajo de muestras DBS de 850 μL). Tenga en cuenta que el flujo de trabajo de muestras de sangre seca para la prueba COBAS® HIV-1/HIV-2 Qualitative se ha evaluado en la función de serie combinada únicamente con el flujo de trabajo de cobas® HIV-1 PSC. El procedimiento de la prueba se describe con detalle en la Asistencia al usuario o la Guía del usuario de los cobas® 6800/8800 Systems. La Ilustración 6 siguiente resume el procedimiento.
Ilustración 6. Procedimiento de la prueba COBAS® HIV-1/HIV-2 QUALITATIVE en los cobas® 6800/8800 Systems
|
1 |
Inicie una sesión en el sistema. Pulse el botón “Iniciar” para preparar el sistema. Solicite las pruebas. |
|
2 |
Cargue los reactivos y el material fungible según las indicaciones del sistema: • Cargue el casete de reactivo específico de la prueba. • Cargue los casetes de control. • Cargue las puntas de pipeta. • Cargue las placas de procesamiento. • Cargue el reactivo MGP. • Cargue las placas de amplificación. • Cargue el diluyente de muestras. • Cargue el reactivo de lisis. • Cargue el reactivo de lavado. |
|
3 |
Cargue las muestras en el sistema: • Cargue los racks de muestras y los racks para puntas obstruidas en el módulo de suministro de muestras. • Confirme que las muestras han sido aceptadas en el módulo de transferencia. |
|
4 |
Inicie la serie analítica mediante el botón “Iniciar manualmente” de la interfaz de usuario o ejecute un inicio automático después de 120 minutos o cuando la serie esté llena. |
|
5 |
Revise y exporte los resultados. |
|
6 |
Retire y tape todos los tubos de muestra que cumplan los requisitos de volumen mínimo para utilizarlos en el futuro, en caso necesario. Limpieza del instrumento: • Descargue los casetes de control vacíos. • Vacíe el cajón de placas de amplificación. • Vacíe los residuos líquidos. • Vacíe los residuos sólidos. |
INFORMACIÓN ADICIONAL:
Características principales de la prueba:
|
Tipo de muestra |
Plasma conservado en EDTA, suero y muestra de sangre seca (DBS) |
||
|
Cantidad mínima de muestra necesaria |
650 μL para muestras de suero y plasma conservado en EDTA o una muestra DBS (70 μL de sangre seca por muestra) en 1.150 μL de cobas® Specimen Pre-Extraction Reagent (SPER) |
||
|
Volumen de procesamiento de muestras |
500 μL para las muestras de plasma conservado en EDTA y suero o 850 μL para las muestras DBS |
||
|
Sensibilidad analítica |
HIV-1M |
HIV-2 |
|
|
Plasma conservado en EDTA |
12,6 cp/mL |
27,9 cp/mL |
|
|
Suero |
12,1 cp/mL |
23,4 cp/mL |
|
|
DBS |
255 cp/mL |
984 cp/mL |
|
|
Especificidad |
100% (intervalo de confianza unilateral del 95%: 99,5%) (plasma en EDTA/suero) 100% (intervalo de confianza unilateral del 95%: 99,5%) (DBS) |
||
|
Grupos/Subtipos - inclusividad |
HIV-1 M (A-D, F-H, J, K, CRF01_AE, CRF02_AG, CRF12_BF, CRF14_BG), HIV-1 O, HIV-1 N, HIV-2 (A y B) |
||
Símbolos:
Los símbolos siguientes se emplean en el rotulado de todos los productos de diagnóstico por PCR de Roche.
Tabla 41. Símbolos utilizados en las etiquetas de los productos de PCR para diagnóstico de Roche

Bibliografía:
1. American Academy of HIV Medicine. Fundamentals of HIV Medicine for the HIV Specialist. Washington, D.C.: American Academy of HIV Medicine, 2007.
2. UNAIDS. Global AIDS Update. Available at: https:// https://www.unaids.org/sites/default/files/media_asset/global-AIDS-update-2016_en.pdf.
3. Hoover DR, Saah AJ, Bacellar H, et al. Clinical manifestations of AIDS in the era of pneumocystis prophylaxis. Multicenter AIDS Cohort Study. N Engl J Med. 1993;329:1922-6. PMID: 7902536.
4. Campbell-Yesufu OT, Gandhi RT. Update on human immunodeficiency virus (HIV)-2 infection. Clin Infect Dis. 2011;52:780-7. PMID: 21367732.
5. Centers for Disease Control and Prevention. Laboratory Testing for the Diagnosis of HIV Infection. Available at: https://stacks.cdc.gov/view/cdc/23447. Accessed December 6, 2020.
6. Gökengin D, Geretti AM, Begovac J, et al. 2014 European Guideline on HIV testing. Int J STD AIDS. 2014;25:695- 704. PMID: 24759563.
7. Masciotra S, McDougal JS, Feldman J, et al. Evaluation of an alternative HIV diagnostic algorithm using specimens from seroconversion panels and persons with established HIV infections. J Clin Virol. 2011;52 Suppl 1:S17-22. PMID: 21981983.
8. O’Brien M, Markowitz M. Should we treat acute HIV infection? Curr HIV/AIDS Rep. 2012;9:101-10. PMID: 22415472.
9. Branson BM, Mermin J. Establishing the diagnosis of HIV infection: new tests and a new algorithm for the United States. J Clin Virol. 2011;52 Suppl 1:S3-4. PMID: 21993308.
10. Cohen MS, Shaw GM, McMichael AJ, Haynes BF. Acute HIV-1 Infection. N Engl J Med. 2011;364:1943-54. PMID: 21591946.
11. World Health Organization. WHO Recommendations on the Diagnosis of HIV Infection in Infants and Children. Available at: https:// www.who.int/hiv/pub/paediatric/diagnosis/en/. Accessed December 6, 2020.
12. Wittek M, Stürmer M, Doerr HW, Berger A. Molecular assays for monitoring HIV infection and antiretroviral therapy. Expert Rev Mol Diagn. 2007;7:237-46. PMID: 17489731.
13. Longo MC, Berninger MS, Hartley JL. Use of uracil DNA glycosylase to control carry-over contamination in polymerase chain reactions. Gene. 1990;93:125-8. PMID: 2227421.
14. Savva R, McAuley-Hecht K, Brown T, Pearl L. The structural basis of specific base-excision repair by uracil-DNA glycosylase. Nature. 1995;373:487-93. PMID: 7845459.
15. Mol CD, Arvai AS, Slupphaug G, et al. Crystal structure and mutational analysis of human uracil-DNA glycosylase: structural basis for specificity and catalysis. Cell. 1995;80:869-78. PMID: 7697717.
16. Higuchi R, Dollinger G, Walsh PS, Griffith R. Simultaneous amplification and detection of specific DNA sequences. Biotechnology (N Y). 1992;10:413-7. PMID: 1368485.
17. Heid CA, Stevens J, Livak KJ, Williams PM. Real time quantitative PCR. Genome Res. 1996;6:986-94. PMID: 8908518.
18. Centers for Disease Control and Prevention. Biosafety in Microbiological and Biomedical Laboratories, 5th ed. https: //www.cdc.gov/labs/pdf/CDC-BiosafetyMicrobiologicalBiomedicalLaboratories-2009-P.PDF. Accessed December 2, 2020.
19. Clinical and Laboratory Standards Institute. Protection of laboratory workers from occupationally acquired infections. Approved Guideline-Fourth Edition. https://clsi.org/media/1459/m29a4_sample.pdf. Accessed December 2, 2020.
Asistencia técnica:
Para obtener asistencia técnica, póngase en contacto con su filial local: https://www.roche.com/about/business/roche_worldwide.htm
Fabricante e importador:
Roche Molecular Systems, Inc.
1080 US Highway 202 South
Branchburg, NJ 08876 USA
Fabricado en los EE. UU.
Roche Diagnostics GmbH
Sandhofer Strasse 116
68305 Mannheim, Germany
Marcas registradas y patentes:
Consulte https://diagnostics.roche.com/us/en/about-us/patents
Copyright
©2022 Roche Molecular Systems, Inc.
Roche Diagnostics GmbH
Sandhofer Str 116
68305 Mannheim
Germany
COBAS® HIV-1/HIV-2 QUALITATIVE: P/N: 09040528190
Para uso en el sistema cobas® 5800:
cobas® HIV-1/HIV-2 Qualitative Control Kit
P/N: 09040536190
cobas® NHP Negative Control Kit: P/N: 09051554190
Para uso en sistemas cobas® 6800/8800:
COBAS® HIV-1/HIV-2 QUALITATIVE Control Kit
P/N: 07862091190 o P/N: 09040536190
cobas® NHP Negative Control Kit
P/N: 07002220190 o P/N: 09051554190
cobas® Specimen Pre-Extraction Reagent
P/N: 08064695190
RESUMEN Y EXPLICACIÓN DE LA PRUEBA:
Información de referencia:
El virus de la inmunodeficiencia humana (HIV) es el agente etiológico del síndrome de inmunodeficiencia adquirida (SIDA).1 El HIV-1 es la causa principal del SIDA en todo el mundo, con más de 35 millones de personas infectadas.2 Tras la infección, las personas infectadas, por lo general, entran en una fase relativamente asintomática, clínicamente estable, que puede prolongarse durante años. Sin un tratamiento antirretroviral, las personas infectadas suelen desarrollar SIDA, que se caracteriza por la disminución de linfocitos CD4+ en el sistema inmune, la propensión a padecer infecciones oportunistas y, finalmente, la muerte.3 El HIV-2, que se detecta básicamente en África occidental, también puede ser causante del SIDA. Se estima que hay entre 1 y 2 millones de personas infectadas por HIV-2 en todo el mundo.
La distinción entre el HIV-1 y el HIV-2 es importante por diversos motivos: (1) el HIV-2 muestra una menor virulencia que el HIV-1, con cargas virales inferiores, un ritmo más lento de pérdida de linfocitos CD4+ y una progresión más pausada a infecciones oportunistas; (2) las cargas virales del HIV-2 pueden no cuantificarse correctamente mediante pruebas de cargas virales del HIV-1; y (3) algunos fármacos para el HIV-1, especialmente los inhibidores de la transcriptasa inversa no nucleósidos, no resultan eficaces contra el HIV-2.4 Asimismo, es posible que se produzca una coinfección por HIV-1 y HIV-2. La coinfección no afecta a la tasa de progresión a SIDA de los sujetos de forma evidente, pero complica la monitorización de la carga viral y el tratamiento antirretroviral.4 Debido a la importancia de distinguir entre una infección por HIV-1 o por HIV-2, las directrices nacionales e internacionales han incluido el diagnóstico y la diferenciación del HIV-1 y el HIV-2 como requisito para el diagnóstico adecuado de una infección por HIV.5,6
Motivos para el uso de la prueba de PCR:
Tradicionalmente, la prueba del HIV se ha basado en la respuesta de los anticuerpos que los pacientes presentan ante el virus. Si bien estos anticuerpos no son eficaces a la hora de combatir el virus, se encuentran en casi todos los pacientes con infecciones crónicas. La principal limitación de la prueba de anticuerpos es el periodo de ventana de varias semanas que tiene lugar durante la fase aguda de la infección, antes del inicio de una respuesta de anticuerpos detectable. El periodo de ventana ha disminuido gracias a las pruebas de inmunoensayo del HIV de cuarta generación, que detectan tanto el antígeno p24 como los anticuerpos del HIV.7 No obstante, las pruebas de amplificación de ácidos nucleicos disponen del potencial necesario para reducir todavía más el periodo de ventana de las pruebas de inmunoensayo de cuarta generación para una mejor detección de infecciones por HIV gracias al aumento de la sensibilidad de los métodos de PCR con respecto a los métodos basados en la detección de proteínas.7
En función del riesgo de infección por HIV de la población analizada, la reducción del periodo de ventana de las pruebas de ácidos nucleicos puede ser sumamente importante tanto para el sujeto como para la comunidad.8 Para un paciente, el diagnóstico de HIV durante una fase de infección aguda ofrece la posibilidad de recibir tratamiento inmediato, lo que podría llegar a retardar la progresión de la enfermedad evitando daños en el sistema inmune y conservando la respuesta inmune celular frente al HIV. Un tratamiento temprano también puede limitar el tamaño y la diversidad genética del depósito viral establecido, lo que facilita la obtención de una cura funcional en pacientes tratados durante la fase aguda de la infección. Para la comunidad, los pacientes con infección aguda desempeñan un papel esencial en la transmisión del HIV, ya que suelen presentar cargas virales muy elevadas y no son conscientes de su estado de infección. La identificación y el tratamiento de estos pacientes pueden ser fundamentales a la hora de detener la propagación de la epidemia del HIV.9,10 La PCR ya se ha convertido en el estándar de atención para el diagnóstico del HIV en bebés; además, su elevada sensibilidad y especificidad permitiría no solo la detección de infecciones agudas en sujetos de todas las edades, sino también la confirmación del diagnóstico del HIV en sujetos seropositivos o de serología indeterminada.11,12
Explicación de la prueba:
La prueba COBAS® HIV-1/HIV-2 QUALITATIVE es una prueba cualitativa que se realiza en el cobas® 5800 System, el cobas® 6800 System o el cobas® 8800 System. La prueba COBAS® HIV-1/HIV-2 QUALITATIVE permite la detección y diferenciación simultáneas de los ácidos nucleicos del HIV-1 y el HIV-2 en plasma conservado en ácido etilendiaminotetraacético (EDTA), suero y muestras DBS de pacientes infectados. Se utilizan dos sondas que detectan el HIV-1 sin discriminar entre los subtipos del grupo M del HIV-1 ni entre los grupos O y N del HIV-1. Se utiliza una tercera sonda que detecta el HIV-2 sin discriminar entre el grupo A y B del HIV-2.
Principios del procedimiento:
La prueba COBAS® HIV-1/HIV-2 QUALITATIVE se basa en la preparación de muestras totalmente automática (extracción y purificación de ácidos nucleicos) seguida de un proceso de amplificación y detección mediante PCR. El cobas® 5800 System se ha diseñado como un único instrumento integrado. Los cobas® 6800/8800 Systems constan del módulo de suministro de muestras, el módulo de transferencia, el módulo de procesamiento y el módulo analítico. La gestión automática de los datos se realiza mediante el software cobas® 5800 o el cobas® 6800/8800 Systems, que asigna los resultados a las pruebas como no reactivos, reactivos o no válidos. Los resultados pueden revisarse directamente en la pantalla del sistema, exportarse o imprimirse como informe PDF.
Los ácidos nucleicos de las muestras de pacientes y las moléculas de control interno (IC) de Armored RNA añadidas (utilizadas como control del proceso de preparación de muestras y amplificación/detección) se extraen simultáneamente. Además, la prueba utiliza tres controles externos: dos positivos y uno negativo. En resumen, los ácidos nucleicos víricos se liberan al añadir proteinasa y reactivo de lisis a la muestra. Los ácidos nucleicos liberados se unen a la superficie de sílice de las partículas de vidrio magnéticas añadidas. Las sustancias sin unir y las impurezas, como las proteínas desnaturali- zadas, los restos celulares y los posibles inhibidores de la PCR se eliminan en los siguientes pasos de lavado, mientras que los ácidos nucleicos purificados se eluyen de las partículas de vidrio magnéticas mediante el buffer de elución a temperatura elevada.
La amplificación selectiva de los ácidos nucleicos de la diana de la muestra se lleva a cabo mediante cebadores que van en un sentido y en sentido contrario específicos del virus para la diana que se seleccionan de regiones altamente conservadas del genoma del HIV-1 y el HIV-2. La prueba COBAS® HIV-1/HIV-2 QUALITATIVE amplifica el gen gag del HIV-1, la región LTR del HIV-1 (diana doble del HIV-1) y la región LTR del HIV-2.
La amplificación selectiva del IC se realiza mediante cebadores que van en un sentido y en sentido contrario específicos de la secuencia que se seleccionan de modo que no presenten homología alguna con los genomas del HIV-1 o el HIV-2. Para los procesos de transcripción inversa y amplificación mediante PCR se utiliza una enzima ADN polimerasa termoestable. La amplificación de las secuencias diana y del IC se realiza simultáneamente mediante un perfil universal de amplificación mediante PCR que incluye unos pasos de temperatura y número de ciclos predefinidos. El reactivo de Master Mix incluye trifosfato de deoxiuridina (dUTP), en lugar de trifosfato desoxitimidina (dTTP), que se incorpora al ADN recién sintetizado (amplicón).13-15 La enzima AmpErase, que se incluye en la Master Mix para PCR, elimina los amplicones contaminados de las series de PCR anteriores durante la primera ciclación térmica. Sin embargo, los amplicones nuevos no se eliminan porque la enzima AmpErase se inactiva cuando se expone a temperaturas superiores a los 55 °C.
El reactivo de Master Mix para la prueba COBAS® HIV-1/HIV-2 QUALITATIVE contiene dos sondas de detección específicas para las secuencias de la diana de HIV-1, una para las secuencias diana del HIV-2 y otra para el IC. Las sondas están marcadas con marcadores emisores fluorescentes específicos para la diana a fin de permitir la detección simultánea de las dianas del HIV-1, HIV-2 e IC en tres canales diana distintos.16,17 Cuando no se une a la secuencia de la diana, la señal fluorescente de las sondas intactas se elimina mediante el marcador silenciador. Durante el paso de amplificación mediante PCR, la hibridación de las sondas con la plantilla específica de ADN monocatenario provoca la escisión de la sonda por la actividad de la exonucleasa 5’ a 3’ de la ADN polimerasa, lo que produce la separación de los marcadores emisor y silenciador y la emisión de una señal fluorescente. Con cada ciclo de PCR, se generan cantidades crecientes de sondas escindidas y la señal acumulada del marcador emisor aumenta concomitantemente. La detección y discriminación en tiempo real de los productos de la PCR se consigue mediante la cuantificación de la fluorescencia generada por los marcadores emisores liberados para las dianas víricas y el IC, respectivamente.




