
BLINCYTO
BLINATUMOMAB
Polvo liofilizado para reconstituir
1 Caja, 1 Frasco(s) ámpula, 35 µg
FORMA FARMACÉUTICA Y FORMULACIÓN:
Cada frasco ámpula de un solo uso contiene: Blinatumomab 35 mcg
Excipientes c.s
INDICACIONES TERAPÉUTICAS:
Leucemia linfoblástica aguda (LLA) de precursores de células B, enfermedad mínima residual (EMR) positiva
BLINCYTO® está indicado para el tratamiento de la leucemia linfoblástica aguda (LLA) de precursores de células B en la primera o segunda remisión completa con una enfermedad mínima residual (EMR) mayor o igual al 0.1% en adultos y niños.
Leucemia linfoblástica aguda (LLA) de precursores de células B en recaída o refractaria: BLINCYTO® está indicado para el tratamiento de leucemia linfoblástica aguda (LLA) de precursores de células B, en recaída o refractaria en adultos y niños.
FARMACOCINÉTICA Y FARMACODINAMIA:
Farmacocinética: La farmacocinética de blinatumomab aparece lineal sobre un intervalo de dosis de 5 a 90 mcg/m2/día (aproximadamente equivalente a 9 mcg/día a 162 mcg/día) en los pacientes adultos. Después de la infusión intravenosa continua, la concentración sérica en estado estable (Css) se logró en el curso de un día y permaneció estable con el tiempo. El aumento en los valores medios de Css fue aproximadamente proporcional a la dosis en el intervalo probado. A las dosis clínicas de 9 mcg/día y 28 mcg/día para el tratamiento de LLA recurrente o refractaria, la Css (DE) media fue 228 (356) pg/mL y 616 (537) pg/mL, respectivamente.
Distribución: El volumen de distribución medio (DE) estimado basado en la fase terminal (Vz) fue 4.35 (2.45) L con la infusión intravenosa continua de blinatumomab.
Metabolismo: La vía metabólica de blinatumomab no se ha caracterizado. Como ocurre con otras proteínas terapéuticas, se espera que BLINCYTO® se degrade en péptidos pequeños y aminoácidos a través de vías catabólicas.
Eliminación: La depuración sistémica media (DE) estimada con infusión intravenosa continua en pacientes recibiendo blinatumomab en estudios clínicos fue 3.11 (2.98) L/hora. La media (DE) de la vida media fue de 2.10 (1.41) horas. Se eliminaron cantidades insignificantes de blinatumomab en la orina en las dosis clínicas probadas.
Género, edad, y peso corporal, superficie corporal: Los resultados de análisis poblacional farmacocinético indican que la edad (0.62 a 80 años de edad), y género no influyen en la farmacocinética de blinatumomab. La superficie corporal (0.4 a 2.70 m2) influye en la farmacocinética de blinatumomab, sin embargo, se desconoce la relevancia clínica de este efecto.
Insuficiencia hepática: No se han llevado a cabo estudios formales de farmacocinética con BLINCYTO® en pacientes con insuficiencia hepática.
Insuficiencia renal: No se han conducido estudios formales de farmacocinética con blinatumomab en pacientes con insuficiencia renal.
Los análisis de farmacocinética mostraron una diferencia aproximada de 2 veces en los valores medios de depuración de blinatumomab entre pacientes con insuficiencia renal moderada (CrCL en el rango de 30 mL/min a 59 mL/min, N = 21) y función renal normal (CrCL de más de 90 mL/min, N = 215). Sin embargo, se percibió una alta variabilidad entre pacientes (CV% de hasta 96.8%), y los valores de depuración en pacientes con insuficiencia renal estuvieron esencialmente dentro del rango observado en pacientes con función renal normal. No hay información disponible de pacientes con insuficiencia renal severa (CrCL de menos de 30 mL/min) o pacientes en hemodiálisis.
Interacciones medicamentosas: La elevación transitoria de las citocinas puede suprimir las actividades de la enzima CYP450 (ver Interacciones medicamentosas y de otro género).
Poblaciones especiales:
Pediátricos: La farmacocinética de blinatumomab se presenta lineal sobre un rango de dosis de 5 mcg/m2/día a 30 mcg/m2/día en pacientes pediátricos. A las dosis recomendadas de 5 mcg/m2/día y 15 mcg/m2/día para el tratamiento de LLA de precursores de células B en recaída o refractaria, los valores de la media (DE) de concentración en estado estable (Css) fueron 162 (179) pg/mL y 533 (392) pg/mL, respectivamente. La media estimada (DE) de volumen de distribución (Vz), depuración (CL) y la vida media terminal (t½, z) en el ciclo 1 fueron 3.91 (3.36) L/m2, 1.88 (1.90) L/hora/m2 y 2.19 (1.53) horas, respectivamente.
Uso en poblaciones específicas:
Uso pediátrico: La seguridad y eficacia de BLINCYTO® ha sido establecida en pacientes pediátricos con LLA de precursores de células B en recaída o refractaria. El uso de BLINCYTO® es respaldado por un estudio de un solo brazo en pacientes pediátricos con LLA de precursores de células B en recaída o refractaria. Este estudio incluyó pacientes pediátricos en los siguientes grupos de edad: 10 infantes (1 mes hasta menos de 2 años), 40 niños (2 años hasta menos de 12 años), y 20 adolescentes (12 años a menos de 18 años). No se observaron diferencias en la eficacia entre los diferentes subgrupos por edad. La eficacia también se ha establecido con base en la extrapolación de estudios adecuados y bien controlados en adultos con LLA de precursores de células B con EMR-positiva.
En general, las reacciones adversas en pacientes pediátricos tratados con BLINCYTO® fueron similares en tipo a aquéllas observadas en pacientes adultos con LLA de precursores de células B recurrente o refractaria (ver Reacciones secundarias y adversas). Las reacciones adversas que fueron observadas con más frecuencia (≥ 10%) en la población pediátrica comparadas con la población adulta fueron pirexia (80% vs. 61%), hipertensión (26% vs. 8%), anemia (41% vs. 24%), reacción relacionada a la infusión (49% vs. 34%), trombocitopenia (34% vs. 21%), leucopenia (24% vs. 11%), e incremento de peso (17% vs. 6%).
En pacientes pediátricos menores de 2 años (infantes), la incidencia de toxicidades neurológicas no fue significativamente diferente para los otros grupos de edad, pero sus manifestaciones fueron diferentes; los únicos términos del evento reportados fueron agitación, cefalea, insomnio, somnolencia e irritabilidad. Los infantes también tuvieron un incremento en la incidencia de hipocalemia (50%) en comparación con otros cohortes de edad pediátrica (15% a 20%) o adultos (17%).
Las concentraciones de estado-estable de blinatumomab fueron comparables en pacientes adultos y pediátricos en niveles de dosis equivalentes en base a regímenes basados en BSA.
Toxicidad por alcohol bencílico en pacientes pediátricos: En neonatos prematuros y lactantes de la unidad de cuidados intensivos neonatales que recibieron medicamentos que contienen alcohol bencílico como conservador, se produjeron reacciones adversas graves, incluidas reacciones fatales y el "síndrome de jadeo". En estos casos, las dosificaciones de alcohol bencílico de 99 mg/kg/día a 234 mg/kg/día produjeron altos niveles de alcohol bencílico y de sus metabolitos en la sangre y la orina (los niveles sanguíneos de alcohol bencílico fueron de 0.61 mmol/L a 1.378 mmol/L). Las reacciones adversas adicionales incluyeron deterioro neurológico gradual, convulsiones, hemorragia intracraneal, anomalías hematológicas, rotura de la piel, insuficiencia hepática y renal, hipotensión, bradicardia y colapso cardiovascular. Los bebés prematuros con bajo peso al nacer pueden tener más probabilidades de desarrollar estas reacciones debido a que pueden tener menor capacidad de metabolizar el alcohol bencílico.
Cuando prescriba BLINCYTO® (con conservador) en pacientes pediátricos, considere la carga metabólica diaria combinada de alcohol bencílico de todas las fuentes, incluyendo BLINCYTO® (con conservador) (contiene 7.4 mg de alcohol bencílico por mL) y otros medicamentos que contengan alcohol bencílico. Se desconoce la cantidad mínima de alcohol bencílico en la que pueden producirse reacciones adversas graves (ver Precauciones generales).
Debido a la adición de bacteriostático salino, la bolsa para infusión de 7 días de solución de BLINCYTO® contiene alcohol bencílico y no es recomendada para su uso en pacientes que pesen menos de 22 kg. Prepare la solución de BLINCYTO® para infusión con solución salina libre de conservadores en bolsas para infusión de 24 horas a 48 horas para pacientes que pesan menos de 22 kg (ver Dosis y vía de administración).
Uso geriátrico: Del número total de pacientes con LLA tratados en estudios clínicos de BLINCYTO®, aproximadamente el 12% tenían 65 años y más mientras que el 2% tenían 75 años y más. No se observaron diferencias generales de seguridad o efectividad entre estos pacientes y pacientes más jóvenes y otra experiencia clínica reportada no ha identificado diferencias en las respuestas entre los pacientes de la tercera edad y pacientes jóvenes. Sin embargo, los pacientes de la tercera edad experimentaron una mayor tasa de infecciones graves y toxicidades neurológicas, incluyendo trastorno cognitivo, encefalopatía y confusión, (ver Precauciones generales).
Farmacodinamia: Durante la infusión intravenosa continua de 4 semanas, la respuesta farmacodinámica se caracterizó por la activación de los linfocitos T y la redistribución inicial, disminución de las células B periféricas y la elevación transitoria de las citocinas.
Después del inicio de la infusión conBLINCYTO® o el escalamiento de la dosis se observó redistribución de las células T periféricas (es decir, adición de células T al endotelio de los vasos sanguíneos y/o trasmigración al tejido). Inicialmente disminuyeron los recuentos de células T en los días 1 a 2 y posteriormente retornaron a los niveles basales dentro de los 7 a 14 días en la mayoría de los pacientes. En pocos pacientes se observó aumento de los recuentos de células T mayor a los valores basales (expansión de células T).
En la mayoría de los pacientes, los recuentos de células B periféricos disminuyeron a menos o igual a 10 células/microlitro durante el primer ciclo de tratamiento a una dosis ≥ 5 mcg/m2/día o ≥ 9 mcg/día. No se observó recuperación de los recuentos de las células B periféricas durante el periodo de 2 semanas sin BLINCYTO® entre los ciclos de tratamiento. La disminución incompleta de células B ocurrió a dosis de 0.5 mcg/m2/día y 1.5 mcg/m2/día y en algunos pacientes a dosis mayores.
Las citocinas incluidas IL-2, IL-4, IL-6, IL-8, IL-10, IL-12, TNF-α e IFN-? se midieron y se observó que IL-6, IL-10 e IFN-? estuvieron elevadas. Las elevaciones más altas de citocinas se observaron en los primeros 2 días después del inicio de la infusión con BLINCYTO®. Los niveles elevados de citocinas retornaron a los valores basales dentro de las 24 a 48 horas durante la infusión. En los ciclos de tratamiento subsiguientes, se observó elevación de las citocinas en menos pacientes con menor intensidad en comparación con las primeras 48 horas del primer ciclo de tratamiento.
Mecanismo de acción: Blinatumomab es un acoplador biespecífico de células T CD3, dirigido a CD19 y que se une al CD19, expresado sobre la superficie de células de linaje B con CD3 expresado en la superficie de las células T. Éste activa las células T endógenas mediante la conexión de CD3 en el complejo de receptores de células T (TCR, por sus siglas en inglés) con CD19 sobre las células B benignas y malignas. Blinatumomab actúa como mediador en la formación de una sinapsis entre la célula T y la célula tumoral, regulación positiva de las moléculas de adhesión celular, la producción de proteínas citolíticas, la liberación de citocinas inflamatorias y la proliferación de células T, que dan lugar a la lisis redirigida de las células CD19+.
Estudios clínicos:
LLA de precursores de células B recurrente o refractario:
Estudio tOWER: La eficacia de BLINCYTO® se comparó con la quimioterapia estándar (SOC, por sus siglas en inglés) en un estudio aleatorizado, de etiqueta abierta, multicéntrico (Estudio TOWER) [NCT02013167]. Los pacientes elegibles tenían ≥ 18 años de edad con LLA de precursores de células B recurrente o refractario [> 5% de blastos en la médula ósea y refractaria a la terapia de inducción primaria o refractaria a la última terapia, primera recaída no tratada con primera duración de remisión < 12 meses, segunda recaída o posterior no tratada, o recaída en cualquier momento después del trasplante alogénico de células madre hematopoyéticas [alloHSCT, por sus siglas en inglés]). BLINCYTO® se administró a 9 mcg/día en los Días 1 a 7 y 28 mcg/día en los días 8 a 28 para el Ciclo 1, y 28 mcg/día en los días 1 a 28 para los Ciclos 2 a 5 en ciclos de 42 días y para los Ciclos 6 a 9 en ciclos de 84-días. El ajuste de la dosis fue posible en caso de eventos adversos. La quimioterapia estándar SOC incluyó fludarabina, citarabina arabinósido y factor estimulante de colonias de granulocitos (FLAG); dosis altas de arabinósido de citarabina (HiDAC); combinación a base de dosis altas de metotrexato (HDMTX); o regímenes basados en clofarabina/clofarabina.
Hubo 405 pacientes aleatorizados 2:1 para recibir BLINCYTO® o quimioterapia estándar SOC seleccionada por el investigador. La aleatorización se estratificó por edad (< 35 años frente a ≥ 35 años de edad), terapia previa de rescate (sí vs. no) y previo alloHSCT (sí vs. no), según se evaluó en el momento del consentimiento. La demografía y las características basales estuvieron bien equilibradas entre los dos brazos (ver Tabla 1).
Tabla 1. Características basales y demográficas en el estudio TOWER
|
Características |
BLINCYTO® (n = 271) |
Quimioterapia estándar (SOC) (n = 134) |
|
Edad |
||
|
Mediana, años (mín, máx) |
37 (18, 80) |
37 (18, 78) |
|
< 35 años, n (%) |
124 (46) |
60 (45) |
|
≥ 35 años, n (%) |
147 (54) |
74 (55) |
|
≥ 65 años, n (%) |
33 (12) |
15 (11) |
|
≥ 75 años, n (%) |
10 (4) |
2 (2) |
|
Hombres, n (%) |
162 (60) |
77 (58) |
|
Raza, n (%) |
||
|
Indio Americano o nativo de Alaska |
4 (2) |
1 (1) |
|
Asiático |
19 (7) |
9 (7) |
|
Negro (o Afroamericano) |
5 (2) |
3 (2) |
|
Múltiple |
2 (1) |
0 |
|
Hawaiano nativo u otra isla del Pacífico |
1 (0) |
1 (1) |
|
Otro |
12 (4) |
8 (6) |
|
Blanco |
228 (84) |
112 (84) |
|
Terapia previa de rescate |
171 (63) |
70 (52) |
|
AlloHSCT1 previa |
94 (35) |
46 (34) |
|
Estado del grupo cooperativo del Este n (%) |
||
|
0 |
96 (35) |
52 (39) |
|
1 |
134 (49) |
61 (46) |
|
2 |
41 (15) |
20 (15) |
|
Desconocido |
0 |
1 (1) |
|
Refractario a tratamiento de rescate - n (%) |
||
|
Sí |
87 (32) |
34 (25) |
|
No |
182 (67) |
99 (74) |
|
Desconocido |
2 (1) |
1 (1) |
|
Máximo de blastos en médula ósea central/local - n (%) |
||
|
≤ 5% |
0 |
0 |
|
> 5 a < 10% |
9 (3) |
7 (5) |
|
10 a < 50% |
60 (22) |
23 (17) |
|
≥ 50% |
201 (74) |
104 (78) |
|
Desconocido |
1 (0) |
0 |
1 alloHSCT = trasplante alogénico de células madre hematopoyéticas.
De los 271 pacientes asignados al azar al brazo de BLINCYTO®, 267 pacientes recibieron tratamiento con BLINCYTO®. La mediana del número de ciclos de tratamiento fue dos (rango: 1 a 9 ciclos); 267 (99%) recibieron Ciclos 1 a 2 (inducción), 86 (32%) recibieron Ciclos 3 a 5 (consolidación) y 27 (10%) recibieron ciclos 6 a 9 (terapia continua). De los 134 pacientes en el brazo de SOC, 25 abandonaron previo al inicio del tratamiento del estudio, y 109 pacientes recibieron una mediana de 1 ciclo de tratamiento (rango: 1 a 4 ciclos).
La determinación de la eficacia se basó en la supervivencia global (SG). El estudio demostró una mejoría estadísticamente significativa en la SG para los pacientes tratados con BLINCYTO® en comparación con la quimioterapia estándar SOC.
Consulte la Figura 1 y la Tabla 2 a continuación para conocer los resultados de eficacia del Estudio TOWER.
Figura 1. Curva de Kaplan-Meier de supervivencia global en el estudio TOWER
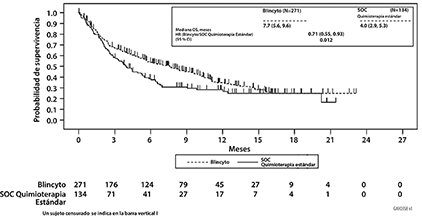
Tabla 2. Resultados de eficacia en pacientes ≥ 18 años de edad con LLA de precursores de células B cromosoma filadelfia negativo en recaída o refractaria (estudio TOWER)
|
BLINCYTO® (n = 271) |
Quimioterapia estándar SOC (n = 134) |
|
|
Supervivencia global |
||
|
Número de muertes (%) |
164 (61) |
87 (65) |
|
Mediana, meses [95% IC] |
7.7 [5.6, 9.6] |
4.0 [2.9, 5.3] |
|
Coeficiente de riesgo [95% IC]1 |
0.71 [0.55, 0.93] |
|
|
Valor-p2 |
0.012 |
|
|
Respuesta global |
||
|
CR4/CRh*5, n (%) [95% IC] |
115 (42) [37, 49] |
27 (20) [14, 28] |
|
Diferencia de tratamiento [95% IC] |
22 [13, 31] |
|
|
Valor-p3 |
< 0.001 |
|
|
CR, n (%) [95% IC] |
91 (34) [28, 40] |
21 (16) [10, 23] |
|
Diferencia de tratamiento [95% IC] |
18 [10, 26] |
|
|
Valor-p3 |
< 0.001 |
|
|
Respuesta6 EMR para CR/CRh* |
||
|
n1/n2 (%)7 [95% IC] |
73/115 (64) [54, 72] |
14/27 (52) [32, 71] |
1 Basado en el modelo estratificado de Cox.
2 El valor de p se obtuvo usando la prueba de rango logarítmico estratificado.
3 El valor de p se obtuvo usando la prueba de Cochran-Mantel-Haenszel.
4 La CR (remisión completa) se definió como ≤ 5% de blastos en la médula ósea, sin evidencia de enfermedad y recuperación completa de los conteos de sangre periférica (plaquetas > 100,000/microlitro y recuentos de neutrófilos absolutos [ANC] > 1,000/microlitro).
5 La CRh* (remisión completa con recuperación hematológica parcial) se definió como ≤ 5% de blastocitos en la médula ósea, sin evidencia de enfermedad y recuperación parcial de los conteos de sangre periférica (plaquetas > 50,000/microlitro y ANC > 500/microlitro).
6 La respuesta de EMR (enfermedad mínima residual) se definió como EMR mediante PCR o citometría de flujo < 1 x 104 (0.01%).
7 n1: número de pacientes que alcanzaron la respuesta de EMR y CR/CRh*; n2: número de pacientes que alcanzaron CR/CRh* y tuvieron una evaluación posterior a la línea de base.
Estudio MT103-211: El estudio MT103-211 [NCT01466179] fue un estudio de etiqueta abierta, multicéntrico, de un solo brazo. Los pacientes elegibles tenían ≥ 18 años de edad con LLA de precursores de células B cromosoma Filadelfia negativo recurrente o refractario (recaída con una duración de la primera remisión de ≤ 12 meses en el primer rescate o recurrente o refractario después de la primera terapia de rescate o recurrente dentro de los 12 meses de alloHSCT y tenían ≥ 10% de blastos en la médula ósea).
BLINCYTO® se administró como una infusión intravenosa continua. La dosis recomendada para este estudio se determinó que era de 9 mcg/día en los Días 1 a 7 y 28 mcg/día en los Días 8 a 28 para el Ciclo 1 y 28 mcg/día en los Días 1 a 28 para los ciclos subsecuentes. El ajuste de la dosis fue posible en caso de eventos adversos. La población tratada incluyó 185 pacientes que recibieron al menos 1 infusión de BLINCYTO®; la mediana del número de ciclos de tratamiento fue 2 (rango: 1 a 5). Los pacientes que respondieron a BLINCYTO®, pero después recayeron tuvieron la opción de ser nuevamente tratados con BLINCYTO®. Entre los pacientes tratados, la mediana de edad fue de 39 años (rango: 18 a 79 años), 63 de 185 (34.1%) se habían sometido a un HSCT antes de recibir blinatumomab, y 32 de 185 (17.3%) habían recibido más de 2 terapias de rescate previas.
La eficacia se basó en la tasa de remisión completa (CR), la duración de la CR y la proporción de pacientes con una EMR-negativa CR/CR con recuperación hematológica parcial (CR/CRh*) en 2 ciclos de tratamiento con BLINCYTO®. La Tabla 3 muestra los resultados de eficacia de este estudio. La tasa de HSCT entre aquellos que alcanzaron CR/CRh* fue del 39% (30 de 77).
Tabla 3. Resultados de eficacia en pacientes ≥ 18 años de edad con LLA de precursores de células B cromosoma filadelfia negativo en recaída o refractario (estudio MT103-211)
|
N = 185 |
|||
|
CR1 |
CRh*2 |
CR/CRh* |
|
|
n (%) [95% IC] |
60 (32.4) [25.7-39.7] |
17 (9.2) [5.4-14.3] |
77 (41.6) [34.4-49.1] |
|
Respuesta a EMR3 |
|||
|
n1/n2 (%)4 [95% IC] |
48/60 (80.0) [67.7-89.2] |
10/17 (58.8) [32.9-81.6] |
58/77 (75.3) [64.2-84.4] |
|
DOR/RFS5 |
|||
|
Mediana (meses) (rango) |
6.7 (0.46-16.5) |
5.0 (0.13-8.8) |
5.9 (0.13-16.5) |
1 La CR (remisión completa) se definió como ≤ 5% de blastos en la médula ósea, sin evidencia de enfermedad y recuperación completa de conteos sanguíneos periféricos (plaquetas > 100,000/microlitro y recuentos de neutrófilos absolutos [ANC] > 1,000/microlitro).
2 La CRh* (remisión completa con recuperación hematológica parcial) se definió como ≤ 5% de blastos en la médula ósea, sin evidencia de enfermedad y recuperación parcial de los conteos de sangre periférica (plaquetas > 50,000/microlitro y ANC > 500/microlitro).
3 La respuesta de EMR (enfermedad mínima residual) se definió como EMR mediante PCR < 1 x 10-4 (0.01%).
4 n1: número de pacientes que alcanzaron la respuesta EMR y el estado de remisión respectivo; n2: número de pacientes que alcanzaron el estado de remisión respectivo. Seis CR/CRh* respondedores con datos faltantes de EMR se consideraron como EMR-no respondedores.
5 La DOR (duración de la respuesta)/RFS (supervivencia libre de recaída) se definió como el tiempo desde la primera respuesta de CR o CRh* a recaída o muerte, lo que ocurra primero. La recaída se definió como una recaída hematológica (blastocitos en la médula ósea mayor al 5% después de CR) o una recaída extramedular.
Estudio ALCANTARA: La eficacia de BLINCYTO® para el tratamiento de la LLA de precursores de células B cromosoma Filadelfia positivo, se evaluó en un estudio abierto, multicéntrico, de brazo único (estudio ALCANTARA) [NCT02000427]. Los pacientes elegibles tenían ≥ 18 años de edad con LLA de precursores de células B cromosoma Filadelfia positivo, recurrentes o refractarios a al menos 1 inhibidor de tirosina cinasa (TKI) de segunda generación o posterior, o intolerantes a TKI de segunda generación e intolerantes o refractarios al mesilato de imatinib.
BLINCYTO® se administró a 9 mcg/día en los Días 1 a 7 y 28 mcg/día en los Días 8 a 28 para el Ciclo 1 y 28 mcg/día en los Días 1 a 28 para los ciclos subsecuentes. El ajuste de la dosis fue posible en caso de eventos adversos.
La población tratada incluyó 45 pacientes que recibieron al menos una infusión de BLINCYTO®; la mediana del número de ciclos de tratamiento fue 2 (rango: 1 a 5). Los datos demográficos y las características iniciales se muestran en la Tabla 4.
Tabla 4. Características y datos demográficos basales en el estudio ALCANTARA
|
Característica |
BLINCYTO® (n = 45) |
|
Edad |
|
|
Mediana, años (mín, máx) |
55 (23, 78) |
|
≥ 65 años y < 75 años, n (%) |
10 (22) |
|
≥ 75 años, n (%) |
2 (4) |
|
Hombres, n (%) |
24 (53) |
|
Raza, n (%) |
|
|
Asiático |
1 (2) |
|
Negro (o Afroamericano) |
3 (7) |
|
Otro |
2 (4) |
|
Blanco |
39 (87) |
|
Historia de la enfermedad |
|
|
Tratamiento previo con TKI1, n (%) |
|
|
1 |
7 (16) |
|
2 |
21 (47) |
|
≥ 3 |
17 (38) |
|
Terapia previa de rescate |
31 (62) |
|
alloHSCT2 previo |
20 (44) |
|
Blastocitos de médula ósea3 |
|
|
≥ 50% a < 75% |
6 (13) |
|
≥ 75% |
28 (62) |
1 Número de pacientes que fracasaron a ponatinib = 23 (51%).
2 alloHSCT = trasplante alogénico de células madre hematopoyéticas.
3 Evaluados centralmente.
La eficacia se basó en la tasa de remisión completa (CR), la duración de la CR y la proporción de pacientes con CR/CR con EMR-negativa y recuperación hematológica parcial (CR/CRh*) en 2 ciclos de tratamiento con BLINCYTO®. La Tabla 5 muestra los resultados de eficacia del estudio ALCANTARA. Cinco de los 16 pacientes que respondieron (31%) se sometieron a HSCT alogénico en CR/CRh* inducido con BLINCYTO®. Hubo 10 pacientes con mutación documentada de T3151; cuatro lograron CR en 2 ciclos de tratamiento con BLINCYTO®.
Tabla 5. Resultados de eficacia en pacientes ≥ 18 años de edad con LLA de precursores de células B cromosoma filadelfia positivo en recaída o refractario (estudio ALCANTARA)
|
N = 45 |
|||
|
CR1 |
CRh*2 |
CR/CRh* |
|
|
n (%) [95% IC] |
14 (31) [18 a 47] |
2 (4) [1 a 15] |
16 (36) [22 a 51] |
|
Respuesta a EMR3 |
|||
|
n1/n2 (%)4 [95% IC] |
12/14 (86) [57 a 98] |
2/2 (100) [16, 100] |
14/16 (88) [62 a 98] |
|
DOR/RFS5 |
|||
|
Mediana (meses) (rango) |
6.7 (3.6 a 12.0) |
NE6 (3.7 a 9.0) |
6.7 (3.6 a 12.0) |
1 La CR (remisión completa) se definió como ≤ 5% de blastos en la médula ósea, sin evidencia de enfermedad y recuperación completa de conteos sanguíneos periféricos (plaquetas > 100,000/microlitro y recuentos de neutrófilos absolutos [ANC] > 1,000/microlitro.
2 CRh* (remisión completa con recuperación hematológica parcial) se definió como ≤ 5% de blastos en la médula ósea, sin evidencia de enfermedad y recuperación parcial de los conteos de sangre periférica (plaquetas > 50,000/microlitro y ANC > 500/microlitro).
3 La respuesta de EMR (enfermedad mínima residual) se definió como EMR mediante PCR <1x10-4 (0.01%).
4 n1: número de pacientes que alcanzaron la respuesta EMR y el estado de remisión respectivo; n2: número de pacientes que alcanzaron el estado de remisión respectivo. Seis CR/CRh* respondedores con datos faltantes de EMR se consideraron como EMR-no respondedores.
5 La DOR (duración de la respuesta)/RFS (supervivencia libre de recaída) se definió como el tiempo desde la primera respuesta de CR o CRh* a recaída o muerte, lo que ocurra primero. La recaída se definió como una recaída hematológica (blastocitos en la médula ósea mayor al 5% después de CR) o una recaída extramedular.
6 NE = no estimable.
Estudio MT103-205: El estudio MT103-205 [NCT01471782] fue un estudio abierto, multicéntrico, de un solo brazo en pacientes pediátricos con LLA de precursores de células B recurrente o refractaria (segunda recaída o posterior de médula ósea, cualquier recaída de médula después de HSCT alogénico o refractaria a otros tratamientos, y > 25% de blastos en la médula ósea). BLINCYTO® se administró a 5 mcg/m2/día en los Días 1 a 7 y 15 mcg/m2/día en los Días 8 a 28 para el Ciclo 1, y 15 mcg/m2/día en los Días 1 a 28 para los ciclos subsecuentes. El ajuste de la dosis fue posible en caso de eventos adversos. Los pacientes que respondieron a blinatumomab pero que después recayeron tuvieron la opción de ser retratados con blinatumomab.
Entre los 70 pacientes tratados, la mediana de edad fue de 8 años (rango: 7 meses a 17 años), 40 de 70 (57.1%) habían sido sometidos a HSCT alogénico antes de recibir BLINCYTO®, y 39 de 70 (55.7%) tenían enfermedad refractaria. La mediana del número de ciclos de tratamiento fue 1 (rango: 1 a 5).
Veintitrés de 70 (32.9%) pacientes alcanzaron CR/CRh* dentro de los primeros 2 ciclos de tratamiento con 17 de 23 (73.9%) ocurriendo dentro del Ciclo 1 de tratamiento. Consulte la Tabla 6 para conocer los resultados de eficacia del estudio. La tasa de HSCT entre los que alcanzaron CR/CRh* fue del 48% (11 de 23).
Tabla 6. Resultados de eficacia en pacientes < 18 años de edad con LLA de precursores de células B en recaída o refractario (estudio MT103 205)
|
n = 70 |
|||
|
CR1 |
CRh*2 |
CR/CRh* |
|
|
n (%) [95% IC] |
12 (17.1) [9.2-28.0] |
11 (15.7) [8.1-26.4] |
23 (32.9) [22.1-45.1] |
|
Respuesta a EMR3 |
|||
|
n1/n2 (%)4 [95% IC] |
6/12 (50.0) [21.1-78.9] |
4/11 (36.4) [10.9-69.2] |
10/23 (43.5) [23.2-65.5] |
|
DOR/RFS5 |
|||
|
Mediana (meses) (rango) |
6.0 (0.5-12.1) |
3.5 (0.5-16.4) |
6.0 (0.5-16.4) |
1 La CR (remisión completa) se definió como ≤ 5% de blastos en la médula ósea, sin evidencia de blastos circulantes o enfermedad medular extra, y recuperación completa de conteos sanguíneos periféricos (plaquetas > 100,000/microlitro y recuentos de neutrófilos absolutos [ANC] > 1,000/microlitro).
2 La CRh* (remisión completa con recuperación hematológica parcial) se definió como ≤ 5% de blastos en la médula ósea, sin evidencia de blastos circulantes o enfermedad medular extra, y recuperación parcial de conteos sanguíneos periféricos (plaquetas >50,000/microlitro y ANC > 500/microlitro).
3 La respuesta de EMR (enfermedad mínima residual) se definió como EMR mediante PCR o citometría de flujo < 1 x 10-4 (0.01%).
4 n1: número de pacientes que alcanzaron la respuesta de EMR y el estado de remisión respectivo; n2: número de pacientes que alcanzaron el estado de remisión respectivo. Un respondedor CR/CRh* con datos EMR faltantes fue considerado como un EMR-no respondedor.
5 La DOR (duración de la respuesta)/RFS (supervivencia libre de recaída) se definió como el tiempo transcurrido desde la primera respuesta de CR o CRh* a recaída o muerte, lo que ocurra primero. La recaída se definió como una recaída hematológica (blastocitos en la médula ósea mayor al 5% después de la CR) o una recaída extramedular.
LLA de Precursores de células B, EMR-positiva:
Estudio BLAST: La eficacia de blinatumomab se evaluó en un estudio de etiqueta abierta, multicéntrico, de un solo brazo (Estudio BLAST) [NCT01207388] que incluyó pacientes que tenían ≥ 18 años de edad, que habían recibido al menos 3 bloques de quimioterapia estándar de LLA, se encontraban en remisión completa hematológica (definida como < 5% de blastos en la médula ósea, recuento absoluto de neutrófilos > 1 Gi/L, plaquetas > 100 Gi/L) y tenía una EMR en un nivel de ≥ 0.1% usando un ensayo con una sensibilidad mínima de 0.01%. BLINCYTO® se administró a una dosis constante de 15 mcg/m2/día (equivalente a la dosis recomendada de 28 mcg/día) por vía intravenosa para todos los ciclos de tratamiento. Los pacientes recibieron hasta 4 ciclos de tratamiento. El ajuste de la dosis fue posible en caso de eventos adversos.
La población tratada incluyó 86 pacientes en la primera o segunda remisión completa hematológica (CR1 o CR2). Las características demográficas y basales se muestran en la Tabla 7. La mediana del número de ciclos de tratamiento fue 2 (rango: 1 a 4). Después del tratamiento con BLINCYTO®, 45 de 61 (73.8%) pacientes en CR1 y 14 de 25 (56.0%) pacientes en CR2 se sometieron a trasplante de células madre hematopoyéticas alogénicas en remisión completa hematológica continua.
Tabla 7. Características basales y demográficas en el estudio BLAST
|
Características |
Blinatumomab (n = 86) |
|
Edad |
|
|
Mediana, años (mín, máx) |
43 (18,76) |
|
≥ 65 años, n (%) |
10 (12) |
|
Hombres, n (%) |
50 (58) |
|
Raza, n (%) |
|
|
Asiáticos |
1 (1) |
|
Otro (mixto) |
0 (0) |
|
Blanco |
76 (88) |
|
Desconocido |
9 (11) |
|
Estado de la enfermedad del cromosoma Filadelfia, n (%) |
|
|
Positivo |
1 (1) |
|
Negativo |
85 (99) |
|
Historial de recaída, n (%) |
|
|
Pacientes en 1o CR |
61 (71) |
|
Pacientes en 2o CR |
25 (29) |
|
Nivel de EMR al inicio del estudio*, n (%) |
|
|
≥ 10% |
7 (8) |
|
≥ 1% y < 10% |
34 (40) |
|
≥ 0.1% y < 1% |
45 (52) |
* Evaluado centralmente usando un ensayo con sensibilidad mínima de 0.01%.
La eficacia se basó en el logro de una EMR indetectable dentro de un ciclo de tratamiento con BLINCYTO®. y supervivencia libre de recaída hematológica (RFS). El ensayo utilizado para evaluar la respuesta de EMR tuvo una sensibilidad de 0.01% para 6 pacientes y < 0.005% para 80 pacientes. En general, 70 pacientes lograron una EMR indetectable (81.4%: IC 95%: 71.6%, 89.0%). La mediana de RFS hematológico fue de 22.3 meses. La Tabla 8 muestra la respuesta de EMR y RFS hematológica por número de remisión.
Tabla 8. Resultados de eficacia en pacientes ≥ 18 años de edad con LLA de precursores de células B y EMR-positiva (Estudio BLAST)
|
Pacientes en CR1 (n = 61) |
Pacientes en CR2 (n = 25) |
|
|
Respuesta completa de EMR1, n (%), [95% IC] |
52 (85.2) [73.8, 93.0] |
18 (72.0) [50.6, 87.9] |
|
Mediana de supervivencia libre de recaída hematológica2 en meses (rango) |
35.2 (0.4, 53.5) |
12.3 (0.7, 42.3) |
1 La respuesta completa de EMR se definió como la ausencia de EMR detectable confirmada en un ensayo con sensibilidad mínima de 0.01%.
2 La recaída se definió como recaída hematológica o extramedular, leucemia secundaria o muerte por cualquier causa; incluye tiempo después del trasplante; estimación de Kaplan-Meier.
La EMR indetectable se logró en 65 de 80 pacientes (81.3%: IC 95%: 71.0%, 89.1%) con una sensibilidad de ensayo de al menos 0.005%. La mediana estimada de RFS hematológica entre los 80 pacientes que utilizaron el ensayo de sensibilidad más alta fue de 24.2 meses (IC 95%: 17.9, NE).
CONTRAINDICACIONES:
Hipersensibilidad: BLINCYTO® está contraindicado en pacientes con hipersensibilidad conocida al blinatumomab u otro componente de la formulación del producto.
Embarazo.
Lactancia.
RESTRICCIONES DE USO DURANTE EL EMBARAZO Y LA LACTANCIA:
Embarazo:
Resumen de riesgo: Con base a su mecanismo de acción, BLINCYTO® puede causar daño fetal incluyendo linfocitopenia de células B cuando se administra a mujeres embarazadas. No existen datos sobre el uso de BLINCYTO® en mujeres embarazadas. En estudios de reproducción animal, una molécula murina equivalente administrada a ratonas preñadas cruzó la barrera placentaria (ver Datos). Aconsejar a mujeres embarazadas del riesgo potencial para el feto.
Se desconoce la tasa de antecedentes de las principales anomalías congénitas y abortos espontáneos para la población indicada. Todos los embarazos tienen un riesgo asociado de defectos de nacimiento, interrupción del embarazo u otros resultados adversos.
Consideraciones clínicas:
Reacciones adversas fetales/neonatos: Los linfocitos B deben ser monitorizados en lactantes antes del inicio de la vacunación con virus vivos debido al potencial de reducción de linfocitos B después de la exposición a BLINCYTO® in utero (ver Precauciones generales).
Datos:
Datos en animales: No se han conducido estudios de reproducción en animales con blinatumomab. En estudios de toxicidad de desarrollo embrio-fetal, se administró intravenosamente una molécula murina equivalente a ratonas preñadas durante el periodo de organogénesis. La molécula equivalente cruzó la barrera placentaria y no causó teratogenicidad o toxicidad embrio-fetal. Se observaron las disminuciones esperadas de células B y células T en las ratonas preñadas, pero los efectos hematológicos no fueron evaluados en los fetos.
Lactancia:
Resumen de riesgo: No se cuenta con información relacionada a la presencia de blinatumomab en la leche materna, los efectos en el lactante, o efectos en la producción de leche. Dado que muchos fármacos son excretados en leche humana y debido al potencial de reacciones adversas serias de BLINCYTO® en lactantes, incluyendo linfocitopenia de células B, aconsejar a los pacientes sobre no amamantar durante el tratamiento con BLINCYTO® y por 48 horas después de la última dosis.
Mujeres y varones con potencial reproductivo: BLINCYTO® puede causar daño fetal cuando se administra a una mujer embarazada (ver Uso en poblaciones específicas).
Prueba de embarazo: Antes de iniciar el tratamiento con BLINCYTO®, verificar el estatus de embarazo en mujeres con potencial reproductivo.
Anticoncepción:
Mujeres: Aconsejar a las mujeres con potencial reproductivo del uso de anticonceptivos efectivos durante el tratamiento BLINCYTO® y 48 horas después de la última dosis.
REACCIONES SECUNDARIAS Y ADVERSAS:
Las siguientes reacciones adversas clínicamente significativas se describen en la sección de Precauciones generales con la etiqueta:
• Síndrome de liberación de citocinas.
• Toxicidades neurológicas.
• Infecciones.
• Síndrome de lisis tumoral.
• Neutropenia y neutropenia febril.
• Efectos sobre la capacidad para conducir y operar máquinas.
• Elevación de las enzimas hepáticas.
• Pancreatitis.
• Leucoencefalopatía.
Experiencia en estudios clínicos: Debido a que los estudios clínicos se llevan a cabo bajo condiciones que varían ampliamente, las tasas de reacciones adversas observadas en los ensayos clínicos de un fármaco no pueden compararse directamente con las tasas en los estudios clínicos de otro fármaco, y pueden no reflejar las tasas observadas en la práctica.
LLA de Precursores de células B, EMR-positivo: La seguridad de BLINCYTO® en pacientes con LLA de precursores de células B con EMR-positiva se evaluó en dos estudios clínicos de un solo brazo en los que 137 pacientes fueron tratados con BLINCYTO®. La mediana de edad de la población de estudio fue de 45 años (rango: 18 a 77 años).
Las reacciones adversas más comunes (≥20%) fueron pirexia, reacciones relacionadas con la infusión, dolor de cabeza, infecciones (patógenos no especificados), temblor y escalofríos. Se informaron reacciones adversas graves en el 61% de los pacientes. Las reacciones adversas graves más comunes (≥2%) incluyeron pirexia, temblor, encefalopatía, afasia, linfopenia, neutropenia, sobredosis, infección relacionada con el dispositivo, convulsiones e infección por estafilococos. Reacciones adversas grado 3 o superiores se reportaron en 64% de los pacientes. La descontinuación de la terapia debido a reacciones adversas ocurrió en el 17% de los pacientes; los eventos neurológicos fueron las razones reportadas con mayor frecuencia para la interrupción. Hubo 2 reacciones adversas fatales que ocurrieron dentro de los 30 días posteriores al final del tratamiento con BLINCYTO® (neumonía atípica y hemorragia subdural).
La Tabla 9 resume las reacciones adversas que ocurren con una incidencia ≥ 10% para cualquier grado o ≥ 5% de incidencia para el grado 3 o superior.
Tabla 9. Reacciones adversas que ocurren con una incidencia ≥ 10% para cualquier grado o ≥ 5% de incidencia para el grado 3 o superior en pacientes tratados con BLINCYTO® con LLA de precursores de células B, EMR-positiva
|
Reacción adversa |
BLINCYTO® (n = 137) |
|
|
Cualquier grado* n (%) |
Grado ≥ 3* n (%) |
|
|
Trastornos de la sangre y del sistema linfático |
||
|
Neutropenia1 |
21 (15) |
21 (15) |
|
Leucopenia2 |
19 (14) |
13 (9) |
|
Trombocitopenia3 |
14 (10) |
8 (6) |
|
Trastornos cardiacos |
||
|
Arritmia4 |
17 (12) |
3 (2) |
|
Trastornos generales y condiciones del sitio de administración |
||
|
Pirexia5 |
125 (91) |
9 (7) |
|
Escalofrío |
39 (28) |
0 (0) |
|
Infecciones e infestaciones |
||
|
Infecciones-patógeno no especificado |
53 (39) |
11 (8) |
|
Lesión, envenenamiento y complicaciones del procedimiento |
||
|
Reacción relacionada a la infusión6 |
105 (77) |
7 (5) |
|
Investigaciones |
||
|
Disminución de inmunoglobulinas7 |
25 (18) |
7 (5) |
|
Peso aumentado |
14 (10) |
1 (< 1) |
|
Hipertransaminasemia8 |
13 (9) |
9 (7) |
|
Trastornos musculoesqueléticos y del tejido conectivo |
||
|
Dolor de espalda |
16 (12) |
1 (< 1) |
|
Trastornos del sistema nervioso |
||
|
Dolor de cabeza |
54 (39) |
5 (4) |
|
Temblor9 |
43 (31) |
6 (4) |
|
Afasia |
16 (12) |
1 (< 1) |
|
Mareo |
14 (10) |
1 (< 1) |
|
Encefalopatía10 |
14 (10) |
6 (4) |
|
Trastornos psiquiátricos |
||
|
Insomnio11 |
24 (18) |
1 (< 1) |
|
Trastornos respiratorios, torácicos y mediastínicos |
||
|
Tos |
18 (13) |
0 (0) |
|
Trastornos de la piel y tejido subcutáneo |
||
|
Erupción12 |
22 (16) |
1 (< 1) |
|
Trastornos vasculares |
||
|
Hipotensión |
19 (14) |
1 (< 1) |
* Calificación basada en los criterios de la terminología común del NCI para eventos adversos (CTCAE) versión 4.0.
1 La neutropenia incluye neutropenia febril, neutropenia y recuento de neutrófilos disminuido.
2 La leucopenia incluye leucopenia y disminución del conteo de glóbulos blancos.
3 La trombocitopenia incluye disminución del recuento de plaquetas y trombocitopenia.
4 La arritmia incluye bradicardia, arritmia sinusal, bradicardia sinusal, taquicardia sinusal, taquicardia y extrasístoles ventriculares.
5 La pirexia incluye aumento de la temperatura corporal y pirexia.
6 La reacción relacionada con la infusión es un término compuesto que incluye el término reacción relacionada con la infusión y los siguientes eventos que ocurren en las primeras 48 horas de infusión y el evento duró ≤2 días: síndrome de liberación de citocinas, edema ocular, hipertensión, hipotensión, mialgia, edema periorbitario, prurito generalizado, pirexia y erupción.
7 Disminución de inmunoglobulinas, incluye disminución de inmunoglobulina A sanguínea, disminución de inmunoglobulina G sanguínea, disminución de inmunoglobulina M sanguínea, hipogammaglobulinemia, hipoglobulinemia e inmunoglobulinas disminuidas.
8 La hipertransaminasemia incluye aumento de alanina-aminotransferasa, aumento de aspartato aminotransferasa aumento de enzima hepática.
9 El temblor incluye temblor esencial, temblor intencional y temblor.
10 La encefalopatía incluye trastorno cognitivo, nivel de conciencia deprimido, alteración de la atención, encefalopatía, letargia, leucoencefalopatía, trastorno de la memoria, somnolencia y encefalopatía tóxica.
11 El insomnio incluye insomnio inicial, insomnio e insomnio terminal.
12 Erupción incluye dermatitis por contacto, eccema, eritema, erupción y erupción maculopapular.
Las reacciones adversas adicionales en pacientes con LLA con EMR-positiva que no cumplieron los criterios de umbral para la inclusión en la Tabla 9 fueron:
Trastornos de la sangre y del sistema linfático: Anemia.
Trastornos generales y condiciones del sitio de administración: Edema periférico, dolor y dolor en el pecho (incluye dolor en el pecho y dolor en el pecho musculoesquelético).
Trastornos hepatobiliares: Aumento de la bilirrubina en la sangre.
Trastornos del sistema inmunitario: Hipersensibilidad y síndrome de liberación de citocinas.
Infecciones e infestaciones: Trastornos infecciosos virales, trastornos infecciosos bacterianos y trastornos de infecciones fúngicas.
Lesión, envenenamiento y complicaciones del procedimiento: Error de medicación y sobredosis (incluye sobredosis y sobredosis accidental).
Investigaciones: Aumento de la fosfatasa alcalina en la sangre.
Trastornos musculoesqueléticos y del tejido conectivo: Dolor en las extremidades y dolor óseo.
Trastornos del sistema nervioso: Convulsiones (incluye convulsiones y crisis tónico-clónicas generalizadas), trastornos del habla e hipoestesia.
Trastornos psiquiátricos: Estado confusional, desorientación y depresión.
Trastornos respiratorios, torácicos y mediastínicos: Disnea y tos productiva.
Trastornos vasculares: Hipertensión (incluye aumento de la presión arterial e hipertensión), rubor (incluye rubor y sofocos) y síndrome de fuga capilar.
LLA de precursores de células B cromosoma Filadelfia-negativo en recaída o refractaria: La seguridad de BLINCYTO® se evaluó en un estudio clínico aleatorizado, de etiqueta abierta, controlado con activo (estudio TOWER) en el que 376 pacientes con LLA de precursores de células B en recaída o refractaria cromosoma Filadelfia negativo fueron tratados con BLINCYTO® (n = 267) o quimioterapia estándar (SOC) (n = 109). La mediana de edad de los pacientes tratados con BLINCYTO® fue de 37 años (rango: 18 a 80 años), 60% hombres, 84% blancos, 7% asiáticos, 2% negros o afroamericanos, 2% indios americanos o nativos de Alaska, y 5% múltiples/otros.
Las reacciones adversas más comunes (≥ 20%) en el brazo de BLINCYTO® fueron infecciones (bacterianas y patógenas no especificadas), pirexia, dolor de cabeza, reacciones relacionadas con la infusión, anemia, neutropenia febril, trombocitopenia y neutropenia. Se informaron reacciones adversas graves en el 62% de los pacientes. Las reacciones adversas graves más comunes (≥ 2%) incluyeron neutropenia febril, pirexia, sepsis, neumonía, sobredosis, shock séptico, CRS, sepsis bacteriana, infección relacionada con el dispositivo y bacteriemia. Las reacciones adversas de grado 3 o superiores se reportaron en el 87% de los pacientes. La descontinuación del tratamiento debido a reacciones adversas ocurrió en el 12% de los pacientes tratados con BLINCYTO®; los eventos neurológicos y las infecciones fueron las razones informadas con mayor frecuencia para la interrupción del tratamiento debido a una reacción adversa. Los eventos adversos fatales ocurrieron en el 16% de los pacientes. La mayoría de los eventos fatales fueron infecciones.
Las reacciones adversas que ocurren con una incidencia ≥ 10% para cualquier grado o ≥ 5% de incidencia para el grado 3 o superior en los pacientes tratados con BLINCYTO® en el primer ciclo de terapia se resumen en la Tabla 10.
Tabla 10. Reacciones adversas que ocurren con una incidencia ≥ 10% para cualquier grado o ≥ 5% de incidencia para el grado 3 o superior en pacientes tratados con BLINCYTO® en el primer ciclo de terapia
|
Reacción adversa |
BLINCYTO® (n = 267) |
Quimioterapia estándar (SOC) (n = 109) |
||
|
Cualquier grado* n (%) |
Grado ≥ 3* n (%) |
Cualquier grado* n (%) |
Grado ≥ 3* n (%) |
|
|
Trastornos de la sangre y sistema linfático |
||||
|
Neutropenia1 |
84 (31) |
76 (28) |
67 (61) |
61 (56) |
|
Anemia2 |
68 (25) |
52 (19) |
45 (41) |
37 (34) |
|
Trombocitopenia3 |
57 (21) |
47 (18) |
42 (39) |
40 (37) |
|
Leucopenia4 |
21 (8) |
18 (7) |
9 (8) |
9 (8) |
|
Trastornos cardiacos |
||||
|
Arritmia5 |
37 (14) |
5 (2) |
18 (17) |
0 (0) |
|
Trastornos generales y condiciones del sitio de administración |
||||
|
Pirexia |
147 (55) |
15 (6) |
43 (39) |
4 (4) |
|
Edema6 |
48 (18) |
3 (1) |
20 (18) |
1 (1) |
|
Trastornos del sistema inmune |
||||
|
Síndrome de liberación de citocina7 |
37 (14) |
8 (3) |
0 (0) |
0 (0) |
|
Infecciones e infestaciones |
||||
|
Infecciones-patógeno no especificado |
74 (28) |
40 (15) |
50 (46) |
35 (32) |
|
Trastornos infecciosos bacterianos |
38 (14) |
19 (7) |
35 (32) |
21 (19) |
|
Trastornos infecciosos virales |
30 (11) |
4 (1) |
14 (13) |
0 (0) |
|
Trastornos infecciosos fúngicos |
27 (10) |
13 (5) |
15 (14) |
9 (8) |
|
Lesiones, envenenamiento y complicaciones de procedimiento |
||||
|
Reacción relacionada a la infusión8 |
79 (30) |
9 (3) |
9 (8) |
1 (1) |
|
Investigaciones |
||||
|
Hipertransaminasemia9 |
40 (15) |
22 (8) |
13 (12) |
7 (6) |
|
Trastorno del sistema nervioso |
||||
|
Dolor de cabeza |
61 (23) |
1 (< 1) |
30 (28) |
3 (3) |
|
Trastornos de la piel y del tejido subcutáneo |
||||
|
Erupción10 |
31 (12) |
2 (1) |
21 (19) |
0 (0) |
* Calificación basada en la terminología común del NCI para eventos adversos (CTCAE) versión 4.0.
1 La neutropenia incluye agranulocitosis, neutropenia febril, neutropenia y recuento de neutrófilos.
2 La anemia incluye anemia y disminución de la hemoglobina.
3 La trombocitopenia incluye disminución del recuento de plaquetas y trombocitopenia.
4 La leucopenia incluye leucopenia y disminución del conteo de glóbulos blancos.
5 La arritmia incluye arritmia, fibrilación auricular, aleteo auricular, bradicardia, bradicardia sinusal, taquicardia sinusal, taquicardia supraventricular y taquicardia.
6 El edema incluye edema facial, retención de líquidos, edema, edema periférico, inflamación periférica e hinchazón de la cara.
7 El síndrome de liberación de citocinas incluye el síndrome de liberación de citocinas y la tormenta de citocinas.
8 La reacción relacionada con la infusión es un término compuesto que incluye el término reacción relacionada con la infusión y los siguientes eventos que ocurren con las primeras 48 horas de infusión y el evento duró ≤ 2 días: pirexia, síndrome de liberación de citoquina, hipotensión, mialgia, lesión renal aguda, hipertensión y erupción eritematosa.
9 La hipertransaminasemia incluye aumento de alanina aminotransferasa, aumento de aspartato aminotransferasa, aumento de enzima hepática y aumento de transaminasas.
10 Erupción incluye eritema, erupción cutánea, erupción eritematosa, erupción generalizada, erupción macular, erupción maculo papular, erupción cutánea pruriginosa, exfoliación de la piel y erupción cutánea tóxica.
Las anomalías de laboratorio seleccionadas que empeoraron desde la basal grado 0-2 hasta el grado 3-4 máximo relacionado con el tratamiento en el primer ciclo de terapia se muestran en la Tabla 11.
Tabla 11. Empeoramiento de anormalidades de laboratorio seleccionadas a partir del grado 0-2 en la línea basal hasta el grado 3-4* máximo relacionado con el tratamiento en el primer ciclo de terapia
|
BLINCYTO® Grado 3 o 4 (%) |
Quimioterapia estándar SOC Grado 3 o 4 (%) |
|
|
Hematología |
||
|
Disminución del recuento de linfocitos |
80 |
83 |
|
Disminución del recuento de glóbulos blancos |
53 |
97 |
|
Disminución de hemoglobina |
29 |
43 |
|
Disminución del recuento de neutrófilos |
57 |
68 |
|
Disminución del recuento de plaquetas |
47 |
85 |
|
Química |
||
|
Incremento de ALT |
11 |
11 |
|
Incremento de bilirrubina |
5 |
4 |
|
Incremento de AST |
8 |
4 |
* Incluye sólo pacientes que tuvieron una medición de laboratorio basal y al menos una medición de laboratorio disponible durante el primer ciclo de terapia.
LLA de Precursores de células B en recaída o refractaria: Otras reacciones adversas importantes de los estudios de LLA de precursor de células B en recaída o refractaria agrupadas fueron:
Trastornos de la sangre y del sistema linfático: Linfadenopatía, histiocitosis hematofágica y leucocitosis (incluye leucocitosis y recuento de glóbulos blancos aumentado).
Trastornos generales y condiciones del sitio de administración: Escalofríos, dolor de pecho (incluye molestias en el pecho, dolor en el pecho, dolor de pecho musculoesquelético y dolor de pecho no cardiaco), dolor, aumento de la temperatura corporal, hipertermia y síndrome de respuesta inflamatoria sistémica.
Trastornos hepatobiliares: Hiperbilirrubinemia (incluye aumento de la bilirrubina en sangre e hiperbilirrubinemia).
Trastornos del sistema inmune: Hipersensibilidad (incluye hipersensibilidad, reacción anafiláctica, angioedema, dermatitis alérgica, erupción medicamentosa, hipersensibilidad a medicamentos, eritema multiforme y urticaria).
Lesiones, envenenamiento y complicaciones de procedimiento: Error de medicación y sobredosis (incluye sobredosis, error de medicación y sobredosis accidental).
Investigaciones: Aumento de peso, disminución de inmunoglobulinas (incluye disminución de inmunoglobulinas, reducción de inmunoglobulina A en sangre, disminución de inmunoglobulina G en sangre, disminución de inmunoglobulina M en sangre e hipogammaglobulinemia), aumento de fosfatasa alcalina en sangre e hipertransaminasemia.
Trastornos del metabolismo y la nutrición: Síndrome de lisis tumoral.
Trastornos musculoesqueléticos y del tejido conectivo: Dolor de espalda, dolor de huesos y dolor en las extremidades.
Trastornos del sistema nervioso: Temblor (temblor de reposo, temblor intencional, temblor esencial y temblor), estado de conciencia alterado (incluye estado de conciencia alterado, nivel de conciencia disminuido, alteración de la atención, letargia, cambios en el estado mental, estupor y somnolencia), mareo, deterioro de la memoria, convulsión (incluye convulsión y crisis atónicas), afasia, trastorno cognitivo, trastorno del habla, hipostesia, encefalopatía, parestesia y trastornos de los pares craneales (neuralgia del trigémino, trastorno del nervio trigémino, parálisis del sexto par, trastorno del par craneal, trastorno del nervio facial y paresis facial).
Trastornos psiquiátricos: Insomnio, desorientación, estado de confusión y depresión (incluye estado de ánimo deprimido, depresión, ideación suicida y suicidio consumado).
Trastornos respiratorios, torácicos y mediastínicos: Disnea (incluye insuficiencia respiratoria aguda, disnea, disnea de esfuerzo, insuficiencia respiratoria, dificultad respiratoria, broncospasmo, hiperreactividad bronquial, taquipnea y sibilancias), tos y tos productiva.
Trastornos vasculares: Hipotensión (incluye presión arterial disminuida, hipotensión, shock hipovolémico y colapso circulatorio), hipertensión (incluye aumento de la presión arterial, hipertensión y crisis hipertensiva), rubor (incluye rubor y sofocos) y síndrome de fuga capilar.
Inmunogenicidad: Como ocurre con todas las proteínas terapéuticas, existe potencial de inmunogenicidad.
La detección de la formación de anticuerpos es altamente dependiente de la sensibilidad y la especificidad del ensayo. Adicionalmente, la incidencia de positividad para anticuerpos observada (incluyendo anticuerpos neutralizantes) en un ensayo puede estar influenciada por varios factores, incluida la metodología del ensayo, el manejo de la muestra, el tiempo de recolección de la muestra, los medicamentos concomitantes y la enfermedad subyacente. Por estas razones, la comparación de la incidencia de anticuerpos contra blinatumomab con la incidencia de anticuerpos contra otros productos, podría ser engañosa.
La inmunogenicidad de BLINCYTO® se ha evaluado utilizando una tecnología de detección de electroquimioluminiscencia (ECL) o un inmunoensayo por inmunoabsorción ligado a enzimas (ELISA) para la detección de anticuerpos de unión anti-blinatumomab. Para los pacientes cuyo suero dio positivo en el inmunoensayo, se realizó un ensayo biológico in vitro para detectar los anticuerpos neutralizantes.
En estudios clínicos, menos del 2% de pacientes tratados con BLINCYTO® dieron positivo para anticuerpos de unión anti-blinatumomab. De los pacientes que desarrollaron anticuerpos anti-blinatumomab, 7 de 9 (78%) presentaron actividad neutralizante in vitro.La formación de anticuerpos anti-blinatumomab puede afectar la farmacocinética de BLINCYTO®.
Experiencia Post-comercialización: Las siguientes reacciones adversas se han identificado durante el uso posterior a la aprobación de BLINCYTO®. Debido a que estas reacciones se reportan voluntariamente a partir de una población de tamaño incierto, no siempre es posible estimar confiablemente su frecuencia o establecer una relación causal con la exposición al fármaco.
• Se ha reportado pancreatitis fatal en pacientes tratados con BLINCYTO® en combinación con dexametasona.
PRECAUCIONES EN RELACIÓN CON EFECTOS DE CARCINOGÉNESIS, MUTAGÉNESIS, TERATOGÉNESIS Y SOBRE LA FERTILIDAD:
Carcinogénesis, mutagénesis, daño en la fertilidad: No se han conducido estudios de carcinogenicidad o genotoxicidad con blinatumomab.
No se han conducido estudios para evaluar los efectos de blinatumomab sobre la fertilidad. No se presentaron efectos en los órganos reproductores de los ratones macho y hembra en 13 semanas de estudio de toxicidad de dosis repetida con la molécula de murino equivalente.
INTERACCIONES MEDICAMENTOSAS Y DE OTRO GÉNERO: No se han conducido estudios formales de interacción del fármaco con BLINCYTO®. El inicio del tratamiento con BLINCYTO® causa liberación transitoria de citocinas que puede suprimir enzimas CYP450. El riesgo más alto de interacción fármaco-fármaco es durante los primeros 9 días del primer ciclo y los primeros 2 días del segundo ciclo en pacientes que están recibiendo sustratos concomitantes de CYP450, particularmente aquellos con un índice terapéutico estrecho. En estos pacientes, monitorizar para toxicidad (por ejemplo., warfarina) o concentraciones de fármaco (por ejemplo, ciclosporina). Ajustar la dosis del fármaco concomitante según se requiera (ver Farmacocinética y farmacodinamia).
ALTERACIONES EN LOS RESULTADOS DE PRUEBAS DE LABORATORIO: Se ha observado neutropenia y neutropenia febril, incluidos casos mortales, en pacientes que reciben BLINCYTO®. Monitoree los parámetros de laboratorio (incluidos, entre otros, el recuento de glóbulos blancos y el recuento absoluto de neutrófilos) durante la infusión de BLINCYTO®. En caso de que ocurra neutropenia prolongada, interrumpir BLINCYTO®.
El tratamiento con BLINCYTO® se asoció con elevaciones transitorias de las enzimas hepáticas. Monitoree alanina aminotransferasa (ALT), aspartato aminotransferasa (AST), gamma-glutamil transferasa (GGT) y bilirrubina total en sangre antes del inicio y durante el tratamiento con BLINCYTO®.
PRECAUCIONES GENERALES:
Síndrome de liberación de citocinas: El síndrome de liberación de citocinas (CRS, por sus siglas en inglés), que puede ser potencialmente mortal o fatal, ocurrió en pacientes que recibían BLINCYTO®. La mediana de tiempo hasta el inicio del CRS fue 2 días después del inicio de la infusión y la mediana de tiempo hasta la resolución del CRS fue de 5 días entre los casos que se resolvieron. Las manifestaciones de CRS incluyen fiebre, cefalea, náusea, astenia, hipotensión, incremento de la alanina-aminotransferasa (ALT), incremento de la aspartato-aminotransferasa (AST), incremento en la bilirrubina total y coagulación intravascular diseminada (CID). Las manifestaciones de CRS después del tratamiento con BLINCYTO® se traslapan con aquéllas de reacciones a la infusión, síndrome de fuga capilar (CLS) e histiocitosis hemofagocítica/síndrome de activación de macrófagos (MAS). Usando todos estos términos para definir el CRS en los ensayos clínicos de BLINCYTO®, se informó CRS en 15% de los pacientes con LLA recurrente o refractaria y en 7% de los pacientes con LLA con EMR-positiva (ver Reacciones secundarias y adversas.
Monitorizar a los pacientes para detectar signos o síntomas de estos eventos. Aconsejar a los pacientes ambulatorios que reciben BLINCYTO® que contacten a su profesional de la salud por signos y síntomas asociados con CRS. Si se produce un CRS grave, interrumpa BLINCYTO® hasta que se resuelva el CRS. Suspenda BLINCYTO® permanentemente si ocurre un CRS potencialmente mortal. Administre corticosteroides para CRS graves o potencialmente mortales (ver Dosificación y vía de administración).
Toxicidades neurológicas: Han ocurrido toxicidades neurológicas en estudios clínicos en aproximadamente 65% de pacientes con LLA que reciben BLINCYTO® (ver Reacciones secundarias y adversas). Entre los pacientes que experimentaron un evento neurológico, la mediana de tiempo hasta la aparición del primer evento estuvo dentro de las primeras 2 semanas del tratamiento con BLINCYTO® y la mayoría de los eventos se resolvieron. Las manifestaciones de toxicidades neurológicas más comunes (≥ 10%) fueron cefalea y temblor: el perfil de toxicidad neurológica varió por grupo de edad (ver Uso en poblaciones específicas). Las toxicidades neurológicas de grado 3 o superior (severas, potencialmente mortales o fatales) después del inicio de la administración de BLINCYTO® ocurrieron en aproximadamente 13% de los pacientes e incluyeron encefalopatía, convulsiones, trastornos del habla, alteraciones de la conciencia, confusión y desorientación, y alteraciones de la coordinación y el equilibrio. Las manifestaciones de toxicidad neurológica incluyeron trastornos de los pares craneales. La mayoría de los eventos neurológicos se resolvieron después de la interrupción del tratamiento con BLINCYTO®, pero algunos llevaron a la discontinuación del tratamiento.
Existe experiencia limitada con BLINCYTO® en pacientes con LLA activa en el sistema nervioso central (CNS, por sus siglas en inglés) o con historia de eventos neurológicos. Los pacientes con una historia o presencia de patología clínicamente relevante en el CNS fueron excluidos de los estudios clínicos.
Monitorizar a los pacientes que reciben BLINCYTO® para detectar signos y síntomas de toxicidades neurológicas. Aconseje a los pacientes ambulatorios con BLINCYTO® que se pongan en contacto con su profesional de la salud si desarrollan signos y síntomas de toxicidad neurológica. Interrumpir temporal o definitivamente BLINCYTO® como se recomienda (ver Dosis y vía de administración).
Infecciones: Se observaron infecciones serias tales como sepsis, neumonía, bacteriemia, infecciones oportunistas e infecciones en el sitio del catéter, de las cuales algunas fueron potencialmente mortales o fatales, en aproximadamente el 25% de los pacientes con LLA que recibieron BLINCYTO® en estudios clínicos (ver Reacciones secundarias y adversas). Según sea apropiado, administrar antibióticos de forma profiláctica y emplear pruebas de vigilancia durante el tratamiento con BLINCYTO®. Monitorizar a los pacientes para signos y síntomas de infección y tratar apropiadamente.
Síndrome de lisis tumoral: El síndrome de lisis tumoral (SLT), que puede ser potencialmente mortal o fatal, se ha observado en pacientes que reciben BLINCYTO® (ver Reacciones secundarias y adversas). Las medidas profilácticas adecuadas que incluyen pretratamiento de citorreducción no tóxica y la hidratación durante el tratamiento, deben ser utilizados para la prevención del SLT durante el tratamiento con BLINCYTO®. Monitorizar a los pacientes por signos o síntomas del SLT. El manejo de estos eventos puede requerir la interrupción temporal o permanente de BLINCYTO® (ver Dosis y vía de administración).
Neutropenia y neutropenia febril: La neutropenia y la neutropenia febril, incluyendo casos potencialmente mortales, se han observado en pacientes que reciben BLINCYTO® (ver Reacciones secundarias y adversas). Monitorizar los parámetros de laboratorio (incluyendo, pero no limitados a, el recuento de leucocitos y el recuento absoluto de neutrófilos) durante la infusión con BLINCYTO®. En caso de que ocurra neutropenia prolongada, interrumpir BLINCYTO®.
Efectos sobre la capacidad para conducir y operar máquinas: Debido al potencial de eventos neurológicos, incluyendo convulsiones, los pacientes que reciben BLINCYTO® están en riesgo de perder la conciencia (ver Reacciones secundarias y adversas). Aconsejar a los pacientes sobre abstenerse de manejar o comprometerse en actividades peligrosas o acciones tales como operar maquinaria pesada o potencialmente peligrosa mientras BLINCYTO® está siendo administrado.
Elevación de las enzimas hepáticas: El tratamiento con BLINCYTO® estuvo asociado con elevaciones transitorias de las enzimas hepáticas (ver Reacciones secundarias y adversas). En pacientes con LLA que recibieron BLINCYTO® en estudios clínicos, la mediana del tiempo para el inicio de las enzimas hepáticas elevadas fue de 3 días.
La mayoría de estas elevaciones transitorias de las enzimas hepáticas se observaron en el escenario de CRS. Para los eventos que se observaron fuera del escenario de CRS, la mediana de tiempo para el inicio fue de 19 días. Las elevaciones de enzimas grado 3 o más, ocurrieron en aproximadamente el 7% de los pacientes fuera del escenario con CRS y resultó en una descontinuación del tratamiento en menos del 1% de los pacientes.
Monitorizar la alanina-aminotransferasa (ALT), aspartato-aminotransferasa (AST), la gamma glutamil transferasa (GGT), y la bilirrubina total en sangre antes de iniciar y durante el tratamiento con BLINCYTO®. Interrumpir BLINCYTO® si las transaminasas se elevan a más de 5 veces el límite superior del normal o si la bilirrubina total se eleva a más de 3 veces el límite superior del normal.
Pancreatitis: Se ha reportado pancreatitis fatal, en pacientes que están recibiendo BLINCYTO® en combinación con dexametasona en estudios clínicos y en la etapa de post-comercialización (ver Reacciones secundarias y adversas).
Evaluar a los pacientes que desarrollen signos y síntomas de pancreatitis. El manejo de pancreatitis puede requerir ya sea interrupción temporal o descontinuación de BLINCYTO® y dexametasona (ver Dosis y vía de administración).
Leucoencefalopatía: Se han observado cambios en las imágenes de resonancia magnética (IRM) craneal que muestran leucoencefalopatía en pacientes recibiendo BLINCYTO®, especialmente en pacientes con tratamiento previo con irradiación craneal y quimioterapia antileucémica (incluyendo metotrexato sistémico a dosis altas o la citarabina intratecal). Se desconoce la importancia clínica de estos cambios en dichas imágenes.
Errores en la preparación y administración: Se han producido errores de preparación y administración con el tratamiento con BLINCYTO®. Siga las instrucciones de preparación (incluida la mezcla) y la administración estrictamente para minimizar los errores de medicación (incluyendo subdosis y sobredosis) (ver Dosis y vía de administración).
Inmunización: No se ha estudiado la seguridad de la inmunización con vacunas de virus vivos durante o después de la terapia con BLINCYTO®. No se recomienda la vacunación con vacunas de virus vivos por lo menos 2 semanas antes de iniciar el tratamiento con BLINCYTO®, durante el tratamiento, y hasta la recuperación inmune después del último ciclo de BLINCYTO®.
Riesgo de reacciones adversas graves en pacientes pediátricos debido al conservador de alcohol bencílico: Las reacciones adversas graves y fatales, incluido el "síndrome de jadeo", pueden ocurrir en neonatos y lactantes tratados con medicamentos conservados con alcohol bencílico, incluyendo BLINCYTO® (con conservador). El "síndrome de jadeo" se caracteriza por depresión del sistema nervioso central, acidosis metabólica y respiración jadeante.
Al prescribir BLINCYTO® (con conservador) para pacientes pediátricos, considere la carga metabólica diaria combinada de alcohol bencílico de todas las fuentes, incluido BLINCYTO® (con conservador) (contiene 7.4 mg de alcohol bencílico por mL) y otros medicamentos que contienen alcohol bencílico. Se desconoce la cantidad mínima de alcohol bencílico a la que pueden producirse reacciones adversas graves (ver Uso en poblaciones específicas).
Debido a la adición de solución salina bacteriostática, las bolsas de 7 días de solución de BLINCYTO® contienen alcohol bencílico y no se recomienda su uso en pacientes que pesen menos de 22 kg (ver Dosis y vía de administración y Uso en poblaciones específicas).
DOSIS Y VÍA DE ADMINISTRACIÓN:
Tratamiento de LLA de precursor de células B, EMR-Positiva.
• Un tratamiento en curso consiste en 1 ciclo de BLINCYTO® para inducción seguido de hasta 3 ciclos adicionales para consolidación.
• Un ciclo único de tratamiento con BLINCYTO® para inducción o consolidación consiste de 28 días de infusión intravenosa continua seguida por un intervalo de 14 días libres de tratamiento (42 días en total).
• Consulte la Tabla 12 para conocer la dosis recomendada según el peso y el programa del paciente. Los pacientes con un peso de 45 kg o más reciben una dosis fija. Para pacientes que pesan menos de 45 kg, la dosis se calcula utilizando el área de superficie corporal del paciente (BSA).
Tabla 12. Dosis recomendada de BLINCYTO® y cronograma para el tratamiento de la LLA de precursores de células B, EMR-positiva
|
Ciclo |
Paciente 45 kg o más (dosis fija) |
Pacientes que pesan menos de 45 kg (dosis basada en BSA) |
|
Ciclo 1 inducción |
||
|
Días 1 a 28 |
28 mcg/día |
15 mcg/m2/día (sin exceder 28 mcg/día) |
|
Días 29 a 42 |
14 días libres de tratamiento |
14 días libres de tratamiento |
|
Ciclos 2 a 4 consolidación |
||
|
Días 1 a 28 |
28 mcg/día |
15 mcg/m2/día (sin exceder 28 mcg/día) |
|
Días 29 a 42 |
14 días libres de tratamiento |
14 días libres de tratamiento |
• Se recomienda hospitalización durante los primeros 3 días del primer ciclo y los primeros 2 días del segundo ciclo. Para todos los inicios y reinicios de ciclos subsecuentes (por ejemplo, si el tratamiento es interrumpido durante 4 horas o más), se recomienda la supervisión de un profesional de la salud u hospitalización.
• Premedicar con prednisona o equivalente para LLA de precursores de células B, EMR-positiva.
o Para pacientes adultos, premedicar con 100 mg de prednisona intravenosa o equivalente (p. ej., dexametasona 16 mg) 1 hora antes de la primera dosis de BLINCYTO® de cada ciclo.
o Para pacientes pediátricos, premedicar con 5 mg/m2 de dexametasona, a una dosis máxima de 20 mg antes de la primera dosis de BLINCYTO® en el primer ciclo y cuando se reinicie una infusión después de una interrupción de 4 horas o más en el primer ciclo.
Para administración de BLINCYTO®:
o Consulte la sección Infusión de 24 horas o 48 horas.
o Consulte la sección Infusión de 7 días utilizando cloruro de sodio bacteriostático inyectable al 0.9%, (que contiene 0.9% de alcohol bencílico). Esta opción está disponible para pacientes con un peso de 22 kg o más. No se recomienda para pacientes que pesan menos de 22 kg.
Tratamiento de LLA de precursores de células B en recaída o refractaria:
• Un curso de tratamiento consiste en hasta 2 ciclos de BLINCYTO® para inducción seguido de 3 ciclos adicionales para consolidación y hasta 4 ciclos adicionales de terapia continua.
• Un ciclo único de tratamiento de inducción o consolidación con BLINCYTO® consiste en 28 días de infusión intravenosa continua seguida por un intervalo de 14 días libres de tratamiento (42 días en total).
• Un ciclo único de tratamiento de terapia continua con BLINCYTO® consiste en 28 días de infusión intravenosa continua seguida por un intervalo de 56 días libres de tratamiento (84 días en total).
• Consulte la Tabla 13 para conocer la dosis recomendada según el peso y el programa del paciente. Los pacientes con un peso de 45 kg o más reciben una dosis fija y, para los pacientes que pesan menos de 45 kg, la dosis se calcula utilizando el BSA del paciente.
Tabla 13. Dosis recomendada de BLINCYTO® y cronograma para el tratamiento de la LLA de precursores de células B, en recaída o refractaria
|
Ciclo |
Pacientes que pesan 45 kg o más (dosis fija) |
Paciente que pesan menos de 45 kg (dosis basada en BSA) |
|
Ciclo 1 inducción |
||
|
Días 1 a 7 |
9 mcg/día |
5 mcg/m2/día (sin exceder 9 mcg/día) |
|
Días 8 a 28 |
28 mcg/día |
15 mcg/m2/día (sin exceder 28 mcg/día) |
|
Días 29 a 42 |
Intervalo de 14 días libres de tratamiento |
Intervalo de 14 días libres de tratamiento |
|
Ciclo 2 inducción |
||
|
Días 1 a 28 |
28 mcg/día |
15 mcg/m2/día (sin exceder 28 mcg/día) |
|
Días 29 a 42 |
Intervalo de 14 días libres de tratamiento |
Intervalo de 14 días libres de tratamiento |
|
Ciclos 3 a 5 consolidación |
||
|
Días 1 a 28 |
28 mcg/día |
15 mcg/m2/día (sin exceder 28 mcg/día) |
|
Días 29 a 42 |
Intervalo de 14 días libres de tratamiento |
Intervalo de 14 días libres de tratamiento |
|
Ciclos 6 a 9 terapia continua |
||
|
Días 1 a 28 |
28 mcg/día |
15 mcg/m2/día (sin exceder 28 mcg/día) |
|
Días 29 a 84 |
Intervalo de 56 días libres de tratamiento |
Intervalo de 56 días libres de tratamiento |
• Se recomienda hospitalización durante los primeros 9 días del primer ciclo y los primeros 2 días del segundo ciclo. En todos los inicios y reinicios de ciclos subsecuentes (por ejemplo, si el tratamiento se interrumpe durante 4 horas o más), se recomienda la supervisión de un profesional de la salud u hospitalización.
• Pre-medicar con dexametasona.
o Para pacientes adultos, pre medicar con 20 mg de dexametasona 1 hora antes de la primera dosis de BLINCYTO® de cada ciclo, antes de un incremento de dosis (tal como el día 8 del Ciclo 1), y cuando se reinicia una infusión después de una interrupción de 4 o más horas.
o Para pacientes pediátricos, pre medicar con 5 mg/m2 de dexametasona, a una dosis máxima de 20 mg antes de la primera dosis de BLINCYTO® en el primer ciclo, antes de una dosis escalonada (como el día 8 del Ciclo 1) y cuando se reinicie una infusión después de una interrupción de 4 horas o más en el primer ciclo.
• Para la administración de BLINCYTO®.
o Consultar la sección Infusión de 24 horas o 48 horas:
o Consultar la sección Infusión de 7 días utilizando cloruro de sodio bacteriostático inyectable al 0.9%, (que contiene 0.9% de alcohol bencílico). Esta opción está disponible para pacientes que pesen 22 kg o más. No se recomienda para pacientes que pesan menos de 22 kg.
Modificaciones de la dosis por reacciones adversas: Si la interrupción después de una reacción adversa no es mayor a 7 días, continuar el mismo ciclo para un total de 28 días de infusión inclusive de días antes y después de la interrupción de dicho ciclo. Si una interrupción debida a un evento adverso es mayor a 7 días, iniciar un nuevo ciclo.
Tabla 14. Modificación de dosis por reacciones adversas
|
Reacciones adversas |
Grado* |
Pacientes que pesan 45 kg o más |
Pacientes que pesan menos de 45 kg |
|
Síndrome de liberación de citocina (CRS) |
Grado 3 |
Interrumpir BLINCYTO® Administrar 8 mg de dexametasona cada 8 horas por vía intravenosa u oral por hasta 3 días, e ir disminuyendo la dosis durante 4 días Cuando el CRS se resuelva, reiniciar BLINCYTO® a dosis de 9 mcg/día después de 7 días si la reacción adversa no reincide |
Interrumpir BLINCYTO® Administrar 5 mg/m2 de dexametasona (máximo 8 mg) cada 8 horas por vía intravenosa u oral por hasta 3 días, e ir disminuyendo la dosis durante 4 días Cuando el CRS se resuelva, reiniciar BLINCYTO® a dosis de 5 mcg/m2/día y escalar a 15 mcg/m2/día después de 7 días si la reacción adversa no reincide. |
|
Grado 4 |
Suspender BLINCYTO® permanentemente. Administrar dexametasona como se indicó para el CRS de grado 3. |
||
|
Toxicidad neurológica |
Convulsión |
Suspender BLINCYTO® permanentemente si ocurre más de una convulsión. |
|
|
Grado 3 |
Suspender BLINCYTO® hasta que no sea mayor a grado 1 (leve) y durante al menos 3 días, después reiniciar BLINCYTO® en dosis de 9 mcg/día. Escalar a 28 mcg/día después de 7 días si la reacción adversa no reincide. Si ocurrió reacción adversa a dosis de 9 mcg/día, o si la reacción adversa toma más de 7 días para resolverse, descontinuar BLINCYTO® permanentemente. |
Suspender BLINCYTO® hasta que no sea mayor a grado 1 (leve) y durante al menos 3 días, después reiniciar BLINCYTO® a dosis de 5 mcg/m2/día. Escalar a 15 mcg/m2/día después de 7 días si la reacción adversa no reincide. Si ocurrió la reacción adversa a dosis de 5 mcg/m2/día o si la reacción adversa toma más de 7 días para resolverse, descontinuar BLINCYTO® permanentemente. |
|
|
Grado 4 |
Suspender BLINCYTO® permanentemente. |
||
|
Otras reacciones adversas clínicamente relevantes |
Grado 3 |
Suspender BLINCYTO® hasta que no sea mayor de grado 1 (leve), después reiniciar BLINCYTO® a dosis de 9 mcg/día. Escalar a 28 mcg/día después de 7 días si la reacción adversa no reincide. Si la reacción adversa toma más de 14 días para resolverse, descontinuar BLINCYTO® permanentemente. |
Suspender BLINCYTO® hasta que no sea mayor de grado 1 (leve), después reiniciar BLINCYTO® a dosis de 5 mcg/m2/día. Escalar a 15 mcg/m2/día después de 7 días si la reacción adversa no reincide. Si la reacción adversa toma más de 14 días para resolverse, descontinuar BLINCYTO® permanentemente. |
|
Grado 4 |
Considerar suspender BLINCYTO® permanentemente. |
||
* Basado en los criterios de terminología común para eventos adversos (CTCAE). grado 3 es severo, y grado 4 es potencialmente mortal.
Preparación: Es de gran importancia que las instrucciones para la preparación (incluida la mezcla) y la administración proporcionadas en esta sección, sean seguidas de manera estricta para minimizar errores de preparación (incluidas dosis menores o sobredosis) (ver Precauciones generales).
BLINCYTO® se puede infundir durante 24 horas (sin conservador) o 48 horas (sin conservador), o 7 días (con conservador). La elección entre estas opciones para la duración de la infusión debe hacerla el médico tratante teniendo en cuenta la frecuencia de los cambios en la bolsa de infusión y el peso del paciente. La administración de BLINCYTO® como una infusión de 7 días no se recomienda para pacientes que pesan menos de 22 kg.
Para la preparación, reconstitución, y administración de BLINCYTO®:
• Consulte la sección Infusión de 24 horas o 48 horas.
• Consulte la sección Infusión de 7 días utilizando cloruro de sodio bacteriostático inyectable al 0.9%, (que contiene 0.9% de alcohol bencílico). Esta opción está disponible para pacientes con un peso de 22 kg o más. No se recomienda para pacientes que pesan menos de 22 kg.
Llame al 55 44-24-45-55 si tiene alguna pregunta acerca de la reconstitución y preparación de BLINCYTO®.
Preparación aséptica: Observe estrictamente la técnica aséptica al preparar la solución para infusión dado que los frascos ámpula de BLINCYTO® no contienen conservadores antimicrobianos. Para prevenir contaminación accidental, preparar BLINCYTO® acorde a los estándares asépticos, incluyendo pero no limitado a:
• Preparar BLINCYTO® en una campana de flujo laminar. o superior.
• Asegurar que el área de mezclado cumpla con las especificaciones ambientales apropiadas, confirmadas mediante una monitorización periódica.
• Asegurar que el personal esté capacitado de manera apropiada en manipulaciones asépticas y en el mezclado de fármacos oncológicos.
• Asegurar que el personal use la vestimenta y los guantes de protección apropiados.
• Asegurar que los guantes y las superficies estén desinfectados.
Contenido del empaque:
1 empaque de BLINCYTO® incluye 1 frasco ámpula de BLINCYTO® y 1 frasco ámpula de solución estabilizadora IV.
• No utilice la solución estabilizadora IV para reconstituir BLINCYTO®. La solución estabilizadora IV está incluida en el empaque de BLINCYTO® y se utiliza para recubrir la bolsa intravenosa antes de la adición de BLINCYTO® reconstituido para evitar la adhesión de BLINCYTO® a la bolsa intravenosa y a la línea intravenosa.
• Es posible que se pueda necesitar más de 1 empaque de BLINCYTO® para preparar la dosis recomendada.
Información de incompatibilidad: BLINCYTO® es incompatible con el di-etilhexilftalato (DEHP) debido a la posibilidad de formación de partículas, lo que lleva a una solución turbia.
• Use bolsas/cartuchos de bomba de infusión de poliolefina, PVC sin DEHP, o acetato de etil vinilo (EVA).
• Use juegos de líneas de infusión intravenosa EVA de poliolefina, PVC sin DEHP.
Preparación y administración de BLINCYTO® como una infusión de 24 horas o 48 horas:
Reconstituya BLINCYTO® con agua estéril sin conservadores, inyección. No reconstituya los frascos ámpula de BLINCYTO® con la solución estabilizadora IV.
Para purgar la línea de infusión intravenosa, utilice sólo la solución de la bolsa que contiene la solución de BLINCYTO® FINAL preparada para infusión. No lo purgue con la solución de cloruro de sodio al 0.9%.
Reconstitución de BLINCYTO® para infusión de 24 horas o 48 horas:
1. Determine la cantidad de frascos ámpula de BLINCYTO® necesarios para una dosis y la duración de la infusión.
2. Reconstituya cada frasco ámpula de BLINCYTO® con 3 ml de agua estéril inyectable libre de conservadores, dirigiendo el agua a lo largo de las paredes del frasco ámpula de BLINCYTO® y no directamente sobre el polvo liofilizado. La concentración resultante por frasco ámpula de BLINCYTO® es de 12.5 mcg/mL.
• No reconstituya los frascos ámpula de BLINCYTO® con la solución estabilizadora IV.
3 Agitar suavemente el contenido para evitar el exceso de espuma.
• No sacudir.
4. Inspeccionar visualmente la solución reconstituida en busca de partículas y decoloración durante la reconstitución y antes de la infusión. La solución resultante debe ser clara a ligeramente opalescente, incolora a ligeramente amarilla.
• No use la solución si está turbia o si se ha precipitado.
Preparación de bolsa de infusión de BLINCYTO® de 24 horas o 48 horas: Verifique la dosis prescrita y la duración de la infusión para cada bolsa de infusión de BLINCYTO®. A fin de minimizar los errores, use los volúmenes específicos descritos en las Tablas 15 y 16 para preparar la bolsa de infusión BLINCYTO®.
• La Tabla 15 corresponde a pacientes con un peso de 45 kg o más.
• La Tablas 16 corresponde a pacientes que pesan menos de 45 kg.
1. Añadir asépticamente 270 mL de cloruro de sodio bacteriostático inyectable al 0.9% a la bolsa intravenosa vacía.
2. Transfiera asépticamente 5.5 mL de solución estabilizadora IV en la bolsa intravenosa que contiene cloruro de sodio inyectable al 0.9%. Mezcle suavemente el contenido de la bolsa para evitar la formación de espuma. Deseche el frasco ámpula que contiene la solución estabilizadora IV sin utilizar.
3. Transfiera asépticamente el volumen requerido de solución de BLINCYTO® reconstituido a la bolsa de infusión intravenosa que contiene solución inyectable de cloruro de sodio al 0.9% y solución estabilizadora IV. Mezcle suavemente el contenido de la bolsa para evitar la formación de espuma.
• Consulte la Tabla 15 correspondiente a pacientes con un peso de 45 kg o más para conocer el volumen específico de BLINCYTO® reconstituido requerido.
• Consulte la Tabla 16 correspondiente a pacientes que pesan menos de 45 kg (dosis basada en el BSA) para conocer el volumen específico de BLINCYTO® reconstituido requerido.
• Deseche el frasco ámpula que contiene BLINCYTO® no utilizado.
4. Bajo condiciones asépticas, coloque la línea de infusión intravenosa a la bolsa intravenosa con un filtro estéril de 0.2 micrones. Asegúrese que la línea de infusión intravenosa es compatible con la bomba de infusión.
5. Extraiga el aire de la bolsa intravenosa. Esto es particularmente importante para el uso de una bomba de infusión ambulatoria.
6. Purgue la línea de infusión intravenosa sólo con la bolsa que contiene la solución FINAL de BLINCYTO® preparada para infusión.
6. Almacene de 2 °C a 8 °C si no se usa inmediatamente.
Tabla 15. Para pacientes que pesan 45 kg o más: Volúmenes para agregar a la bolsa intravenosa
|
Cloruro de sodio inyectable al 0.9% (volumen inicial) |
270 mL |
|||
|
Solución estabilizadora IV (volumen fijo para infusiones de 24 y 48 horas) |
5.5 mL |
|||
|
Duración de la infusión |
Dosis |
Velocidad de infusión |
BLINCYTO® reconstituido |
|
|
Volumen |
Frascos ámpula |
|||
|
24 horas |
9 mcg/día |
10 mL/hora |
0.83 mL |
1 |
|
28 mcg/día |
10 mL/hora |
2.6 mL |
1 |
|
|
48 horas |
9 mcg/día |
5 mL/hora |
1.7 mL |
1 |
|
28 mcg/día |
5 mL/hora |
5.2 mL |
2 |
|
Tabla 16. Para pacientes que pesan menos de 45 kg: volúmenes para agregar a la bolsa intravenosa
|
Cloruro de sodio inyectable al 0.9% (volumen inicial) |
270 mL |
||||
|
Solución estabilizadora IV (volumen fijo para infusiones de 24 y 48 horas) |
5.5 mL |
||||
|
Duración de la infusión |
Dosis |
Velocidad de infusión |
BSA (m2) |
BLINCYTO® reconstituido |
|
|
Volumen |
Frascos ámpula |
||||
|
24 horas |
5 mcg/m2/día |
10 mL/hora |
1.5 a 1.59 |
0.7 mL |
1 |
|
1.4 a 1.49 |
0.66 mL |
1 |
|||
|
1.3 a 1.39 |
0.61 mL |
1 |
|||
|
1.2 a 1.29 |
0.56 mL |
1 |
|||
|
1.1 a 1.19 |
0.52 mL |
1 |
|||
|
1 a 1.09 |
0.47 mL |
1 |
|||
|
0.9 a 0.99 |
0.43 mL |
1 |
|||
|
0.8 a 0.89 |
0.38 mL |
1 |
|||
|
0.7 a 0.79 |
0.33 mL |
1 |
|||
|
0.6 a 0.69 |
0.29 mL |
1 |
|||
|
0.5 a 0.59 |
0.24 mL |
1 |
|||
|
0.4 a 0.49 |
0.2 mL |
1 |
|||
|
24 horas |
15 mcg/m2/día |
10 mL/hora |
1.5 a 1.59 |
2.1 mL |
1 |
|
1.4 a 1.49 |
2 mL |
1 |
|||
|
1.3 a 1.39 |
1.8 mL |
1 |
|||
|
1.2 a 1.29 |
1.7 mL |
1 |
|||
|
1.1 a 1.19 |
1.6 mL |
1 |
|||
|
1 a 1.09 |
1.4 mL |
1 |
|||
|
0.9 a 0.99 |
1.3 mL |
1 |
|||
|
0.8 a 0.89 |
1.1 mL |
1 |
|||
|
0.7 a 0.79 |
1 mL |
1 |
|||
|
0.6 a 0.69 |
0.86 mL |
1 |
|||
|
0.5 a 0.59 |
0.72 mL |
1 |
|||
|
0.4 a 0.49 |
0.59 mL |
1 |
|||
|
48 horas |
5 mcg/m2/día |
5 mL/hora |
1.5 a 1.59 |
1.4 mL |
1 |
|
1.4 a 1.49 |
1.3 mL |
1 |
|||
|
1.3 a 1.39 |
1.2 mL |
1 |
|||
|
1.2 a 1.29 |
1.1 mL |
1 |
|||
|
1.1 a 1.19 |
1 mL |
1 |
|||
|
1 a 1.09 |
0.94 mL |
1 |
|||
|
0.9 a 0.99 |
0.85 mL |
1 |
|||
|
0.8 a 0.89 |
0.76 mL |
1 |
|||
|
0.7 a 0.79 |
0.67 mL |
1 |
|||
|
0.6 a 0.69 |
0.57 mL |
1 |
|||
|
0.5 a 0.59 |
0.48 mL |
1 |
|||
|
0.4 a 0.49 |
0.39 mL |
1 |
|||
|
48 horas |
15 mcg/m2/día |
5 mL/hora |
1.5 a 1.59 |
4.2 mL |
2 |
|
1.4 a 1.49 |
3.9 mL |
2 |
|||
|
1.3 a 1.39 |
3.7 mL |
2 |
|||
|
1.2 a 1.29 |
3.4 mL |
2 |
|||
|
1.1 a 1.19 |
3.1 mL |
2 |
|||
|
1 a 1.09 |
2.8 mL |
1 |
|||
|
0.9 a 0.99 |
2.6 mL |
1 |
|||
|
0.8 a 0.89 |
2.3 mL |
1 |
|||
|
0.7 a 0.79 |
2 mL |
1 |
|||
|
0.6 a 0.69 |
1.7 mL |
1 |
|||
|
0.5 a 0.59 |
1.4 mL |
1 |
|||
|
0.4 a 0.49 |
1.2 mL |
1 |
|||
Administración de BLINCYTO® para infusión de 24 horas o 48 horas:
• Administrar BLINCYTO® como una infusión intravenosa continua con una velocidad de flujo constante usando una bomba de infusión. La bomba deberá ser programable, que se pueda cerrar, no-elastomérica, y tener una alarma.
• El volumen inicial (270 mL) es mayor que el volumen administrado al paciente (240 mL) para considerar el purgado de las líneas de infusión intravenosa y para asegurar que el paciente recibirá la dosis completa de BLINCYTO®.
• Infundir la solución de infusión final de BLINCYTO® preparada acorde a las instrucciones en la etiqueta de la bolsa preparada en una de las siguientes velocidades de infusión constantes:
- Velocidad de infusión de 10 mL/hora para una duración de 24 horas, o
- Velocidad de infusión de 5 mL/hora para una duración de 48 horas.
• Administre la solución de infusión final de BLINCYTO® preparada usando líneas de infusión intravenosa que contengan un filtro en línea de 0.2 micrones, estéril, no-pirogénico, de baja unión a proteína. Para obtener información acerca de la administración de la bolsa de 7 días, consulte la sección Administración de BLINCYTO® como infusión de 7 días.
• Nota importante: No purgar la línea de infusión de BLINCYTO®, o el catéter intravenoso especialmente cuando se cambian las bolsas de infusión. Purgar cuando se cambien las bolsas o al finalizar la infusión puede resultar en exceso de dosificación y complicaciones. Cuando se administra a través de un catéter venoso de múltiples vías, infunda BLINCYTO® a través de un lumen exclusivo.
• Al final de la infusión, deseche cualquier solución no usada de BLINCYTO® en la bolsa intravenosa y en las líneas intravenosas de acuerdo con los requerimientos locales.
Preparación y administración de BLINCYTO® como una infusión de 7 días usando solución bacteriostática inyectable de cloruro de sodio al 0.9% (conservante):
La administración de BLINCYTO® como una infusión de 7 días no se recomienda para pacientes que pesen menos de 22 kg (ver Precauciones generales y Uso en poblaciones especiales).
Utilice agua estéril sin conservador parala inyección a fin de reconstituir BLINCYTO®. No reconstituya los frascos ámpula de BLINCYTO® con la solución estabilizadora IV. No utilice un filtro en línea con la bolsa de infusión de 7 días.
Purgue la línea de infusión intravenosa solo con la solución de la bolsa que contiene la solución FINAL preparada para infusión. No la purgue con la solución de cloruro de sodio al 0.9%.
Reconstitución de BLINCYTO® para la infusión de 7 días:
1. Determine la cantidad de frascos ámpula de BLINCYTO® necesarios para una dosis.
2. Reconstituya cada frasco ámpula de BLINCYTO® con 3 mL de agua estéril inyectable sin conservadores, dirigiendo el agua a lo largo de las paredes del frasco ámpula de BLINCYTO® y no directamente sobre el polvo liofilizado. Lo que da como resultado una concentración final de BLINCYTO® de 12.5 mcg/mL.
• No reconstituya los viales con BLINCYTO® con la solución estabilizadora IV.
3. Agitar suavemente el contenido para evitar el exceso de espuma.
• No sacudir.
4. Inspeccionar visualmente la solución reconstituida en busca de partículas y decoloración durante la reconstitución y antes de la infusión. La solución resultante debe ser clara a ligeramente opalescente, incolora a ligeramente amarilla.
• No use la solución si está turbia o si se ha precipitado.
Preparación de la bolsa de infusión de BLINCYTO® para infusión de 7 días.
Verifique la dosis prescrita y la duración de la infusión para cada bolsa de infusión de BLINCYTO®. A fin de minimizar los errores, use los volúmenes específicos descritos en la Tabla 17 para preparar la bolsa de infusión BLINCYTO®.
1. Añada asépticamente 90 mL de cloruro de sodio bacteriostático inyectable al 0.9% a la bolsa intravenosa vacía.
2. Transfiera asépticamente 2.2 mL de solución estabilizadora IV en la bolsa intravenosa que contiene 0.9% de cloruro de sodio inyectable. Mezcle suavemente el contenido de la bolsa para evitar la formación de espuma. Deseche el frasco ámpula que contiene la solución estabilizadora IV sin utilizar.
3. Transfiera asépticamente el volumen requerido de solución de BLINCYTO® reconstituido a la bolsa de infusión intravenosa que contiene solución bacteriostática inyectable de cloruro de sodio al 0.9% y solución estabilizadora IV. Mezcle suavemente el contenido de la bolsa para evitar la formación de espuma.
• Consulte la Tabla 17 para el volumen específico de BLINCYTO® reconstituido. Deseche el frasco ámpula que contiene BLINCYTO® no utilizado.
4. Añada asépticamente el volumen requerido de solución inyectable de cloruro de sodio al 0.9% a la bolsa intravenosa para obtener un volumen final de 110 mL. Mezcle suavemente el contenido de la bolsa para evitar la formación de espuma.
• Consulte la Tabla 17 para el volumen específico de cloruro de sodio inyectable al 0.9%.
5. Bajo condiciones asépticas, coloque la línea de infusión intravenosa a la bolsa intravenosa.
• Asegúrese de que la línea de infusión intravenosa es compatible con la bomba de infusión.
• No utilice un filtro en línea para una bolsa de 7 días.
6. Extraiga el aire de la bolsa intravenosa. Esto es particularmente importante para el uso de una bomba de infusión ambulatoria.
7. Purgue la línea de infusión intravenosa sólo con la bolsa que contiene la solución FINAL de BLINCYTO® preparada para infusión.
8. Almacene de 2 ºC a 8 ºC si no se usa inmediatamente.
Tabla 17. Para la infusión de 7 días: volúmenes para agregar a la bolsa intravenosa para 28 mcg/día y 15 mcg/m2/día
|
Cloruro de sodio bacteriostático al 0.9% inyectable (volumen inicial) |
90 mL |
||||
|
Solución estabilizadora IV (volumen fijo para infusión de 7 días) |
2.2 mL |
||||
|
BLINCYTO® reconstituido |
El volumen específico se enlista en la tabla |
||||
|
Cantidad suficiente (c.s.) de cloruro de sodio inyectable al 0.9%, a un volumen final de 110 mL |
El volumen específico se enlista en la tabla |
||||
|
Duración de la infusión |
7 días |
||||
|
Velocidad de infusión |
0.6 mL/hora |
||||
|
Peso del paciente |
Dosis |
BSA (m2) |
BLINCYTO® reconstituido |
Volumen de cloruro de sodio inyectable al 0.9% necesario como c.s. para un volumen final de 110 mL |
|
|
Volumen |
Frasco ámpula |
||||
|
Dosis fija |
|||||
|
45 kg o más |
28 mcg/día |
N/A |
16.8 mL |
6 |
1 mL |
|
Dosis basada en BSA |
|||||
|
22 kg a menos de t kg |
15 mcg/m2/día |
1.5 a 1.59 |
14 mL |
5 |
3.8 mL |
|
1.4 a 1.49 |
13.1 mL |
5 |
4.7 mL |
||
|
1.30 a 1.39 |
12.2 mL |
5 |
5.6 mL |
||
|
1.20 a 1.29 |
11.3 mL |
5 |
6.5 mL |
||
|
1.10 a 1.19 |
10.4 mL |
4 |
7.4 mL |
||
|
1 a 1.09 |
9.5 mL |
4 |
8.3 mL |
||
|
0.9 a 0.99 |
8.6 mL |
4 |
9.2 mL |
||
|
Menor que 22 kg |
No se recomienda la infusión de 7 días |
||||
Administración de BLINCYTO® como infusión de 7 días:
• Administrar BLINCYTO® como una infusión intravenosa continua con una velocidad de flujo constante usando una bomba de infusión. La bomba deberá ser programable, que se pueda cerrar, no-elastomérica, y tener una alarma.
• El volumen final de la solución de infusión (110 mL) será mayor que el volumen administrado al paciente (100 mL) para considerar el purgado de la línea de infusión intravenosa y para asegurar que el paciente recibirá la dosis completa de BLINCYTO®.
• No utilice un filtro en línea para una bolsa de 7 días.
• Infundir la solución de infusión final de BLINCYTO® preparada acorde a las instrucciones en la etiqueta de la bolsa preparada a una velocidad de infusión de 0.6 mL/hora por una duración de 7 días.
• Nota Importante: No purgar la línea de infusión de BLINCYTO®, o el catéter intravenoso especialmente cuando se cambian las bolsas de infusión. Purgar cuando se cambien las bolsas o al finalizar la infusión puede resultar en exceso de dosificación y complicaciones. Cuando se administra a través de un catéter venoso de múltiples vías, infunda BLINCYTO® a través de un lumen exclusivo.
• Al final de la infusión, deseche cualquier solución no usada de BLINCYTO® en la bolsa intravenosa y en las líneas intravenosas de acuerdo con los requerimientos locales.
MANIFESTACIONES Y MANEJO DE LA SOBREDOSIFICACIÓN O INGESTA ACCIDENTAL: Se han observado sobredosis las cuales incluye un paciente adulto que recibió 133 veces la dosis terapéutica recomendada de BLINCYTO® administrado durante un corto periodo.
En la fase de evaluación de la dosis de un estudio en pacientes pediátricos y adolescentes con LLA de precursores de células B en recaída o refractaria, un paciente experimentó un evento de insuficiencia cardiaca fatal en el contexto de síndrome de liberación de citocinas (CRS) potencialmente mortal a una dosis de 30 mcg/m2/día (superior a la dosis máxima tolerada/recomendada) (ver Precauciones generales y Reacciones secundarias y adversas).
Las sobredosis resultaron en reacciones adversas consistentes con las reacciones observadas a la dosis recomendada, e incluyeron fiebre, temblor y cefalea. En caso de sobredosis interrumpir la infusión y monitorizar al paciente para signos de reacciones adversa, y proporcionar cuidados de soporte (ver Precauciones generales). Considerar el reinicio de BLINCYTO® a la dosis recomendada cuando todas las reacciones adversas se han resuelto y no antes de 12 horas después de la interrupción de la infusión (ver Dosis y vía de administración).
PRESENTACIÓN: Cada empaque de BLINCYTO® contiene un frasco ámpula de blinatumomab (35 mcg) y un frasco ámpula con solución estabilizadora IV.
RECOMENDACIONES SOBRE ALMACENAMIENTO: La información en la Tabla 18 indica el tiempo de almacenamiento para el frasco ámpula de BLINCYTO® reconstituido y la bolsa de infusión preparada.
Tabla 18. Tiempo de almacenamiento para el frasco ámpula de BLINCYTO® reconstituido y la bolsa de infusión preparada de BLINCYTO®
|
Tiempo máximo de almacenamiento |
||
|
Temperatura ambiente 23 °C a 27 °C (73 °F a 81 °F) |
Refrigerado 2 °C a 8 °C (36 °F a 46 °F) |
|
|
Frasco ámpula de BLINCYTO® reconstituido |
4 horas |
24 horas |
|
Bolsa de infusión preparada de BLINCYTO® (sin conservador) |
48 horas* |
8 días |
|
Bolsa de infusión preparada de BLINCYTO® (con conservador) |
7 días* |
14 días |
* El tiempo de almacenamiento incluye el tiempo de infusión. Si la bolsa IV que contiene la solución de BLINCYTO® para infusión no es administrada dentro de los marcos de tiempo y temperaturas indicados, debe ser desechada; no deberá ser refrigerada nuevamente.
Almacenar los frascos ámpula de BLINCYTO® y la solución estabilizadora IV en el empaque original refrigerado entre 2 °C a 8 °C y protegerlos de la luz hasta el momento de usarlos. No congelar.
Los frascos ámpula de BLINCYTO® y la solución estabilizadora IV se puede almacenar por un máximo de 8 horas a temperatura ambiente (23 °C a 27 °C). en la caja original para protegerlos de la luz.
LEYENDAS DE PROTECCIÓN:
Manténgase fuera del alcance de los niños. Su venta requiere receta médica. No se use durante el embarazo y la lactancia. Literatura exclusiva para el médico. Este producto deberá ser prescrito por médicos especialistas en oncología o hematología.
Reporte las sospechas de reacción adversa al correo: farmacovigilancia@cofepris.gob.mx o
farmacovigilanciamx@amgen.com
Representante legal:
AMGEN MÉXICO, S.A. de C.V.
Av. Vasco de Quiroga No. 3000, piso 4, Col. Santa Fe,
C.P. 01210, Álvaro Obregón, Ciudad de México, México.
233300EL870270


