
BENLYSTIA SC - Solución inyectable
Sustancia(s):
- Belimumab
Presentaciones:
- 1 Caja, 1 Jeringa(s) prellenada(s), 1 mL,
- 1 Caja, 4 Jeringa(s) prellenada(s), 1 mL,
- 1 Caja, 12 Jeringa(s) prellenada(s), 1 mL,
FORMA FARMACÉUTICA Y FORMULACIÓN:
La jeringa prellenada o la pluma precargada contiene:
Belimumab1 200 mg
Vehículo cbp 1 mL
1 Anticuerpo monoclonal humano IgG1? de origen ADN recombinante expresado en células murinas (NS0)
Cada mL contiene 200 mg de belimumab.
INDICACIONES TERAPÉUTICAS:
BENLYSTIA® SC (belimumab) está indicado para el tratamiento de:
• Pacientes adultos con lupus eritematoso sistémico (LES) activo, con autoanticuerpos positivos, que están recibiendo tratamiento estándar.
• Pacientes adultos con nefritis lúpica activa que estén recibiendo tratamiento estándar.
FARMACOCINÉTICA Y FARMACODINAMIA:
Propiedades farmacodinámicas:
Mecanismo de acción: BENLYSTIA® SC es un inhibidor específico de BLyS que bloquea la unión de BLyS soluble, un factor de supervivencia de las células B, a sus receptores en las células B. BENLYSTIA® SC no se une a las células B directamente, pero al unirse a BLyS, BENLYSTIA® SC inhibe la supervivencia de las células B, incluidas las células B autorreactivas, y reduce la diferenciación de las células B en células plasmáticas productoras de inmunoglobulinas.
Efecto farmacodinámico: El tratamiento con BENLYSTIA® SC en pacientes adultos redujo significativamente las células CD19+, CD20+ naïve circulantes, y células B activadas, y el subconjunto de células B LES en la semana 52. Se observaron reducciones en células naïve y el subconjunto de células B LES desde la semana 8 y se mantuvieron hasta la semana 52. Las células de memoria aumentaron inicialmente y disminuyeron lentamente hacia los niveles de referencia en la semana 52.
El tratamiento con BENLYSTIA® SC en pacientes adultos dio lugar a reducciones en los anticuerpos IgG y anti-ADN de doble cadena (anti-dsDNA) que se observaron desde la semana 8 y se mantuvieron hasta la semana 52. En pacientes con niveles bajos de complemento a nivel basal, el tratamiento produjo aumentos en el complemento C3 y C4 desde la semana 12 y se mantuvieron hasta la semana 52.
La respuesta farmacodinámica observada en pacientes de raza negra (Estudio 4) fue consistente con los estudios anteriores.
En pacientes con nefritis lúpica activa (Estudio 5), después del tratamiento con BENLYSTIA® SC, hubo una disminución en los niveles séricos de IgG desde la semana 4, y posteriormente hubo un aumento en los niveles séricos de IgG que se asoció con una disminución de la proteinuria. Las reducciones en los autoanticuerpos, los aumentos en el complemento y las reducciones en las células B totales circulantes y los subconjuntos de células B observadas fueron consistentes con los estudios de LES.
No se ha establecido la relevancia clínica de los biomarcadores farmacodinámicos mencionados anteriormente.
Propiedades farmacocinéticas:
Inyección subcutánea en adultos:
Lupus eritematoso sistémico: Los parámetros farmacocinéticos que se muestran en la Tabla 1 se basan en estimaciones de parámetros poblacionales de 661 sujetos después de la administración subcutánea de belimumab 200 mg una vez a la semana. El tiempo para alcanzar la concentración sérica máxima (Cmáx) fue de 2.6 días (Tmáx) después de la administración en estado estacionario. La biodisponibilidad de belimumab fue aproximadamente del 74%. Con la administración subcutánea semanal hubo fluctuaciones menores alrededor de la concentración promedio (Cavg 104 μg/mL), con Cmín (97 μg/mL) estando sólo ligeramente por debajo de Cavg.
Tabla 1. Parámetros farmacocinéticos poblacionales en adultos después de la administración subcutánea de BENLYSTIA® SC
|
Parámetro farmacocinético |
Estimaciones de población (n = 661) |
|
Concentración máxima (Cmáx, μg/mL) |
108 |
|
Área bajo la curva (AUC00-∞, día• μg/mL) |
726 |
|
Vida media de distribución (t½, días) |
1.1 |
|
Vida media terminal (t½, días) |
18.3 |
|
Aclaramiento sistémico (CL, mL/día) |
204 |
|
Volumen de distribución (Vss, L) |
5 |
Nefritis lúpica: Basado en modelos farmacocinéticos poblacionales y de simulación de la dosis de carga subcutánea semanal de 400 mg, la concentración promedio de belimumab durante las primeras 12 semanas se pronosticó en 78 μg/mL, que es similar a la concentración estimada de 89 μg/mL para administración intravenosa. La dosis de carga de 400 mg semanales, proporciona concentraciones en estado estacionario a partir de la segunda semana de dosificación. Se prevé que las concentraciones promedio en el estado estacionario de la administración subcutánea de belimumab 200 mg una vez a la semana en adultos con nefritis lúpica sean similares a las observadas en adultos con nefritis lúpica que reciben belimumab 10 mg/kg por vía intravenosa cada 4 semanas.
Poblaciones específicas: La siguiente información se basa en los análisis farmacocinéticos poblacionales de la administración subcutánea de BENLYSTIA® SC.
Edad: La edad no influyó significativamente en la farmacocinética de belimumab, donde la mayoría de los sujetos tenían entre 18 y 45 años (74% con dosificación subcutánea).
Pacientes geriátricos: Se dispone de datos farmacocinéticos limitados para pacientes de edad avanzada, ya que menos del 2% de los sujetos incluidos en el análisis farmacocinético tenían 65 años o más (véase Dosis y vía de administración - Uso en poblaciones específicas - Uso geriátrico).
Pacientes masculinos y femeninos: El género no influyó significativamente en la farmacocinética de belimumab en el estudio poblacional mayoritariamente femenino (85% con dosis subcutánea).
Grupos raciales: La raza no influyó significativamente en la farmacocinética de belimumab. La distribución racial con la administración subcutánea (Estudio 7) fue 61% blanca, 20% asiáticos, 11% negros y 6% nativos de Alaska/indoamericanos.
Peso: El peso corporal y el índice de masa corporal (IMC) no tuvieron ningún efecto clínicamente relevante sobre la farmacocinética de belimumab administrado por vía subcutánea en adultos. No se recomienda un ajuste de dosis basado en el peso o el IMC para la administración subcutánea.
Pacientes con insuficiencia renal: No se realizaron estudios formales para examinar los efectos de la insuficiencia renal en la farmacocinética de belimumab. BENLYSTIA® SC se estudió en un número limitado de pacientes adultos con LES que tenían insuficiencia renal leve (CrCl ≥ 60 y < 90 mL/min), moderada (CrCl ≥ 30 y < 60 mL/min) o grave (CrCl ≥ 15 y < 30 mL/min): 121 pacientes con insuficiencia renal leve y 30 pacientes con insuficiencia renal moderada recibieron belimumab por vía subcutánea (véase Uso en poblaciones específicas).
Pacientes con insuficiencia hepática: No se realizaron estudios formales para examinar los efectos de la insuficiencia hepática en la farmacocinética de belimumab. Los niveles basales de alanina aminotransferasa (ALT) y aspartato aminotransferasa (AST) no influyeron significativamente en la farmacocinética de belimumab. (véase Dosis y vía de administración - Uso en poblaciones específicas).
Estudios de interacción farmacológica: No se han realizado estudios formales de interacciones medicamentosas con BENLYSTIA® SC. El uso concomitante de micofenolato, ciclofosfamida, azatioprina, metotrexato, antimaláricos, AINE, aspirina y/o inhibidores de la HMG-CoA reductasa no influenciaron significativamente en la farmacocinética de belimumab. La coadministración de esteroides e inhibidores de la enzima convertidora de angiotensina (ECA) resultó en un aumento del aclaramiento sistémico de belimumab que no fue clínicamente significativo porque la magnitud estuvo dentro del rango de variabilidad normal del aclaramiento. El efecto de belimumab sobre la farmacocinética de otros fármacos no ha sido evaluado.
Estudios clínicos:
Estudios clínicos para la administración intravenosa en adultos con nefritis lúpica:
Estudio 5: Nefritis lúpica – BENLYSTIA® SC 10 mg/kg - Intravenoso:
La seguridad y eficacia de BENLYSTIA® SC 10 mg/kg administrado por vía intravenosa durante 1 hora los días 0, 14, 28 y después cada 28 días más la terapia estándar se evaluaron en un estudio aleatorizado, doble ciego y controlado con placebo de 104 semanas en 448 pacientes con nefritis lúpica activa proliferativa y/o nefritis lúpica membranosa (Estudio 5). Los pacientes tenían un diagnóstico clínico de LES según los criterios de clasificación del American College of Rheumatology; nefritis lúpica de clase III, IV y/o V comprobada por biopsia; y enfermedad renal activa en la selección que requería tratamiento estándar: corticosteroides con: 1) micofenolato para inducción seguido de micofenolato para mantenimiento, o 2) ciclofosfamida para inducción seguida de azatioprina para mantenimiento. Este estudio se realizó en Asia, América del Norte, América del Sur y Europa. La edad media de los pacientes fue de 33 años (rango: 18 a 77); la mayoría (88%) eran mujeres.
El criterio de validación principal de eficacia fue la respuesta renal de eficacia primaria (PERR) en la semana 104, definida como una respuesta en la semana 100 confirmada por una medición repetida en la semana 104 de los siguientes parámetros: proteína en orina: correlación de creatinina uPCR ≤ 0.7 g/g y tasa de filtración glomerular estimada (eGFR) ≥ 60 mL/min/1.73 m2 o sin disminución de la eGFR > 20% del valor anterior al brote.
Los principales criterios de valoración secundarios incluyeron:
• Respuesta renal completa (CRR) definida como una respuesta en la semana 100 confirmada por una medición repetida en la semana 104 de los siguientes parámetros: uPCR < 0.5 g/g y eGFR ≥ 90 mL/ min/1.73m2 o ninguna disminución en eGFR > 10% del valor anterior al brote.
• PERR en la semana 52.
• Tiempo hasta desenlace renal o la muerte (desenlace renal definido como el primer evento de enfermedad renal en etapa terminal, duplicación de la creatinina sérica, empeoramiento renal (definido como aumento cuantificado en la proteinuria y/o deterioro de la función renal) o recepción de la terapia prohibida relacionada a la enfermedad renal debido al inadecuado control de la nefritis lúpica o al tratamiento de la exacerbación renal).
La proporción de pacientes que lograron PERR en la semana 104 fue significativamente mayor en los pacientes que recibieron BENLYSTIA® SC más terapia estándar en comparación con placebo más terapia estándar (Tabla 2). Los principales criterios de valoración secundarios también mostraron una mejora significativa con BENLYSTIA® más terapia estándar en comparación con placebo más terapia estándar (Tabla 2 y Tabla 3).
Tabla 2. Resultados de eficacia en adultos con nefritis lúpica (Estudio 5)
|
Criterio de valoración de eficaciaa |
Placebo + terapia estándar n = 223 |
BENLYSTIA® SC + terapia estándar n = 223 |
Razón de probabilidades (OR) vs. placebo (IC 95%) |
|---|---|---|---|
|
Respuesta renal de eficacia primaria (PERR) en la semana 104b,c con respuesta |
32% |
43% |
1.6 (1.0, 2.3) p = 0.031 |
|
Componentes de PERR |
|||
|
Relación proteína en orina: creatinina ≤ 0.7 g/g |
34% |
44% |
1.5 (1.0, 2.3) |
|
eGFR ≥ 60 mL/min/1.73 m2 o sin disminución de la eGFR desde el valor anterior al brote de > 20% |
50% |
57% |
1.3 (0.9, 1.9) |
|
Respuesta renal completa (CRR) en la semana 104b,c con respuesta |
20% |
30% |
1.7 (1.1, 2.7) p = 0.017 |
|
Componentes de CRR |
|||
|
Relación proteína en orina: creatinina < 0.5 g/g |
29% |
39% |
1.6 (1.1, 2.4) |
|
eGFR ≥ 90 mL/min/1.73 m2 o sin disminución de la eGFR desde el valor anterior al brote de > 10% |
40% |
47% |
1.3 (0.9, 2.0) |
|
PERR en la semana 52b con respuesta |
35% |
47% |
1.6 (1.1, 2.4) p = 0.025 |
eGFR = Tasa de filtración glomerular estimada.
a PERR en la semana 104 fue el análisis de eficacia principal; CRR en la semana 104, PERR en la semana 52, fueron incluidos en la jerarquía de pruebas pre-especificada.
b Con el propósito de ser considerado una respuesta, el tratamiento con esteroides tuvo que reducirse a ≤ 10 mg/día a partir de la semana 24. Los pacientes que interrumpieron el tratamiento prematuramente, recibieron medicamentos prohibidos o incrementos de la terapia estándar inicial o se retiraron del estudio cuando fueron considerados como sujetos sin respuesta. Se definieron los medicamentos prohibidos y los incrementos en la terapia estándar inicial como: 1) uso de corticosteroides por encima de lo permitido por el protocolo; 2) agente inmunosupresor adicional (excepto tópicos) más allá de sus regímenes de inducción/mantenimiento;3) inhibidores de la enzima convertidora de angiotensina (IECA), bloqueadores del receptor de angiotensina II (BRA), o antipalúdicos iniciados después de la semana 24; 4) exceder las dosis permitidas por el protocolo para la terapia estándar (ciclofosfamida, azatioprina, micofenolato) o 5) otros biológicos, inmunoglobulina IV o plasmaféresis.
c El porcentaje de pacientes que no tomaron medicamentos prohibidos o presentan un incremento en la terapia estándar inicial en la semana 104 fue del 83% para BENLYSTIA® SC y del 74% para placebo.
En el análisis descriptivo de subgrupos, las tasas PERR, CRR se examinaron mediante terapias de inducción (micofenolato o ciclofosfamida), clase de biopsia (clase III o IV, clase III + V o clase IV + V o clase V) y niveles de uPCR al inicio (< 3 g/g o ≥ 3 g/g; análisis post-hoc). Figura 1.
Figura 1. Razón de probabilidad de PERR y CRR en la semana 104 en subgruposa,b (Estudio 5)
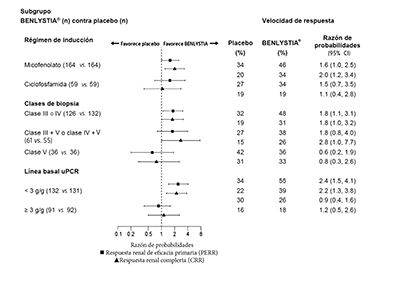
a Clase III = nefritis lúpica proliferativa focal; clase IV = nefritis lúpica proliferativa difusa; clase V = nefritis lúpica membranosa, clase III + V = nefritis lúpica mixta proliferativa focal-membranosa; clase IV + V = nefritis lúpica mixta proliferativa membranosa-difusa.
b La relación basal proteína urinaria: creatinina (uPCR) fue un análisis post-hoc.
La relación de respuestas para PERR por visita hasta la semana 104 se muestra en la Figura 2.
Figura 2. Respuesta renal de eficacia primaria (PERR) en adultos con nefritis lúpica (+/- error estándar) por visitaa (Estudio 5)
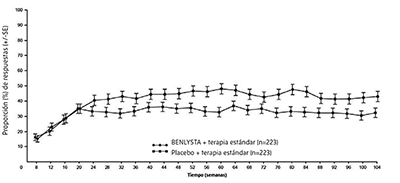
a Análisis descriptivo; las barras representan el error estándar. Los mismos pacientes pueden no tener respuesta en cada periodo de tiempo.
En el Estudio 5, los sujetos que recibieron BENLYSTIA® SC fueron significativamente menos propensos a experimentar desenlace renal o muerte en comparación con placebo (Tabla 3).
Tabla 3. Tiempo hasta desenlace renal o muerte en adultos con nefritis lúpica (Estudio 5)
|
Criterio de valoración de eficacia |
Placebo + terapia estándar n = 223 |
BENLYSTIA® SC + terapia estándar n = 223 |
Razón de riesgo (HR) vs. placebo (IC 95%) |
|
Tiempo hasta desenlace renal o muertea |
|||
|
Porcentaje de pacientes con eventob Número de pacientes con evento |
28% 63 |
16% 35 |
|
|
Tiempo para el eventoc |
0.5 (0.3, 0.8) p = 0.001 |
||
|
Componentes punto finald |
|||
|
Porcentaje de pacientes con evento |
|||
|
Enfermedad renal en etapa terminal (ESRD) |
0.4% |
1% |
|
|
Duplicación de la creatinina sérica inicial |
4% |
1% |
|
|
Empeoramiento renale |
18% |
8% |
|
|
Falla del tratamiento durante el desenlace renalf |
16% |
9% |
|
|
Muerte |
1% |
0.4% |
|
a Tiempo hasta desenlace renal o la muerte incluido en la jerarquía de pruebas preespecificadas.
b Al excluir las muertes del análisis (1 para BENLYSTIA® SC; 2 para placebo), el porcentaje de pacientes con desenlace renal fue del 15% para BENLYSTIA® SC comparado con el 27% para placebo (HR = 0.5; 95% IC: 0.3; 0.8).
c Los sujetos que interrumpieron el tratamiento, fueron eliminados del estudio, la pérdida de seguimiento, o fracasos del tratamiento no relacionados con la enfermedad renal fueron censurados en la fecha del evento. Los sujetos que completaron el periodo de tratamiento de 104 semanas fueron censurados en la visita de la semana 104. El tiempo transcurrido hasta el evento se definió como (fecha del evento menos fecha de inicio del tratamiento más 1 día).
d Los pacientes podrían haber tenido más de un evento; el primer evento contribuyó al criterio de valoración general.
e El empeoramiento renal se definió prospectivamente como el desarrollo de aumento de proteinuria y/o deterioro de la función renal definida como: 1) Aumento de la proteinuria (usando una gota de orina): el aumento reproducible de los niveles de proteína en orina de 24 horas a > 1 g/g si el valor inicial era < 0.2 g/g o > 2 g/g si el valor inicial estaba entre 0.2 g/g y 1 g / g o más del doble del valor basal si el valor inicial fue > 1 g/g; y 2) deterioro de la función renal: una disminución reproducible de la eGFR de > 20% acompañado de al menos 1 de los siguientes: proteinuria (> 1 g/g), cilindros de glóbulos rojos o cilindros de glóbulos blancos.
f La falla del tratamiento durante el desenlace renal se definió prospectivamente como la ingesta de medicamentos prohibidos para el control inadecuado de la nefritis lúpica adjudicada o el tratamiento de la exacerbación renal.
En análisis descriptivos de subgrupos del tiempo transcurrido hasta desenlace renal o muerte, los resultados fueron consistentes con el criterio de valoración general independientemente de la terapia de inducción (micofenolato o ciclofosfamida), clase de biopsia (clase III o IV, clase III + V o clase IV + V, o clase V; análisis post-hoc), y proteinuria basal (< 3 g/g o ≥ 3 g/g; análisis post-hoc). La diferencia de tratamiento se debió principalmente al empeoramiento renal y a los componentes de la falla del tratamiento durante el desenlace renal en las condiciones finales.
Administración subcutánea en adultos con LES:
Estudio 7. LES- BENLYSTIA® SC 200 mg – subcutánea: La seguridad y eficacia de BENLYSTIA® SC administrado por vía subcutánea se evaluó en un estudio aleatorizado, doble ciego y controlado con placebo en el que participaron 836 pacientes adultos con LES de acuerdo con los criterios del American College of Rheumatology (Estudio 7, NCT 01484496). Se excluyeron los pacientes con nefritis lúpica activa grave y lupus activo grave del SNC.
El estudio (aleatorización 2:1) evaluó BENLYSTIA® SC 200 mg una vez a la semana más terapia estándar (n = 556) en comparación con placebo una vez a la semana más terapia estándar (n = 280) durante 52 semanas en pacientes con LES activo. Los pacientes debían tener una puntuación SELENA-SLEDAI de ≥ 8 y resultados de la prueba de autoanticuerpos positivos (anticuerpo antinuclear [ANA] y/o ADN anti-ADN de doble cadena [anti-dsDNA]) en la selección.
No se observaron diferencias significativas en las características iniciales de los pacientes entre los grupos de tratamiento. En algunos países, se permitió el tratamiento con un agente dirigido a células B si se recibió un año o más antes de la línea de base; de lo contrario, no se permitió el tratamiento con un agente dirigido a células B. Los pacientes fueron excluidos del estudio si estaban recibiendo otros agentes biológicos. No se permitió la terapia anti-factor de necrosis tumoral, ciclofosfamida intravenosa, antagonista del receptor de interleucina 1, inmunoglobulina intravenosa (IVIG), prednisona > 100 mg/día, y plasmaféresis en los 3 meses anteriores o durante el estudio. El estudio se realizó en América del Norte, América del Sur, Europa y Asia. Los medicamentos concomitantes iniciales incluyeron corticosteroides (86%), antimaláricos (69%) e inmunosupresores (46%, incluidos azatioprina, metotrexato y micofenolato). La mayoría de los pacientes (aproximadamente el 80%) estaban recibiendo 2 o más clases de medicamentos para el LES.
Más del 50% de los pacientes tenían 3 o más sistemas de órganos activos involucrados al inicio del estudio. Los sistemas de órganos activos más comunes al inicio del estudio basados en SELENA-SLEDAI fueron mucocutáneos (88%), musculoesqueléticos (78%) e inmunológicos (76%). En general, el 12% de los pacientes tenían algún grado de actividad renal y menos del 15% de los pacientes tenían actividad en los sistemas vascular, cardiorrespiratorio o del SNC. Los pacientes se estratificaron según la gravedad de la enfermedad de acuerdo a su puntuación SELENA-SLEDAI (≤ 9 frente a ≥ 10), nivel de complemento (C3 y/o C4 bajo frente a otros) y raza (negros frente a otros), y luego asignados al azar para recibir BENLYSTIA® SC 200 mg más terapia estándar o placebo una vez a la semana más terapia estándar.
El criterio principal de valoración de la eficacia fue el índice de respuesta al LES-4 (SRI-4) en la semana 52 como se describió en los estudios de administración intravenosa. Los criterios de valoración secundarios de la eficacia incluyeron el tiempo transcurrido hasta el primer brote grave (medido por el índice de brotes de LES SELENA-SLEDAI modificado) y la proporción de pacientes que recibieron prednisona > 7.5 mg/día al inicio, cuya dosis promedio de prednisona se había reducido en ≥ 25% a ≤ 7.5 mg/día durante las semanas 40 a 52.
La proporción de pacientes que lograron una respuesta SRI-4 fue significativamente mayor en los pacientes que recibieron BENLYSTIA® SC más terapia estándar en comparación con placebo más terapia estándar. Las tendencias que comparan los grupos de tratamiento con respecto a la probabilidad de respuesta para los componentes individuales del criterio de valoración fueron consistentes con las del SRI-4 (Tabla 4).
Tabla 4. Tasa de respuesta clínica en pacientes con LES después de 52 semanas de tratamiento
(Estudio 7)
|
Respuestaa |
Placebo + terapia estándar (n = 279) |
BENLYSTIA® SC + terapia estándar (n = 554) |
|---|---|---|
|
Índice de respuesta al LES- 4 (SRI-4)b |
48% |
61% p = 0.0006 |
|
Razón de probabilidades (IC 95%) frente a placebo |
1.7 (1.3, 2.3) |
|
|
Componentes del índice-4 (SRI-4) de respuesta al LES |
||
|
Porcentaje de pacientes con reducción de SELENA-SLEDAI ≥ 4 |
49% |
62% |
|
Porcentaje de pacientes sin empeoramiento según el índice BILAG |
74% |
81% |
|
Porcentaje de pacientes sin empeoramiento por PGA |
73% |
81% |
a Los análisis excluyeron a cualquier sujeto al que le faltara una evaluación inicial para cualquiera de los componentes (1 para placebo; 2 para belimumab).
b Los pacientes que abandonaron el estudio o experimentaron ciertos aumentos en la medicación de fondo se consideraron fracasos en estos análisis. Una mayor proporción de pacientes que recibieron placebo más terapia estándar se consideraron fracasos por esta razón en comparación con el grupo de pacientes que recibieron BENLYSTIA® SC más terapia estándar.
La reducción en la actividad de la enfermedad observada en el SRI-4 se relacionó principalmente con la mejora en los sistemas de órganos más comúnmente involucrados, a saber, mucocutáneo, musculoesquelético, inmunológico y vascular.
La proporción de sujetos con respuesta SRI-4 por visita hasta la semana 52 se muestra en la Figura 3.
Figura 3. Proporción (%) de sujetos con respuesta SRI-4 (+/- error estándar) por visitaa
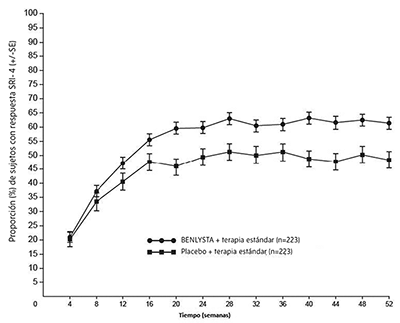
a Es posible que los mismos pacientes no hayan respondido en cada periodo de tiempo.
Efecto en pacientes de raza negra/afroamericanos: Se realizaron análisis de subgrupos exploratorios de la tasa de respuesta SRI-4 en pacientes de raza negra (n = 91). La tasa de respuesta de SRI-4 en pacientes de raza negra que recibieron BENLYSTIA® más terapia estándar fue del 45% (26/58) en comparación con el 39% (13/33) en el grupo que recibió placebo más terapia estándar (véase Uso en poblaciones específicas).
Efecto sobre el tratamiento concomitante con esteroides: Al inicio del estudio, el 60% de los pacientes recibían prednisona en dosis > 7.5 mg/día. Entre estos pacientes, el 18% de los pacientes que recibieron BENLYSTIA® SC más terapia estándar redujeron su dosis promedio de prednisona en al menos un 25% a ≤ 7.5 mg/día durante las semanas 40 a 52 en comparación con el 12% de los pacientes que recibieron placebo más terapia estándar; esta diferencia no fue estadísticamente significativa (OR = 1.65 [IC 95%: 0.95, 2.84]).
Efecto sobre los brotes de LES graves: Se calculó la probabilidad de experimentar un brote de LES grave, medido por el índice de brotes de LES de SELENA-SLEDAI modificado, excluyendo los brotes graves desencadenados sólo por un aumento de la puntuación de SELENA-SLEDAI a > 12. La proporción de pacientes que reportaron al menos un brote grave durante el estudio fue menor en los pacientes tratados con BENLYSTIA® SC más terapia estándar (11%) en comparación con los que recibieron placebo más terapia estándar (18%). Los pacientes tratados con BENLYSTIA® SC más la terapia estándar tuvieron un 49% menos de riesgo de experimentar al menos un brote severo durante las 52 semanas de observación, en comparación con los pacientes que recibieron placebo más la terapia estándar (HR = 0.51 [IC 95%: 0.35, 0.74]). De los pacientes que experimentaron un brote severo, la mediana del tiempo hasta el primer brote severo se retrasó en los pacientes que recibieron BENLYSTIA® SC más la terapia estándar en comparación con placebo más la terapia estándar (171 días frente a 118 días).
CONTRAINDICACIONES:
Hipersensibilidad al principio activo o a cualquiera de sus componentes.
RESTRICCIONES DE USO DURANTE EL EMBARAZO Y LA LACTANCIA:
Embarazo:
Resumen del riesgo: Datos disponibles sobre el uso de BENLYSTIA® SC en mujeres embarazadas, tomados de estudios observacionales, reportes de casos publicados, y vigilancia posterior a la comercialización, son insuficientes para determinar si existe un riesgo asociado con el medicamento para defectos importantes de nacimiento o aborto espontáneo. Existen riesgos para la madre y el feto asociados con el LES (véase Consideraciones clínicas). Los anticuerpos monoclonales, como belimumab, son transportados activamente a través de la placenta durante el tercer trimestre del embarazo y puede afectar la respuesta inmune en el infante expuesto en el útero (véase Consideraciones clínicas).
En un estudio de desarrollo embriofetal y pre y posnatal combinado en animales, con monos que recibieron belimumab por administración intravenosa, no hubo evidencia de daño fetal con exposiciones aproximadamente 20 veces (basado en la administración subcutánea) a la exposición a la dosis máxima recomendada para humanos (MRHD). Los hallazgos relacionados con belimumab en fetos de monos y/o lactantes incluyeron reducciones en el recuento de células B, reducciones en la densidad de linfocitos B del tejido linfoide en el bazo y los ganglios linfáticos y títulos de IgG e IgM alterados. El nivel de efecto no adverso (NOAEL) no se identificó para estos hallazgos; sin embargo, fueron reversibles dentro de los 3 a 12 meses posteriores a la suspensión del medicamento (véanse Datos). Según datos en animales y el mecanismo de acción de belimumab, el sistema inmunológico en bebés de madres tratadas puede verse afectado negativamente. Se desconoce, según los datos disponibles, si los efectos inmunitarios, de ser identificados, son reversibles (véase Farmacocinética y farmacodinamia).
Se desconoce el riesgo de fondo estimado de defectos importantes de nacimiento y aborto espontáneo para la población indicada. Todos los embarazos tienen un riesgo de fondo de defectos de nacimiento, pérdida u otros resultados adversos. En la población general de Estados Unidos, el riesgo de fondo de defectos congénitos importantes y aborto espontáneo en embarazos clínicamente reconocidos es del 2% al 4% y del 15% al 20%, respectivamente.
Consideraciones clínicas:
Riesgo materno y/o embrionario/fetal asociado a la enfermedad: Las mujeres embarazadas con LES tienen un mayor riesgo de resultados adversos en el embarazo, incluyendo empeoramiento de la enfermedad subyacente, el parto prematuro, el aborto espontáneo y la restricción del crecimiento intrauterino. La nefritis lúpica materna aumenta el riesgo de hipertensión y preeclampsia/eclampsia. El paso de los autoanticuerpos de la madre a través de la placenta pueden ocasionar resultados neonatales adversos, incluyendo el lupus neonatal y el bloqueo cardiaco congénito.
Reacciones adversas fetales/neonatales: Conforme progresa el embarazo, cada vez más anticuerpos monoclonales se transportan a través de la placenta, con la mayor cantidad transferida durante el tercer trimestre. Los riesgos y beneficios deben considerarse antes de administrar vacunas vivas o atenuadas a bebés expuestos a BENLYSTIA® en el útero. Se debe monitorear al bebé de una madre tratada para la reducción de células B y otras disfunciones inmunológicas (véase Precauciones generales).
Datos en animales: En un estudio combinado de desarrollo embriofetal y pre y postnatal, monos cynomolgus preñadas recibieron belimumab en dosis de 0.5 o 150 mg/kg cada 2 semanas por vía intravenosa desde la confirmación del embarazo de 20 a 22 días de gestación (GD), durante el periodo de organogénesis (hasta aproximadamente GD 50), y continuando ya sea hasta el día de la cesárea programada (GD 150 [final del tercer trimestre]) o el día del parto. No hubo evidencia de toxicidad materna, efectos sobre la supervivencia embriofetal e infantil, o anomalías estructurales a la exposición aproximadamente de 20 veces la MRHD de 200 mg por vía subcutánea (sobre una base de AUC con dosis intravenosas maternas de animales de hasta 150 mg/kg). Los hallazgos relacionados con belimumab en madres incluyeron reducciones de los recuentos de células B maduras e inmaduras en fetos y/o infantes incluyeron reducciones de los recuentos de células B maduras e inmaduras, reducciones en la densidad de linfocitos B del tejido linfoide en el bazo y los ganglios linfáticos, reducción del peso del bazo, títulos aumentados de IgG y títulos reducidos de IgM. Los recuentos de células B en monos bebés expuestos a belimumab en el útero se recuperaron a los 3 meses de edad y en madres después de 1 año. Los niveles de IgG e IgM en monos bebés se recuperaron a los 6 meses de edad y las reducciones de linfocitos B en los ganglios linfáticos y el bazo se invirtieron al año de edad. belimumab cruzó la placenta, ya que se detectó en sangre de cordón fetal y líquido amniótico en GD 150.
Lactancia:
Resumen del riesgo: No se dispone de información sobre la presencia de belimumab en la leche materna, los efectos del fármaco en el lactante o los efectos del fármaco en la producción de leche. Belimumab se detectó en la leche de monos cynomolgus; sin embargo, debido a diferencias específicas de la especie en la fisiología de la lactancia, los datos en animales pueden no predecir los niveles de fármaco en la leche materna. Se sabe que IgG materna está presente en la leche humana. Si belimumab es transferido a la leche materna en humanos, se desconocen los efectos de la exposición local en el tracto gastrointestinal y la posible exposición sistémica limitada en el lactante a belimumab.
La falta de datos clínicos durante la lactancia impiden una clara determinación del riesgo de BENLYSTIA® SC para un lactante; por lo tanto, el desarrollo y los beneficios para la salud del lactante deben ser considerados junto con la necesidad clínica de la madre de recibir BENLYSTIA® SC, y cualquier efecto potencial adverso en el lactante por BENLYSTIA® SC o por la condición subyacente de la madre.
Mujeres y hombres con potencial reproductivo:
Anticoncepción: Después de una evaluación del riesgo versus el beneficio, si se garantiza la prevención del embarazo, las mujeres en edad reproductiva deben utilizar un método anticonceptivo eficaz durante el tratamiento y durante al menos 4 meses después de finalizar el tratamiento.
REACCIONES SECUNDARIAS Y ADVERSAS:
Se han observado las siguientes reacciones adversas con BENLYSTIA® SC y se comentan en detalle en la sección de Precauciones generales:
• Infecciones graves.
• Reacciones de hipersensibilidad, incluida la anafilaxia.
• Depresión y suicidio.
• Neoplasias malignas.
Debido a que los estudios clínicos se llevan a cabo en condiciones muy variables, las tasas de reacciones adversas observadas en los estudios clínicos de un fármaco no pueden compararse directamente con las tasas de los estudios clínicos de otro fármaco y pueden no reflejar las tasas observadas en la práctica.
Estudios clínicos con administración subcutánea en adultos: Los datos descritos a continuación reflejan la exposición a BENLYSTIA® SC administrado por vía subcutánea más terapia estándar en comparación con placebo más terapia estándar en 836 pacientes con LES en un estudio controlado (Estudio 7). Además de la terapia estándar, los pacientes recibieron BENLYSTIA® SC 200 mg (n = 556) o placebo (n = 280) (aleatorización 2:1) una vez a la semana por hasta 52 semanas (véase Estudios clínicos).
En el ensayo, el 81% de los pacientes tratados con BENLYSTIA® SC más la terapia estándar reportaron un evento adverso en comparación con el 84% de los tratados con placebo más la terapia estándar. La proporción de pacientes que interrumpieron el tratamiento debido a cualquier reacción adversa durante el estudio clínico controlado fue del 7.2% de los pacientes que recibieron BENLYSTIA® SC más la terapia estándar y del 8.9% de los pacientes que recibieron placebo más la terapia estándar.
El perfil de seguridad observado para BENLYSTIA® SC administrado por vía subcutánea más la terapia estándar fue consistente con el perfil de seguridad conocido de BENLYSTIA® SC administrado por vía intravenosa más la terapia estándar, con excepción de las reacciones locales en el lugar de la inyección.
Infecciones: En un estudio controlado de BENLYSTIA® SC administrado por vía subcutánea en adultos con LES (N= 836), la incidencia general de infecciones fue del 55% en los pacientes que recibieron BENLYSTIA® SC en comparación con el 57% en los pacientes que recibieron placebo. Las infecciones reportadas con mayor frecuencia con BENLYSTIA® SC administrado por vía subcutánea fueron similares a las reportadas con BENLYSTIA® SC administrado por vía intravenosa.
Depresión y suicidio: En un estudio controlado de BENLYSTIA® SC administrado por vía subcutánea en adultos con LES (N = 836), que excluyó a pacientes con antecedentes de trastornos psiquiátricos, se reportaron eventos psiquiátricos en el 6% de los pacientes que recibieron BENLYSTIA® SC y el 11% de los pacientes que reciben placebo. Se reportaron eventos relacionados con la depresión en el 2.7% (15/556) de los pacientes que recibieron BENLYSTIA® SC y el 3.6% (10/280) de los pacientes que recibieron placebo. Se reportaron eventos psiquiátricos graves en el 0.2% (1/556) de los pacientes que recibieron BENLYSTIA® SC y en ningún paciente que recibió placebo. No hubo eventos graves relacionados con la depresión ni suicidios reportados en cualquiera de los grupos. En el C-SSRS, el 1.3% (7/554) de los pacientes que recibieron BENLYSTIA® SC reportaron ideación o comportamiento suicida en comparación con el 0.7% (2/277) de los pacientes que recibieron placebo.
Reacciones en el sitio de inyección: En un ensayo clínico controlado de BENLYSTIA® SC administrado por vía subcutánea en adultos con LES (N = 836), la frecuencia de reacciones en el sitio de inyección fue del 6.1% (34/556) para los pacientes que recibieron BENLYSTIA® SC más la terapia estándar y del 2.5% (7/280) para los pacientes que recibieron placebo más la terapia estándar. Estas reacciones en el sitio de la inyección (más comúnmente dolor, eritema, hematoma, prurito y endurecimiento) fueron de intensidad leve a moderada. La mayoría (94%) no requirió la interrupción del tratamiento.
Uso concomitante de rituximab en adultos: BENLYSTIA® SC administrado por vía subcutánea en combinación con rituximab fue estudiado en un estudio de Fase III, aleatorizado, doble ciego, controlado con placebo, de 104 semanas en pacientes adultos con LES. Los pacientes fueron aleatorizados a 1 de los 3 brazos de tratamiento: BENLYSTIA® SC con un solo ciclo de rituximab (n = 144); BENLYSTIA® SC con placebo (n = 72); BENLYSTIA® SC más terapia estándar (n = 76). En general, las reacciones adversas fueron consistente con el perfil de seguridad conocido de BENLYSTIA® SC y rituximab. Cuando se comparan BENLYSTIA® SC y placebo o BENLYSTIA® más terapia estándar, BENLYSTIA® SC en combinación con rituximab se asoció con una mayor frecuencia de eventos adversos graves (13.9%, 19.7%, 22.2%), infecciones graves (2.8 %, 5.3 %, 9.0 %) y reacciones sistémicas posteriores a la inyección (9.7 %, 5.3%, 13.2%).
Experiencia posterior a la comercialización: Se han identificado las siguientes reacciones adversas durante el uso posterior a la aprobación de BENLYSTIA® SC. Debido a que estas reacciones son reportadas voluntariamente por una población de tamaño incierto, no siempre es posible estimar de manera confiable su frecuencia o establecer una relación causal con la exposición a los fármacos.
• Anafilaxia fatal (véase Precauciones generales).
Inmunogenicidad: La incidencia observada de anticuerpos anti-fármaco depende en gran medida de la sensibilidad y especificidad del ensayo. Las diferencias en los métodos de ensayo impiden comparaciones significativas de la incidencia de anticuerpos anti-fármaco, en los estudios descritos a continuación con la incidencia de anticuerpos anti-fármacos en estudios, incluidos estos de BENLYSTIA® SC o de otros productos de belimumab.
En el Estudio 7 (la dosis subcutánea en adultos con LES), no presentó formación de anticuerpos anti-belimumab en 556 pacientes que recibieron BENLYSTIA® SC 200 mg durante el periodo de 52 semanas controlado con placebo.
Se desconoce la relevancia clínica de la presencia de anticuerpos anti-belimumab.
PRECAUCIONES EN RELACIÓN CON EFECTOS DE CARCINOGÉNESIS, MUTAGÉNESIS, TERATOGÉNESIS Y SOBRE LA FERTILIDAD:
No se han realizado estudios en animales a largo plazo para evaluar el potencial carcinogénico de belimumab.
Los efectos sobre la fertilidad masculina y femenina no se han evaluado directamente en estudios con animales.
INTERACCIONES MEDICAMENTOSAS Y DE OTRO GÉNERO:
No se han realizado estudios formales de interacciones medicamentosas con BENLYSTIA® SC. En estudios clínicos el uso de BENLYSTIA® SC concomitantemente con otros medicamentos, incluidos corticosteroides, antimaláricos, agentes inmunomoduladores e inmunosupresores (Incluidos azatioprina, ciclofosfamida, metotrexato y micofenolato), antihipertensivos de la vía de la angiotensina, inhibidores de la HMG-CoA reductasa (estatinas) y/o medicamentos antiinflamatorios no esteroideos (AINE) sin evidencia de un efecto clínicamente significativo de estos medicamentos concomitantes sobre la farmacocinética de belimumab. No se ha evaluado el efecto de belimumab sobre la farmacocinética de otros medicamentos (véase Farmacocinética y farmacodinamia).
Uso concomitante con otras terapias biológicas: Los datos disponibles no respaldan la seguridad y eficacia del uso concomitante de BENLYSTIA® SC con rituximab en pacientes con LES. Se ha observado un incremento en la incidencia de infecciones graves y reacciones sistémicas posterior a la inyección en pacientes que reciben BENLYSTIA® SC concomitantemente con rituximab en comparación con pacientes que reciben BENLYSTIA® solo. No se ha establecido la seguridad y eficacia del uso de BENLYSTIA® SC concomitantemente con otras terapias biológicas, incluidas las terapias dirigidas a las células B. Se debe tener precaución si BENLYSTIA® SC es administrada en combinación con otras terapias biológicas.
ALTERACIONES EN LOS RESULTADOS DE PRUEBAS DE LABORATORIO:
Hasta el momento no hay eventos relacionados con la administración de belimumab.
PRECAUCIONES GENERALES:
Limitaciones de uso: No se ha evaluado la eficacia de BENLYSTIA® en pacientes con lupus activo grave del sistema nervioso central. No se recomienda el uso de BENLYSTIA® SC en estas situaciones.
Infecciones graves: Se han informado infecciones graves y, en ocasiones, fatales en pacientes que reciben terapia inmunosupresora, incluyendo BENLYSTIA® SC. En general, la incidencia de infecciones graves en los estudios controlados fue similar en pacientes que recibieron BENLYSTIA® SC en comparación con placebo, mientras que las infecciones fatales ocurrieron con mayor frecuencia en pacientes que recibieron BENLYSTIA® SC.
En estudios controlados de BENLYSTIA® administrado por vía intravenosa en adultos con LES, la incidencia de infecciones graves fue del 6.0% en los pacientes que recibieron BENLYSTIA® SC en comparación con el 5.2% en los pacientes que recibieron placebo. Las infecciones graves más frecuentes incluyeron neumonía, infecciones del tracto urinario, celulitis y bronquitis. Se produjeron infecciones fatales en el 0.3% (4/1,458) de los pacientes que recibieron BENLYSTIA® SC y en el 0.1% (1/675) de los pacientes que recibieron placebo (véase Reacciones secundarias y adversas).
En un estudio aleatorizado, doble ciego, controlado con placebo, de 104 semanas de duración para nefritis lúpica activa, en adultos que recibieron BENLYSTIA® SC administrado por vía intravenosa se produjeron infecciones graves en el 14% de los pacientes que recibieron BENLYSTIA® SC y en el 17% de los pacientes que recibieron placebo. Se produjeron infecciones fatales en el 0.9% (2/224) de los pacientes que recibieron BENLYSTIA® SC y en el 0.9% (2/224) de los pacientes que recibieron placebo (véase Reacciones secundarias y adversas).
En un estudio de seguridad aleatorizado, doble ciego, controlado con placebo, de 52 semanas, post comercialización de BENLYSTIA® SC administrado por vía intravenosa a adultos con LES (N = 4,003), la incidencia de infecciones graves fue del 3.7% en los pacientes que recibieron BENLYSTIA® SC en comparación con el 4.1% en los pacientes que recibieron placebo. Se produjeron infecciones graves que dieron lugar a la interrupción del tratamiento en el 1.0% de los pacientes que recibieron BENLYSTIA® SC y en el 0.9% de los pacientes que recibieron placebo. Infecciones graves ocurrieron en el 0.45% (9/2,002) de los pacientes que recibieron BENLYSTIA® SC y en el 0.15% (3/2,001) de los pacientes que recibieron placebo donde la incidencia de mortalidad por todas las causas fue del 0.50% (10/2,002) en pacientes que recibieron BENLYSTIA® SC y 0.40% (8/2,001) en pacientes que recibieron placebo (véase Reacciones secundarias y adversas).
En un estudio controlado de BENLYSTIA® SC administrado por vía subcutánea en adultos con LES (N=836), la incidencia de infecciones graves fue del 4.1% en pacientes que recibieron BENLYSTIA® SC y del 5.4% en pacientes que recibieron placebo. Se produjeron infecciones fatales en el 0.5% (3/556) de los pacientes que recibieron BENLYSTIA® SC y en ninguno de los pacientes que recibieron placebo (0/280) (véase Reacciones secundarias y adversas).
Considerar el riesgo y el beneficio antes de iniciar el tratamiento con BENLYSTIA® SC en pacientes con infecciones graves o crónicas. Considerar interrumpir el tratamiento con BENLYSTIA® SC en pacientes que desarrollen una nueva infección mientras lo reciben, monitorear de cerca a estos pacientes.
Leucoencefalopatía multifocal progresiva (PML): Se han notificado casos de PML asociada al virus JC que dieron lugar a déficits neurológicos, incluidos casos fatales, en pacientes con LES que recibieron inmunosupresores, incluyendo BENLYSTIA® SC. Los factores de riesgo de PML incluyen el tratamiento con terapias inmunosupresoras y el deterioro de la función inmunológica. Considerar el diagnóstico de leucoencefalopatía multifocal progresiva en cualquier paciente que presente signos y síntomas neurológicos de nueva aparición o en deterioro y consultar con un neurólogo u otro especialista apropiado según esté clínicamente indicado. En pacientes con sospecha de PML, la terapia inmunosupresora, incluyendo BENLYSTIA® SC, debe suspenderse hasta que se haya eliminado la PML. Si la PML es confirmada, la terapia inmunosupresora, incluyendo BENLYSTIA® SC debe ser suspendida.
Reacciones de hipersensibilidad, incluida anafilaxia:
Se han notificado reacciones de hipersensibilidad aguda, que incluyen anafilaxia y muerte, en asociación con BENLYSTIA® SC. Estos eventos generalmente ocurrieron pocas horas después de la infusión; sin embargo, pueden ocurrir más tarde. Se han reportado reacciones de hipersensibilidad no aguda que incluyen erupción cutánea, náuseas, fatiga, mialgia, dolor de cabeza y edema facial y, por lo general, se presentaron hasta una semana después de la infusión más reciente. Se ha producido hipersensibilidad, incluidas reacciones graves, en pacientes que han tolerado previamente infusiones de BENLYSTIA® SC. Los datos limitados sugieren que los pacientes con antecedentes de alergias a múltiples fármacos o hipersensibilidad significativa pueden tener un mayor riesgo.
En los estudios clínicos controlados de BENLYSTIA® SC administrado por vía intravenosa en adultos con LES, se reportaron reacciones de hipersensibilidad (que ocurrieron el mismo día de la infusión) en el 13% (191/1,458) de los pacientes que recibieron BENLYSTIA® SC y el 11% (76/675) de los pacientes que recibieron placebo. Se observó anafilaxia en el 0.6% (9/1,458) de los pacientes que recibieron BENLYSTIA® SC y en el 0.4% (3/675) de los pacientes que recibieron placebo. Las manifestaciones incluyeron hipotensión, angioedema, urticaria u otra erupción cutánea, prurito y disnea.
Debido a la superposición de signos y síntomas, no fue posible distinguir entre reacciones de hipersensibilidad y reacciones relacionadas a la infusión en todos los casos (véase Precauciones generales). En estudios clínicos controlados de BENLYSTIA® SC administrado por vía intravenosa en adultos con LES, algunos pacientes (13%) recibieron premedicación, que puede haber mitigado o enmascarado una respuesta de hipersensibilidad; sin embargo, no hay pruebas suficientes para determinar si la premedicación disminuye la frecuencia, la gravedad de las reacciones de hipersensibilidad.
En un estudio controlado de BENLYSTIA® SC administrado por vía subcutánea en adultos con LES, las reacciones sistémicas de hipersensibilidad fueron similares a las observadas en los estudios clínicos por vía intravenosa.
BENLYSTIA® SC para uso intravenoso debe ser administrado por médicos o enfermeros preparados para manejar la anafilaxia. En caso de una reacción grave, suspender BENLYSTIA® SC inmediatamente y administrar la terapia médica apropiada.
Informar a los pacientes que reciben BENLYSTIA® SC de los signos y síntomas de reacciones de hipersensibilidad e indicarles que busquen atención médica inmediata si se presenta una reacción.
Depresión y suicidio: En estudios clínicos controlados de BENLYSTIA® SC administrado por vía intravenosa en adultos con LES (2,133), los eventos psiquiátricos se reportaron con más frecuencia en pacientes tratados con BENLYSTIA® SC (16%) que con placebo (12%) y se relacionaron principalmente con eventos relacionados con la depresión (6.3% BENLYSTIA® SC; 4.7% placebo), insomnio (6% BENLYSTIA®; 5.3% placebo) y ansiedad (3.9% BENLYSTIA® SC; 2.8% placebo). Se reportaron eventos psiquiátricos graves en el 0.8% (12/1,458) de los pacientes que recibieron BENLYSTIA® SC, y 0.4% (3/675) de pacientes que recibieron placebo. Se reportó depresión grave en 0.4% (6/1,458) de pacientes que recibieron BENLYSTIA® SC y 0.1% (1/675) de los pacientes que recibieron placebo. Se reportaron dos suicidios (0.1%) en pacientes que recibieron BENLYSTIA® SC (uno con 10 mg/kg y otro con 1 mg/kg) (véase Reacciones secundarias y adversas).
En un estudio de seguridad posterior a la comercialización, aleatorizado, doble ciego controlado con placebo de 52 semanas de duración, de BENLYSTIA® SC administrado por vía intravenosa en adultos con LES (N = 4,003), se reportaron eventos psiquiátricos graves en 1% (20/2,002) de pacientes que recibieron BENLYSTIA® SC y 0.3% (6/2,001) de pacientes que recibieron placebo. Se reportó depresión grave en 0.3% (7/2,002) de los pacientes que recibieron BENLYSTIA® SC y en 0.1% (1/2,001) de los pacientes que recibieron placebo. La incidencia en general de ideación suicida grave o comportamiento de autolesión sin intención suicida fue del 0.7% (15/2,002) de los pacientes que recibieron BENLYSTIA® SC y del 0.2% (5/2,001) de los pacientes que recibieron placebo. En la escala de clasificación de gravedad del suicidio de Columbia (C-SSRS), el 2.4% (48/1,974) de los pacientes que recibieron BENLYSTIA® SC reportaron ideación o comportamiento suicida en comparación con el 2% (39/1,988) de los pacientes que recibieron placebo. No se reportó ningún suicidio en ninguno de los grupos (véase Reacciones secundarias y adversas).
Los estudios por vía intravenosa anteriores no excluyeron a los pacientes con antecedentes de trastornos psiquiátricos.
En estudios clínicos controlados, se reportaron depresión y suicidio en pacientes que recibieron BENLYSTIA®. Evaluar el riesgo de depresión y suicidio considerando el historial médico del paciente y su estado psiquiátrico actual antes del tratamiento con BENLYSTIA® SC y continuar monitoreando a los pacientes durante el tratamiento. Indicar a los pacientes que reciben BENLYSTIA® SC (y a los cuidadores, si corresponde) que contacten a su médico o enfermera si experimentan depresión o ésta empeora, pensamientos o comportamiento suicida u otros cambios de humor. Considerar el riesgo y el beneficio de continuar el tratamiento con BENLYSTIA® SC para los pacientes que desarrollan tales síntomas.
Neoplasias malignas: Existe un mayor riesgo de neoplasias malignas con el uso de inmunosupresores. Se desconoce el impacto del tratamiento con BENLYSTIA® en el desarrollo de neoplasias malignas.
Considerar el riesgo-beneficio individual en pacientes con factores de riesgo conocidos para el desarrollo o recurrencia de neoplasias malignas antes de prescribir BENLYSTIA® SC. En pacientes que desarrollan neoplasias malignas, considere el riesgo y el beneficio de continuar el tratamiento con BENLYSTIA® SC.
En los estudios clínicos controlados de BENLYSTIA® SC administrado por vía intravenosa en adultos con LES, se reportaron neoplasias malignas (incluidos cánceres de piel no melanoma) en 0.4% de los pacientes que recibieron BENLYSTIA® SC y 0.4% de los pacientes que recibieron placebo. En los estudios clínicos controlados por vía intravenosa, se observaron neoplasias malignas, excluyendo los cánceres de piel no melanoma, en el 0.2% (3/1,458) y el 0.3% (2/675) de los pacientes que recibieron BENLYSTIA® SC y placebo, respectivamente.
En un estudio clínico controlado de BENLYSTIA® SC administrado por vía subcutánea en adultos con LES (N = 836), los informes de neoplasias malignas fueron similares a los informados con BENLYSTIA® SC administrado por vía intravenosa.
Inmunización: Debido a su mecanismo de acción, BENLYSTIA® SC puede interferir con la respuesta a las inmunizaciones. No se deben administrar vacunas vivas 30 días antes o al mismo tiempo que BENLYSTIA® SC ya que no se ha establecido la seguridad clínica. No hay datos disponibles sobre la transmisión secundaria de la infección de personas que reciben vacunas vivas a pacientes que reciben BENLYSTIA® SC o el efecto de BENLYSTIA® SC en nuevas inmunizaciones.
DOSIS Y VÍA DE ADMINISTRACIÓN:
Vía de administración: Subcutánea.
BENLYSTIA® SC puede administrarse como inyección subcutánea en pacientes de 18 años o más.
Los autoinyectores y jeringas precargadas están destinados únicamente para uso subcutáneo (no para uso intravenoso).
Instrucciones de dosificación subcutánea para pacientes adultos con LES o nefritis lúpica: La dosis subcutánea de BENLYSTIA® SC no se ha evaluado y no está aprobada para pacientes menores de 18 años.
Pacientes adultos con LES: La dosis recomendada es de 200 mg una vez a la semana administrada como inyección subcutánea en el abdomen o el muslo. La dosificación subcutánea no se basa en el peso.
Si pasa de la terapia intravenosa con BENLYSTIA® SC a la administración subcutánea, administre la primera dosis subcutánea de 1 a 4 semanas después de la última dosis intravenosa.
Pacientes adultos con nefritis lúpica: En pacientes que inician el tratamiento con BENLYSTIA® SC para la nefritis lúpica activa (véase Estudios clínicos), el régimen de dosificación recomendado es una dosis de 400 mg (dos inyecciones de 200 mg) una vez a la semana para 4 dosis, después 200 mg una vez a la semana a partir de entonces. La dosis se administra mediante inyección subcutánea en el abdomen o el muslo. La dosis de 400 mg para la nefritis lúpica activa requiere la administración de 2 autoinyectores o 2 jeringas precargadas como se describe a continuación.
Un paciente con nefritis lúpica puede pasar de la terapia intravenosa con BENLYSTIA® SC a la terapia subcutánea en cualquier momento después de que el paciente complete las 2 primeras dosis intravenosas. Si está en transición, administre la primera dosis subcutánea de 200 mg 1 a 2 semanas después de la última dosis intravenosa.
Instrucciones de administración para inyección subcutánea:
1. Se recomienda que la primera inyección subcutánea de BENLYSTIA® SC® sea bajo la supervisión de un profesional de la salud. El profesional de la salud debe brindar la capacitación adecuada en la técnica subcutánea y la educación sobre los signos y síntomas de las reacciones de hipersensibilidad (véase Precauciones generales). Un paciente puede autoinyectarse o el cuidador del paciente puede administrar BENLYSTIA® SC por vía subcutánea una vez que el profesional de la salud determine que es apropiado.
2. Indicar al paciente o al cuidador del paciente que siga las instrucciones de administración que se proporcionan en las instrucciones de uso.
3. Indicar al paciente que retire el autoinyector o la jeringa precargada del refrigerador y lo deje reposar a temperatura ambiente durante 30 minutos antes de la inyección subcutánea. No calentar BENLYSTIA® SC® de ninguna otra forma.
4. Antes de la administración, indicar al paciente o al cuidador del paciente que inspeccione visualmente la ventana del autoinyector o la jeringa prellenada en busca de partículas o decoloración. BENLYSTIA® SC debe ser de transparente a opalescente e incolora a amarillo pálido. No utilizar BENLYSTIA® SC si el producto presenta decoloración o partículas. Indicar al paciente que no utilice el autoinyector o la jeringa precargada BENLYSTIA® SC si éstas cayeron sobre una superficie dura.
5. Cuando se inyecte en la misma región del cuerpo, aconsejar al paciente que utilice un lugar de inyección diferente para cada inyección; nunca aplique inyecciones en áreas donde la piel esté sensible, amoratada, enrojecida o endurecida. Cuando se administra una dosis de 400 mg en el mismo sitio, se recomienda que las 2 inyecciones individuales de 200 mg se administren al menos a 5 cm (aproximadamente 2 pulgadas) de distancia.
6. Indicar al paciente que se administre BENLYSTIA® SC 200 mg una vez a la semana, preferiblemente el mismo día cada semana.
7. Si se olvida una dosis, indicar al paciente que se administre una dosis tan pronto como se acuerde. A partir de entonces, el paciente puede reanudar la dosificación en su día habitual de administración o comenzar un nuevo programa semanal desde el día en que se administró la dosis omitida.
Uso en poblaciones específicas:
Uso geriátrico: Los estudios clínicos de BENLYSTIA® no incluyeron un número suficiente de sujetos de 65 años o más para determinar si responden de manera diferente a los sujetos más jóvenes. Usar con precaución en pacientes de edad avanzada.
Insuficiencia renal: La seguridad y eficacia de BENLYSTIA® SC se evaluaron en estudios que incluyeron pacientes con LES que presentaban insuficiencia renal leve (aclaramiento creatinina [CrCl] ≥ 60 y < 90 mL/min), moderada (CrCl ≥ 30 y < 60 mL/min) o grave (CrCl ≥ 15 y < 30 mL/min). No se recomienda ajustar la dosis en pacientes con insuficiencia renal.
Insuficiencia hepática: No se realizaron estudios formales para examinar los efectos de la insuficiencia hepática sobre la farmacocinética de belimumab. No se recomienda ajustar la dosis en pacientes con insuficiencia hepática.
Grupos raciales: En el Estudio 7 (dosis subcutánea), la respuesta del SRI-4 fue del 45% (26/58) en pacientes de raza negra que recibieron BENLYSTIA® SC más terapia estándar en comparación con 39% (13/33) en pacientes de raza negra que recibieron placebo más terapia estándar (véase Estudios clínicos - Administración subcutánea).
El perfil de seguridad de BENLYSTIA® SC en pacientes de raza negra fue consistente con el perfil de seguridad conocido de BENLYSTIA® SC administrado en la población en general.
MANIFESTACIONES Y MANEJO DE LA SOBREDOSIFICACIÓN O INGESTA ACCIDENTAL:
Existe una experiencia limitada con la sobredosis de belimumab.
PRESENTACIONES:
Caja de cartón con 1, 4 o 12 jeringa(s) prellenada(s) (jeringa de seguridad) con 1 mL e instructivo anexo.
Caja de cartón con 1, 4 o 12 pluma(s) precargada(s) (autoinyector) con 1 mL e instructivo anexo.
RECOMENDACIONES SOBRE ALMACENAMIENTO:
Antes de administrar:
Consérvese la caja bien cerrada, en refrigeración entre 2 °C y 8 °C. No se congele. Protéjase de la luz. Almacenar en la caja original hasta su uso. No agitar.
Manténgase en refrigeración hasta 30 minutos antes de su uso. No se use si ha estado por más de 12 horas a temperatura ambiente. No se use si éste cae sobre una superficie dura.
LEYENDAS DE PROTECCIÓN:
Literatura exclusiva para médicos. Su venta requiere receta médica. No se deje al alcance de los niños. No se administre si el cierre ha sido violado. No se administre si la solución no es transparente, o si contiene partículas en suspensión o sedimentos. No se use en el embarazo y la lactancia. No se use en menores de 18 años. Retire la tapa hasta justo antes de la inyección.
Reporte las sospechas de reacción adversa a los correos:
farmacovigilancia@cofepris.gob.mx y
farmacovigilancia.mx@gsk.com
Titular del Registro Sanitario:
Glaxo Wellcome, S.A.
Avda. Extremadura, 3, Pol. Ind. Allendeduero,
Aranda de Duero, Burgos, 09400, España.
Representante Legal:
GLAXOSMITHKLINE MÉXICO, S.A. de C.V.
Autopista México-Querétaro km. 41.5 Edif. TR9,
Interior 5-C, Ex. Hacienda San Miguel,
C.P. 54715, Cuautitlán Izcalli, México, México.
Reg. Núm. 164M2022 SSA IV
®Marca registrada