
APLEEK PATCH
ETINILESTRADIOL, GESTODENO
Parche transdérmico
1 Caja, 3 Parches,
1 Caja, 9 Parches,
FORMA FARMACÉUTICA Y FORMULACIÓN:
Cada parche transdérmico de 11 cm2 contiene:
Gestodeno 2.1 mg
Etinilestradiol 0.55 mg
Excipiente, c.b.p. 1 parche.
INDICACIONES TERAPÉUTICAS: Anticonceptivo hormonal.
FARMACOCINÉTICA Y FARMACODINAMIA:
Propiedades farmacodinámicas:
En dos estudios realizados en la Unión Europea, se investigó la inhibición de la ovulación durante 3 ciclos de tratamiento usando un régimen de administración de 3 semanas del parche APLEEK PATCH® en mujeres sanas con índice de masa corporal (IMC) normal. Se demostró el efecto antigonadotrópico supresor adecuado del desarrollo folicular e inhibición confiable de la ovulación. En un tercer estudio sobre inhibición de la ovulación llevado a cabo en los Estados Unidos de América (EUA), se evaluó el parche APLEEK PATCH® en mujeres con IMC normal, con sobrepeso y obesas. No se observó ninguna diferencia clínicamente significativa en la supresión de la actividad ovárica entre los tres grupos.
Adicionalmente se demostró, evaluando los cambios físicos del cuello uterino, la cantidad y filancia del moco cervical, y la ocurrencia del fenómeno de arborización (“helecho”), que el moco cervical se volvía más espeso, lo que evita la penetración de los espermatozoides a través del canal cervical.
Eficacia y seguridad clínicas:
En el estudio clínico llevado a cabo con el parche APLEEK PATCH® en mujeres de la Unión Europea, América Latina y Australia, se obtuvieron en conjunto, los siguientes Índices de Pearl:
Índice de Pearl corregido (de 18 a 35 años de edad, índice de masa corporal [IMC] £ 30 kg/m2): 0.82 (límite superior del intervalo de confianza de 95% 1.55).
Índice de Pearl no corregido (falla del método y de la usuaria): 1.19 (límite superior del intervalo de confianza de 95% 2.00)
Se calcularon los índices de Pearl siguientes solo para la población europea:
Índice de Pearl corregido (de 18 a 35 años de edad, índice de masa corporal [IMC] £ 30 kg/m2): 0.40 (límite superior del intervalo de confianza de 95% 1.18).
Índice de Pearl no corregido 0.76 (límite superior del intervalo de confianza de 95% 1.66)
Se investigó la histología del endometrio en 49 mujeres en un estudio clínico después de 13 ciclos de tratamiento. No hubo resultados anómalos.
Se evaluaron los parámetros de patrón de sangrado y control del ciclo en dos estudios: uno en EUA en un periodo de siete ciclos de tratamiento y otro en la Unión Europea, América Latina y Australia, en un periodo de 13 ciclos de tratamiento. El tratamiento con el parche APLEEK PATCH® proporciona un buen control del ciclo, con presencia de sangrado por privación en 94.3 a 97.6% de las mujeres después de los 3 ciclos de tratamiento iniciales (en estos ciclos iniciales, el sangrado por privación estuvo presente en 91.2 a 93.7% de las mujeres). Solo una minoría de mujeres, alrededor de 1% por ciclo, no tuvieron sangrado por privación. La intensidad del sangrado fue comparable a la producida con un AOC con 100 µg de levonorgestrel y 20 µg de etinilestradiol y con otro parche de control de la fertilidad con 6 mg de norelgestromina y 600 µg de etinilestradiol.
Propiedades farmacocinéticas:
Absorción:
Después de la aplicación dérmica del parche APLEEK PATCH®, el etinilestradiol y el gestodeno se absorben bien a través de la piel. La liberación promedio del etinilestradiol y el gestodeno en el periodo de aplicación de 7 días del parche APLEEK PATCH® tiene como resultado la misma exposición sistémica (área bajo la curva, ABC) durante el estado estable que la observada después de la administración diaria de un anticonceptivo oral combinado con 0.02 mg de etinilestradiol y 0.06 mg de gestodeno.
Se determinaron las concentraciones en suero del etinilestradiol y el gestodeno durante la tercera semana en diferentes ciclos de tratamiento (ciclo 1 a ciclo 7). Se alcanzaron las concentraciones máximas medias de etinilestradiol en el rango de 36 a 51 ng/L alrededor de 1 día después de la aplicación dérmica del parche. Luego, las concentraciones disminuyeron a valores mínimos medios en el rango de 15 a 23 ng/L al final del periodo de aplicación de 1 semana. La concentración promedio durante la semana 3 se ubicó en el rango de 22 a 33 ng/L.
Se alcanzaron las concentraciones máximas medias de gestodeno total en el rango de 4.7 a 7.5 µg/L alrededor de 1 a 1.5 días después de la aplicación dérmica del parche. Luego, las concentraciones disminuyeron a valores mínimos medios en el rango de 2.6 a 4.0 µg/L al final de la primer semana de aplicación. La concentración promedio durante la semana 3 se ubicó en el rango de 3.6 a 5.7 µg/L.
Influencia del índice de masa corporal (IMC):
Las concentraciones en suero de etinilestradiol y gestodeno durante el tratamiento con el parche APLEEK PATCH® dependen del IMC de la mujer. En mujeres obesas con un IMC > 35 kg/m2, las concentraciones promedio de etinilestradiol y gestodeno son 24 y 30% más bajas, respectivamente, que las observadas en mujeres con un IMC normal de £ 30 kg/m2. Esto no condujo a resultados diferentes en la farmacodinámica y la eficacia en las mujeres obesas.
Influencia del calor, la humedad y el ejercicio: Se investigó la farmacocinética del etinilestradiol y el gestodeno después de la aplicación del parche APLEEK PATCH® en condiciones específicas de calor, humedad y ejercicio, es decir, sauna, piscina de hidromasaje, natación y ejercicios físicos diferentes en comparación con la actividad normal. En general, se demostró la bioequivalencia para los parámetros de Cmáx. y ABC del etinilestradiol y el gestodeno en estas condiciones específicas. Los resultados no demuestran una diferencia clínicamente relevante en la exposición del parche con etinilestradiol y gestodeno a las condiciones específicas de un club deportivo tales como sauna, piscina de hidromasaje, natación y diferentes ejercicios físicos en comparación con las actividades normales de la vida cotidiana.
Datos comparativos con otros anticonceptivos hormonales combinados: En un estudio de biodisponibilidad relativa, se compararon las concentraciones en suero en estado estable y los parámetros farmacocinéticos del etinilestradiol y el gestodeno después de la aplicación del parche APLEEK PATCH® con las obtenidas con un anticonceptivo oral combinado con 0.020 mg de etinilestradiol y 0.075 mg de gestodeno. Los valores medios en estado estable de Cmáx. del etinilestradiol y el gestodeno fueron 30 a 40% menores después de la aplicación del parche APLEEK PATCH® en comparación con el anticonceptivo oral combinado. La exposición (ABC y Cav) al etinilestradiol fue comparable entre ambas vías de administración, mientras que la exposición al gestodeno (concentración sin unión) fue 18% menor después de la aplicación del parche APLEEK PATCH®. Estos datos dieron como resultado estimaciones promedio de exposición/dosis para el parche APLEEK PATCH® iguales a la administración oral diaria de 0.020 mg de etinilestradiol y 0.060 mg de gestodeno. La variabilidad interindividual (%CV) de los parámetros farmacocinéticos importantes como Cmáx. y ABC después de la aplicación del parche APLEEK PATCH® fue menor para el etinilestradiol pero mayor para el gestodeno en relación con aquellos determinados después de la administración oral.
En un estudio que investigó el patrón de sangrado después de la aplicación del parche APLEEK PATCH® en comparación con otro parche de control de la fertilidad (con 6 mg de norelgestromina y 600 µg de etinilestradiol) aplicado durante un periodo de 7 ciclos y que usó los tres lugares de aplicación, abdomen, nalgas o parte superior del brazo, se midieron las concentraciones en suero del etinilestradiol mediante la toma de muestras periódicas durante el periodo completo del estudio. La evaluación farmacocinética de las concentraciones de etinilestradiol en la población tuvo como resultado un valor medio de Cav en el rango de 29.7 a 40.7 ng/L para el parche APLEEK PATCH® y más de 2 veces los valores medios más altos en el intervalo de 75.8 a 91.3 ng/L para el otro parche de control de la fertilidad.
Distribución: El etinilestradiol se une ávida pero no específicamente a la albúmina del suero (aprox. 98%) aunque no a la globulina fijadora de hormonas sexuales (GFHS). El gestodeno se une extensamente a la albúmina del suero y la GFHS. Solo alrededor de 1% de las concentraciones en suero totales del fármaco está presente como esteroide libre, 40 a 80% se une a la GFHS. El etinilestradiol induce un notable aumento en las concentraciones en suero de la GFHS mientras que la administración de gestodeno produce un leve descenso de la concentración de GFHS. Después de la aplicación dérmica repetida del parche APLEEK PATCH®, las concentraciones medias en suero de GFHS en estado estable se ubican en un rango de 201 a 237 nmol/L.
Después de la administración intravenosa del etinilestradiol, se determinó un volumen de distribución aparente de 39 L/kg. El volumen de distribución aparente respectivo del gestodeno es de aproximadamente 0.7 L/kg.
Metabolismo/biotransformación: El etinilestradiol se metaboliza principalmente mediante hidroxilación aromática, formando una amplia variedad de metabolitos hidroxilados y metilados, y éstos están presentes como metabolitos libres y como conjugados con glucurónidos y sulfato. La vía de metabolismo más importante del etinilestradiol es la 2-hidroxilación dependiente de CYP450 y la formación de 2-hidroxi-estrógeno catecol del EE. La 2-hidroxilación del EE se cataliza mediante las familias genéticas CYP2C, CYP2E y CYP3A. La tasa de depuración metabólica se ubica en el rango de 2 a 7 ml/min/kg.
El gestodeno se metaboliza completamente en metabolitos generalmente más polares. El metabolismo del gestodeno se caracteriza por la hidroxilación en diversas posiciones del núcleo esteroide y por la reducción de la función 3-ceto y la doble unión delta-4. No se han descrito metabolitos activos. Además de CYP3A4, un grupo de otras enzimas del citocromo P450 puede contribuir en una menor medida al metabolismo del gestodeno.
Eliminación/excreción: El etinilestradiol no se excreta en su forma original -sin cambios- en ningún grado significativo. Los metabolitos del etinilestradiol se excretan en una relación orina:bilis de 4:6. El descenso de la concentración del suero se caracteriza por al menos dos fases de disposición con una vida media terminal de alrededor de 16 horas, determinada después de la administración intravenosa que produce generalmente concentraciones no cuantificables dos días después del retiro del parche.
El gestodeno no se excreta en su forma original -sin cambios-. Sus metabolitos se excretan en una relación orina:bilis de 6:4. Después del retiro del parche, las concentraciones en suero totales del gestodeno disminuyen más lentamente en comparación con el etinilestradiol con una vida media terminal media de alrededor de 26 horas.
Linealidad/no linealidad: La farmacocinética del etinilestradiol es lineal con respecto a la dosis en el rango de 20-100 µg. No se ha observado ningún cambio clínicamente relevante en la farmacocinética del etinilestradiol con el tiempo.
La farmacocinética del gestodeno depende de la concentración de la GFHS que se ve afectada por los estrógenos, andrógenos y también por el gestodeno. Después de la aplicación dérmica repetida del parche APLEEK PATCH®, se observan concentraciones 3 a 4 veces superiores de GFHS en comparación con los valores iniciales. Asimismo, los niveles en suero del gestodeno en estado estable difieren de aquellos posteriores a la aplicación única. Estos cambios dependientes de GFHS producen un cambio no lineal en la farmacocinética del gestodeno con el tiempo. Además, se consideró que la farmacocinética del gestodeno no unido dependía de su concentración según tres estudios que investigaron la farmacocinética del parche APLEEK PATCH® en un periodo de tres ciclos. Así, la farmacocinética del gestodeno se considera no lineal con respecto al tiempo y a la concentración.
Información adicional sobre poblaciones especiales:
Pacientes pediátricas: El parche APLEEK PATCH® está indicado después de la menarca solamente. El parche APLEEK PATCH® no se ha estudiado en mujeres menores de 18 años de edad.
Pacientes geriátricas: No aplica. El parche APLEEK PATCH® no está indicado después de la menopausia.
Pacientes con insuficiencia hepática: El parche APLEEK PATCH® no se ha estudiado en mujeres con insuficiencia hepática. El parche APLEEK PATCH® está contraindicado en mujeres con enfermedades hepáticas severas, véase Contraindicaciones.
Pacientes con insuficiencia renal: El parche APLEEK PATCH® no se ha estudiado en mujeres con insuficiencia renal. Debido a la metabolización completa del etinilestradiol y gestodeno en metabolitos inactivos antes de la eliminación y debido a la disponibilidad de una segunda vía de eliminación a través del hígado, no se espera que aumente el riesgo para las mujeres con insuficiencia renal.
Sexo: El parche APLEEK PATCH® está indicado en mujeres únicamente.
Tabaquismo: No hay ninguna evidencia de que el tabaquismo tenga un impacto en la farmacocinética del etinilestradiol y gestodeno.
Diferencias étnicas: Se estudió la farmacocinética del etinilestradiol en combinación con otra progestina en mujeres caucásicas, chinas y japonesas, y no reveló ninguna diferencia clínicamente significativa. No se ha estudiado la farmacocinética del parche APLEEK PATCH® específicamente en mujeres de etnias diferentes. No se conocen enzimas polimórficas que contribuyan en una medida importante en la metabolización del gestodeno. De este modo, los datos disponibles no indican ninguna diferencia en la farmacocinética del parche APLEEK PATCH® entre mujeres de diferentes etnias.
Otras poblaciones especiales: No aplica.
Relaciones farmacocinéticas/farmacodinámicas: Se estudió la relación farmacocinética/farmacodinámica en un estudio de inhibición de la ovulación que investigó el parche APLEEK PATCH® y dos formulaciones de parches en desarrollo con dosis menores de etinilestradiol y gestodeno en tres ciclos. La evaluación farmacocinética/farmacodinámica poblacional de los datos mostró una relación exposición-respuesta entre el ABC del gestodeno no unido y la probabilidad de no observar ovulación ni folículos luteinizados no rotos (LUF). De esta forma, el gestodeno se reveló como el supresor primario de la actividad ovárica con el aporte adicional del etinilestradiol. En tasas de exposición alrededor de la exposición media del gestodeno no unido durante la aplicación del parche APLEEK PATCH®, la probabilidad de no observar ovulación o LUF es mayor a 98%, y está aún por encima de 95% en el extremo inferior de exposición a gestodeno no unido (percentil 5%) durante la aplicación del parche APLEEK PATCH®.
CONTRAINDICACIONES: No deben usarse anticonceptivos hormonales combinados incluido el parche APLEEK PATCH® en presencia de cualquiera de las afecciones que se enuncian a continuación. Si alguna de las afecciones se presenta por primera vez durante el uso del parche APLEEK PATCH®, debe retirarse el parche de inmediato.
• Presencia o antecedente de eventos trombóticos/tromboembólicos venosos o arteriales (por ejemplo, trombosis venosa profunda, embolismo pulmonar o infarto de miocardio) o de un accidente cerebrovascular.
• Presencia o antecedente de pródromos de trombosis (por ejemplo, ataque isquémico transitorio, angina de pecho).
• Riesgo elevado de trombosis venosa o arterial (véase Precauciones generales).
• Antecedentes de migraña con síntomas neurológicos focales.
• Diabetes mellitus con afectación vascular.
• Hepatopatía severa mientras los valores del funcionamiento hepático no hayan regresado a la normalidad.
• Presencia o antecedente de tumor de hígado (benigno o maligno).
• Enfermedades malignas influidas por esteroides sexuales, conocidas o presuntas (por ejemplo, de los órganos genitales o las mamas).
• Sangrado vaginal no diagnosticado.
• Hipersensibilidad a los principios activos o a cualquiera de los excipientes.
RESTRICCIONES DE USO DURANTE EL EMBARAZO Y LA LACTANCIA:
Embarazo: El parche APLEEK PATCH® no está indicado durante el embarazo. Si se produce un embarazo durante el uso del parche APLEEK PATCH®, hay que retirar el parche y suspender su uso. No obstante, los estudios epidemiológicos exhaustivos no han revelado un riesgo mayor de defectos de nacimiento en niños nacidos de mujeres que utilizaron anticonceptivos hormonales combinados antes del embarazo ni un efecto teratogénico cuando se usaron anticonceptivos hormonales combinados inadvertidamente durante el embarazo temprano.
Lactancia: La lactancia puede verse afectada por anticonceptivos hormonales combinados ya que pueden reducir la cantidad y modificar la composición de la leche materna. Por ende, en general no debe recomendarse el uso de anticonceptivos hormonales combinados hasta que la madre en periodo de lactancia haya destetado por completo a su hijo. Se pueden excretar cantidades pequeñas de esteroides anticonceptivos y/o sus metabolitos en la leche materna.
REACCIONES SECUNDARIAS Y ADVERSAS:
Resumen del perfil de seguridad: Las reacciones adversas informadas con más frecuencia con el parche APLEEK PATCH® son las reacciones en el lugar de aplicación. Se producen en > 10% de las usuarias. Las reacciones adversas severas poco frecuentes son la tromboembolia arterial y venosa.
Lista tabulada de reacciones adversas: Las frecuencias de las reacciones adversas al medicamento informadas en los estudios clínicos con el parche APLEEK PATCH® (n = 3,573) se resumen en la tabla a continuación. Dentro de cada agrupamiento de frecuencia, las reacciones adversas al medicamento se presentan en orden de severidad decreciente. Las frecuencias se definen como muy frecuentes (³ 1/10), frecuentes (³ 1/100 a < 1/10), infrecuentes (³ 1/1,000 a < 1/100) y raras (³ 1/10,000 a < 1/1,000).
Tabla 2
|
Clase de sistema u órgano (MedDRA) |
Muy frecuentes |
Frecuentes |
Infrecuentes |
Raros |
|
Trastornos psiquiátricos |
Labilidad emocional |
Depresión/humor deprimido, disminución y pérdida de la libido |
||
|
Trastornos del sistema nervioso |
Migraña |
|||
|
Trastornos vasculares |
Eventos tromboembólicos venosos y arteriales* |
|||
|
Trastornos gastrointestinales |
Náuseas |
|||
|
Trastornos de la piel y del tejido subcutáneo |
Reacción en el lugar de aplicación |
|||
|
Trastornos del aparato reproductor y de las mamas |
Sangrado del tracto genital**, dolor de mamas |
Los eventos adversos en los estudios clínicos se codificaron usando el diccionario MedDRA (versión 14.1). Se han agrupado los términos diferentes del diccionario MedDRA que representan el mismo fenómeno médico como reacciones adversas únicas para evitar diluir u oscurecer el efecto verdadero.
* Frecuencia estimada de estudios epidemiológicos que incluyen un grupo de anticonceptivos orales combinados. La frecuencia fue limitada a muy raros.
- El término “Eventos tromboembólicos venosos y arteriales” resume las siguientes entidades médicas: oclusión venosa profunda periférica, trombosis y embolismo/oclusión vascular pulmonar, trombosis, embolismo e infarto/infarto de miocardio/infarto cerebral y accidente cerebrovascular no especificado como hemorrágico.
** Contiene las entidades médicas: sangrado del tracto genital femenino, sangrado uterino no programado.
Se utiliza el término preferido del diccionario MedDRA para describir una reacción determinada y sus sinónimos y afecciones relacionadas. La representación de las reacciones adversas al medicamento se basa en la versión 14.1 del MedDRA.
Descripción de reacciones adversas selectas: Las reacciones adversas con muy baja frecuencia o con inicio demorado de síntomas que se consideran relacionadas con el grupo de anticonceptivos hormonales combinados, incluidos los AOC, se enumeran a continuación (véanse Contraindicaciones, Advertencias y precauciones especiales de empleo):
Tumores:
• La frecuencia de diagnóstico de cáncer de mama aumenta muy levemente entre las usuarias de anticonceptivos hormonales combinados. Debido a que el cáncer de mama es poco frecuente en las mujeres menores de 40 años de edad, el exceso en la cantidad de casos diagnosticados es pequeño en comparación con el riesgo general de cáncer de mama. Se desconoce la causalidad con el uso de anticonceptivos hormonales combinados.
• Tumores hepáticos (benignos y malignos).
Otras afecciones:
• Eritema nodoso, eritema multiforme.
• Mujeres con hipertrigliceridemia (riesgo mayor de pancreatitis cuando se usan AOC).
• Hipertensión arterial.
• Ocurrencia o empeoramiento de afecciones cuya asociación con el uso de AOC no es concluyente: ictericia y/o prurito relacionados con colestasis; formación de cálculos biliares; porfiria; lupus eritematoso sistémico; síndrome urémico hemolítico; corea de Sydenham; herpes gestacional; pérdida de la audición asociada con otosclerosis.
• En mujeres con angioedema hereditario, los estrógenos exógenos pueden inducir o exacerbar los síntomas de angioedema.
• Alteraciones de la función hepática.
• Cambios en la tolerancia a la glucosa o efecto en la resistencia periférica a la insulina.
• Enfermedad de Crohn, colitis ulcerosa.
• Cloasma.
• Hipersensibilidad (incluidos síntomas como erupción cutánea, urticaria).
Interacciones:
El sangrado intermenstrual y/o la falla anticonceptiva pueden producirse como consecuencia de interacciones de otros fármacos (inductores de enzimas, algunos antibióticos) con los anticonceptivos hormonales combinados (véase Interacciones medicamentosas y de otro género).
PRECAUCIONES EN RELACIÓN CON EFECTOS DE CARCINOGÉNESIS, MUTAGÉNESIS, TERATOGÉNESIS Y SOBRE LA FERTILIDAD: Los datos preclínicos sobre los principios activos no revelan riesgos especiales para los humanos, teniendo en cuenta estudios convencionales de toxicidad de dosis repetidas, genotoxicidad, tolerabilidad local, potencial carcinogénico y toxicidad para la reproducción. Sin embargo, se debe tener en cuenta que los esteroides sexuales pueden estimular el crecimiento de determinados tejidos y tumores dependientes de hormonas. Los estudios de biocompatibilidad con el parche y sus materiales no revelaron riesgos especiales con respecto al uso pretendido del parche en humanos.
INTERACCIONES MEDICAMENTOSAS Y DE OTRO GÉNERO:
Efectos de otros medicamentos en el parche APLEEK PATCH®:
Las interacciones de otros fármacos (inductores de enzimas, algunos antibióticos) con anticonceptivos hormonales combinados puede conducir a sangrado intermenstrual y/o a la falla anticonceptiva. Las mujeres en tratamiento con alguno de estos fármacos deben usar temporalmente un método de barrera además del parche APLEEK PATCH® o elegir otro método de anticoncepción. Con los fármacos inductores de enzimas microsomales, debe usarse el método de barrera durante el periodo de administración del fármaco concomitante y durante 28 días después de su interrupción.
Sustancias que disminuyen la eficacia de los anticonceptivos hormonales combinados (inductores de enzimas y antibióticos):
Inducción de enzimas (aumento del metabolismo hepático): Se pueden producir interacciones con los fármacos que inducen las enzimas microsomales que pueden producir un aumento de la depuración de hormonas sexuales (por ejemplo, fenitoína, barbitúricos, primidona, carbamazepina, rifampicina y posiblemente además oxcarbazepina, topiramato, felbamato, griseofulvina y productos con Hierba de San Juan).
Asimismo, se ha informado que los inhibidores de la proteasa del VIH (por ejemplo, ritonavir) y los inhibidores no nucleósidos de la transcriptasa inversa (por ejemplo, nevirapina) y combinaciones de los mismos, aumentan potencialmente el metabolismo hepático.
Antibióticos (interferencia con la circulación enterohepática): Algunos informes clínicos sugieren que la circulación enterohepática de estrógenos puede disminuir cuando se administran ciertos agentes antibióticos, que pueden reducir las concentraciones de etinilestradiol (por ejemplo, penicilinas, tetraciclinas).
Las mujeres en tratamiento con antibióticos (excepto los inductores de enzima como rifampicina y griseofulvina) deben usar el método de barrera hasta 7 días después de la interrupción. Si el periodo durante el cual se usa el método de barrera supera el tercer parche de un ciclo de aplicación, el próximo parche debe iniciarse sin el intervalo sin parche.
Sustancias que aumentan las concentraciones de los fármacos del parche APLEEK PATCH® (inhibidores de enzimas):
En dos estudios que investigan el efecto de los inhibidores de CYP3A4 (ketoconazol, eritromicina), las concentraciones séricas de etinilestradiol en estado estable no se vieron afectadas por ninguno de los dos inhibidores. Para el gestodeno, la coadministración de los inhibidores tuvo como resultado un aumento de 11 y de 34% del ABC(0-168) (Área Bajo la Curva) para el ketoconazol y la eritromicina, respectivamente. Este pequeño aumento, que tiene como resultado la exposición dentro del rango de los anticonceptivos orales combinados comercializados, no se considera clínicamente relevante.
Efectos de los anticonceptivos hormonales combinados en otros medicamentos: Los anticonceptivos hormonales combinados pueden afectar el metabolismo de algunos otros fármacos. De manera acorde, las concentraciones en plasma y tejidos pueden aumentar (por ejemplo, ciclosporina) o disminuir (por ejemplo, lamotrigina).
En un estudio que investiga el efecto del parche APLEEK PATCH® en la administración única de midazolam, un sustrato modelo de las sustancias que son metabolizadas por CYP3A4, no se observó ningún aumento clínicamente relevante en las concentraciones plasmáticas del midazolam. La coadministración de midazolam produjo un aumento menor de 7% y de 14% en el ABC(0-últ) y la Cmáx. del midazolam, respectivamente.
ALTERACIONES EN LOS RESULTADOS DE PRUEBAS DE LABORATORIO: El uso de esteroides anticonceptivos puede influir en los resultados de ciertas pruebas de laboratorio, incluidos parámetros bioquímicos de la función hepática, tiroidea, suprarrenal y renal, los niveles plasmáticos de proteínas (transportadoras), por ejemplo, globulina fijadora de corticosteroides y fracciones de lípidos/lipoproteínas, parámetros del metabolismo de carbohidratos y parámetros de coagulación y fibrinólisis. En general, los cambios se mantienen dentro del intervalo normal de laboratorio.
Nota: Se debe consultar la información para prescribir de los medicamentos concomitantes, a fin de identificar posibles interacciones.
PRECAUCIONES GENERALES: La experiencia clínica se limita a población adulta con poca experiencia en población menor de 18 años, por lo que la prescripción de este producto en dicha población deberá ser a criterio del médico tratante quien adicionalmente, deberá alentar sobre la planificación familiar y en la selección del método más adecuado para cada paciente.
La mayor parte de las advertencias y precauciones siguientes se basa en hallazgos de los AOC que, en principio, pueden esperarse para otros anticonceptivos hormonales combinados incluido el parche APLEEK PATCH®. No obstante, no se conoce si el parche APLEEK PATCH® aplicado transdérmicamente influye en el riesgo de eventos tromboembólicos en comparación con los anticonceptivos orales combinados.
Advertencias: Si está presente alguna de las afecciones/factores de riesgo mencionados a continuación, deben sopesarse los beneficios del uso de un anticonceptivo hormonal combinado contra los posibles riesgos para cada mujer en particular, y discutirse con la mujer antes de que decida comenzar a usarlo.
Trastornos circulatorios: Los estudios epidemiológicos han sugerido una asociación entre el uso de AOC y un riesgo mayor de enfermedades trombóticas y tromboembólicas arteriales y venosas como infarto de miocardio, trombosis venosa profunda, embolismo pulmonar y accidentes cerebrovasculares. Estos eventos ocurren raramente.
El riesgo de TEV es mayor durante el primer año de uso. Este mayor riesgo está presente después de comenzar el tratamiento inicial con un AOC o de reanudar el tratamiento (tras un intervalo de 4 semanas o más sin tomarlo) con el mismo AOC u otro distinto. Los datos de un estudio prospectivo grande con una cohorte en 3 grupos sugieren que este mayor riesgo está presente principalmente durante los primeros 3 meses.
El riesgo general de tromboembolia venosa (TEV) en las usuarias de un AOC de baja dosis de estrógeno (< 50 µg de etinilestradiol) es de dos a tres veces mayor que en quienes no usan un AOC y que no están embarazadas, y sigue siendo más bajo que el riesgo asociado con el embarazo y el parto.
La TEV puede poner en riesgo la vida o tener un desenlace mortal (en 1-2% de los casos).
Puede ocurrir tromboembolia venosa (TEV), manifestada en forma de una trombosis venosa profunda y/o embolismo pulmonar, durante el uso de todos los AOC.
Se han registrado casos extremadamente raros de trombosis en otros vasos sanguíneos, por ejemplo, venas y arterias hepáticas, mesentéricas, renales, cerebrales o retinales, en usuarias de AOC. No existe consenso acerca de si estos eventos están asociados con el uso de AOC.
Los síntomas de la trombosis venosa profunda (TVP) pueden incluir: hinchazón de una pierna o a lo largo de una vena de la pierna; dolor o sensibilidad en la pierna que puede sentirse solamente al ponerse de pie o al caminar; aumento del calor en la pierna afectada, y piel enrojecida o descolorida en la pierna.
Los síntomas del embolismo pulmonar (EP) pueden incluir: aparición repentina de falta de aire o respiración rápida sin razón aparente; tos repentina con posible esputo de sangre; dolor agudo en el pecho, que puede intensificarse al respirar profundamente; sensación de ansiedad; mareo o aturdimiento intenso, y frecuencia cardiaca rápida o irregular. Algunos de estos síntomas (por ejemplo, la “falta de aire” y la “tos”) no son específicos y podrían malinterpretarse como eventos más frecuentes o menos graves (por ejemplo, infecciones de las vías respiratorias).
Un evento tromboembólico arterial puede incluir un accidente cerebrovascular, una oclusión vascular o un infarto de miocardio (IM). Los síntomas del accidente cerebrovascular pueden incluir: entumecimiento o debilidad repentina de la cara, un brazo o una pierna, especialmente de un solo lado del cuerpo; confusión, dificultad para hablar o entender que comienza repentinamente; dificultad repentina para ver con un ojo o con ambos; dificultad para caminar; mareo, pérdida repentina del equilibrio o la coordinación; cefalea repentina, intensa o prolongada, sin causa conocida; pérdida del conocimiento o desmayo con o sin convulsión. Otros signos de oclusión vascular pueden incluir: dolor repentino, hinchazón y decoloración azulada en una extremidad; abdomen agudo.
Los síntomas del IM pueden incluir: dolor, malestar, presión, pesadez, sensación de opresión o llenura en el pecho, un brazo, o debajo del esternón; molestia que se irradia a la espalda, la quijada, la garganta, un brazo, el estómago; llenura, indigestión o sensación de atragantamiento; sudor, náuseas, vómitos o mareos; debilidad extrema, ansiedad o falta de aire; frecuencia cardiaca rápida o irregular.
Los eventos tromboembólicos arteriales pueden poner en riesgo la vida o tener un desenlace mortal.
Debe tenerse en cuenta el potencial de un aumento del riesgo sinérgico de trombosis en las mujeres que tienen una combinación de factores de riesgo o que presentan un factor de riesgo individual de mayor seriedad. Este aumento del riesgo puede ser mayor que un simple riesgo acumulado de los factores individuales. No se debe prescribir un anticonceptivo hormonal combinado si la evaluación riesgo-beneficio es negativa, (véase Contraindicaciones).
El riesgo de eventos trombóticos/tromboembólicos venosos o arteriales o de un accidente cerebrovascular aumenta con:
• La edad.
• La obesidad (índice de masa corporal superior a 30 kg/m2).
• Antecedentes familiares positivos (es decir, tromboembolismo arterial o venoso en un hermano o progenitor a una edad relativamente joven). Si se sabe o sospecha de una predisposición hereditaria, se debe remitir a la mujer a un especialista para que aporte su opinión antes de decidir si usar un anticonceptivo hormonal combinado o no.
• Inmovilización prolongada, cirugía mayor, cualquier cirugía en las piernas o traumatismo mayor. En estas situaciones se aconseja interrumpir el uso del parche APLEEK PATCH® (en caso de cirugía programada, al menos cuatro semanas antes) y no reanudar el tratamiento hasta dos semanas después de recuperar por completo la movilidad.
• Tabaquismo (el riesgo aumenta aún más en quienes fuman mucho y con la edad, especialmente en mujeres mayores de 35 años).
• Dislipoproteinemia.
• Hipertensión arterial.
• Migraña.
• Cardiopatía valvular.
• Fibrilación auricular.
No hay consenso acerca del posible papel de las venas varicosas y la tromboflebitis superficial en la tromboembolia venosa.
Debe considerarse el incremento en el riesgo de tromboembolia en el puerperio (para información sobre embarazo y lactancia, véase Restricciones de uso durante el embarazo y la lactancia). Otras afecciones médicas que se han asociado con eventos circulatorios adversos incluyen: diabetes mellitus, lupus eritematoso sistémico, síndrome hemolítico-urémico, enfermedad intestinal inflamatoria crónica (enfermedad de Crohn o colitis ulcerosa) y anemia falciforme.
Un aumento en la frecuencia o intensidad de las migrañas durante el uso de un anticonceptivo hormonal combinado (lo que puede ser prodrómico de un evento cerebrovascular) puede justificar la interrupción inmediata de ese medicamento.
Los factores bioquímicos que pueden ser indicativos de una predisposición hereditaria o adquirida a la trombosis venosa o arterial incluyen: resistencia a la proteína C activada (APC), hiperhomocisteinemia, deficiencia de antitrombina III, deficiencia de proteína C, deficiencia de proteína S, anticuerpos antifosfolípidos (anticuerpos anticardiolipina, anticoagulante lúpico).
Al ponderar el riesgo-beneficio, el médico debe tener en cuenta que el tratamiento adecuado de una enfermedad puede reducir el riesgo asociado de trombosis y que el riesgo asociado con un embarazo es mayor que el asociado con los anticonceptivos hormonales combinados de baja dosis (< 0.05 mg de etinilestradiol).
Tumores: El factor de riesgo más importante de cáncer del cuello uterino es la infección por VPH persistente. Algunos estudios epidemiológicos han indicado que el uso de AOC a largo plazo puede aumentar este riesgo, pero continúa habiendo controversia acerca del grado por el cual este hallazgo es atribuible a efectos de confusión, por ejemplo, la exploración del cuello uterino y el comportamiento sexual, incluido el uso de anticonceptivos de barrera.
Un meta-análisis de 54 estudios epidemiológicos indicó que existe un riesgo relativo ligeramente mayor (RR = 1.24) de diagnóstico de cáncer de mama en mujeres que están usando un AOC. El exceso de riesgo disminuye gradualmente a lo largo de 10 años tras la interrupción del uso de AOC. Debido a que el cáncer de mama es poco frecuente en las mujeres menores de 40 años de edad, el exceso en la cantidad de diagnósticos de cáncer de mama en usuarias actuales y recientes de AOC es pequeño en comparación con el riesgo general de cáncer de mama. Estos estudios no aportan pruebas de la causa. El patrón observado de aumento en el riesgo puede deberse a un diagnóstico anterior de cáncer de mama en usuarias de AOC, a los efectos biológicos de los AOC o a una combinación de ambos factores. Los cánceres de mama diagnosticados en las mujeres que usaron AOC tienden a ser menos clínicamente avanzados que los cánceres diagnosticados en las mujeres que nunca los usaron.
Se ha informado de casos poco frecuentes de tumores hepáticos benignos y de casos aún más infrecuentes de tumores hepáticos malignos en usuarias de AOC. En casos aislados, esos tumores produjeron hemorragia intraabdominal que puso en riesgo la vida. Debe considerarse un tumor hepático en el diagnóstico diferencial ante la presencia de dolor intenso en la parte superior del abdomen, agrandamiento del hígado o signos de hemorragia intraabdominal en mujeres que usan anticonceptivos hormonales combinados.
Las enfermedades malignas pueden poner en riesgo la vida o tener un desenlace mortal.
Otras afecciones: Si hay irritaciones de la piel persistentes, repetidas (por ejemplo, prurito o eritema persistente en el lugar de aplicación) a pesar de que los lugares de aplicación se cambien según las indicaciones, se debe considerar la interrupción del tratamiento transdérmico.
Las mujeres con hipertrigliceridemia, o antecedentes familiares de esta afección, pueden estar expuestas a un incremento en el riesgo de pancreatitis con el uso de AOC.
Si bien se han informado aumentos pequeños de la presión arterial en algunas mujeres que reciben AOC, los aumentos clínicamente relevantes son poco frecuentes. Sin embargo, si aparece una hipertensión arterial clínicamente significativa sostenida durante el uso de un parche APLEEK PATCH®, será prudente que el médico suspenda la preparación y trate la hipertensión arterial. Cuando se considere adecuado, se puede reanudar el uso del parche APLEEK PATCH® si se pueden alcanzar valores normales con el tratamiento antihipertensivo.
Se ha informado que las siguientes afecciones han ocurrido o empeorado con el embarazo y con el uso de AOC, pero no se cuenta con pruebas concluyentes de una asociación con el uso de AOC: ictericia y/o prurito relacionados con colestasis; formación de cálculos biliares; porfiria; lupus eritematoso sistémico; síndrome urémico hemolítico; corea de Sydenham; herpes gestacional; pérdida de la audición asociada con otosclerosis.
En mujeres con angioedema hereditario, los estrógenos exógenos pueden inducir o exacerbar los síntomas de angioedema.
Las alteraciones agudas o crónicas de la función hepática pueden requerir la interrupción del uso del parche APLEEK PATCH® hasta que los marcadores de la función hepática regresen a la normalidad. La recurrencia de ictericia colestásica que se produjo por primera vez durante el embarazo o el uso previo de esteroides sexuales requiere la interrupción del parche APLEEK PATCH®.
Si bien los AOC pueden tener un efecto en la resistencia a la insulina periférica y la tolerancia a la glucosa, no hay evidencia de la necesidad de modificar el régimen terapéutico en las mujeres diabéticas usando AOC de baja dosis (con < 0.05 mg de etinilestradiol). No obstante, las mujeres diabéticas deben observarse estrechamente mientras usan los anticonceptivos hormonales combinados.
La enfermedad de Crohn y la colitis ulcerosa han sido asociadas con el uso de AOC.
En ocasiones puede producirse cloasma, en especial en mujeres con antecedentes de cloasma gravídico. Las mujeres que tienen tendencia a desarrollar cloasma deben evitar exponerse al sol o a radiación ultravioleta mientras reciban anticonceptivos hormonales combinados.
Exploración/consulta médica: Deben registrarse los antecedentes médicos completos y debe realizarse un examen físico antes del inicio o restablecimiento del parche APLEEK PATCH®, teniendo en cuenta las contraindicaciones (véase Contraindicaciones) y advertencias (véase Advertencias), y esto se debe repetir periódicamente. La evaluación médica periódica es también de importancia debido a las contraindicaciones (por ejemplo, un ataque isquémico transitorio, etc.) o factores de riesgo (por ejemplo, antecedentes familiares de trombosis venosa o arterial) que pueden aparecer por primera vez durante el uso de un anticonceptivo hormonal combinado. La frecuencia y naturaleza de estas evaluaciones deben basarse en las pautas de la práctica establecida y adaptarse a cada mujer en particular pero, en general, deben tener especialmente en cuenta la presión arterial, mamas, abdomen y órganos pélvicos, incluida la citología cervical.
Se debe indicar a las mujeres que los anticonceptivos hormonales no protegen contra la infección por el VIH (SIDA) y otras enfermedades de transmisión sexual.
Eficacia reducida:
La eficacia del parche APLEEK PATCH® puede verse disminuida, por ejemplo, en caso de:
• Que se no se realice la aplicación programada del parche.
• Que se desprenda el parche.
• Que se olvide realizar el cambio del parche (véase Manejo de los parches desprendidos, olvidados o no reemplazados).
• Medicamento concomitante (véase Interacciones medicamentosas y de otro género).
Control del ciclo reducido: Con todos los anticonceptivos hormonales combinados, se puede producir sangrado irregular (manchado o sangrado intermenstrual), especialmente durante los primeros meses de uso. En tales eventos, debe continuarse la aplicación del parche APLEEK PATCH®. La evaluación de cualquier sangrado irregular es únicamente significativa después de un intervalo de adaptación de alrededor de tres ciclos de uso del parche APLEEK PATCH®.
Si persisten las irregularidades en el sangrado o se producen después de ciclos regulares de uso del parche APLEEK PATCH®, deben considerarse causas no hormonales y se indican medidas diagnósticas adecuadas para descartar embarazo o malignidad, que pueden incluir legrado.
Es probable que en algunas mujeres no se produzca el sangrado por privación durante el intervalo sin parche. Si se ha usado el parche APLEEK PATCH® de conformidad con las indicaciones descritas (véase Dosis y vía de administración), es poco probable que la mujer esté embarazada. No obstante, si no se ha usado el parche APLEEK PATCH® de conformidad con estas indicaciones ante un primer sangrado por privación faltante o si faltaron dos sangrados por privación, se debe descartar un embarazo antes de continuar con el uso del parche APLEEK PATCH®.
DOSIS Y VÍA DE ADMINISTRACIÓN: Uso transdérmico.
El parche APLEEK PATCH® se usa en un ciclo de 28 días (cuatro semanas):
Durante tres semanas consecutivas (21 días), se aplica un parche nuevo por semana (cada siete días) y se retira el parche usado en cada ocasión que se aplica un parche nuevo. Durante la semana cuatro no se usa ningún parche. Se espera que comience el sangrado por privación durante ese periodo. Una semana después de que se retire el último parche, se inicia un ciclo nuevo de cuatro semanas aplicando un parche nuevo (el mismo día de la semana que antes, el “Día de cambio del parche”), independientemente de si el sangrado por privación continúa o se ha detenido. Consulte la sección “Control del ciclo reducido” en caso de que no se produzca el sangrado por privación. Si desea conocer el cronograma preciso de aplicación/retiro del parche, consulte la sección “Día de cambio del parche”.
Cuándo comenzar con el parche APLEEK PATCH® por primera vez:
Sin uso de anticonceptivo hormonal previo (en el último mes):
El parche debe ser aplicado el primer día del ciclo natural de la mujer (es decir, el primer día de su sangrado menstrual). Está permitido comenzar los días 2 a 5, pero durante el primer ciclo se requiere un método de barrera por un lapso de 7 días después de haber aplicado el primer parche.
Cambio de un anticonceptivo hormonal combinado (anticonceptivo oral combinado/AOC, inyección, anillo vaginal o un parche transdérmico diferente):
Se debe aplicar el parche preferentemente el día posterior al último comprimido AOC con hormona, y a más tardar el día siguiente al intervalo habitual sin comprimidos o sin hormonas del AOC.
Cuando cambia de un anillo vaginal o un parche transdérmico diferente, la mujer debe aplicarse el parche preferentemente el día en que retira el anillo vaginal o el parche previo, y a más tardar cuando correspondería la próxima aplicación del método previo. Si se ha usado un anticonceptivo hormonal combinado inyectable, la mujer debe comenzar a usar el parche APLEEK PATCH® cuando correspondería la próxima inyección.
Cambio a partir de otro método con progestágenos solamente (minipíldora, inyección, implante) o de un sistema intrauterino (SIU) liberador de progestágenos.
La mujer puede cambiar cualquier día si reemplaza a la minipíldora (el día del retiro del implante o del SIU, o en el momento en que debe administrarse la próxima inyección en el caso de que reemplace a un anticonceptivo inyectable). En todos estos casos, se debe aconsejar a la mujer que use, además, un método de barrera por un lapso de 7 días después de aplicado el primer parche.
Después de un aborto del primer trimestre:
La mujer puede comenzar de inmediato. Al hacerlo, no es necesario que tome otras medidas anticonceptivas.
Después de un parto o aborto en el segundo trimestre:
Para mujeres en periodo de lactancia, véase Restricciones de uso durante el embarazo y la lactancia.
Se les debe aconsejar a las mujeres que comiencen durante los días 21 a 28 después del parto o del aborto del segundo trimestre. Cuando comience más tarde, se le debe aconsejar a la mujer que además use un método de barrera por un lapso de 7 días después de aplicado el primer parche. No obstante, si ya ha tenido relaciones sexuales, debería descartarse un embarazo antes de iniciar el uso del parche APLEEK PATCH® o la mujer tendría que esperar su primer periodo menstrual.
Cómo usar el parche APLEEK PATCH®:
El parche APLEEK PATCH® se usa en un ciclo de 28 días (cuatro semanas) (1 parche por semana durante 3 semanas seguidas de un intervalo sin parche de 7 días). Se debe usar solamente un parche por vez. Cada ciclo subsiguiente comienza inmediatamente después del intervalo sin parche del ciclo anterior, independientemente de si el sangrado por privación continúa o se ha detenido.
Se llevaron a cabo dos estudios para evaluar la eficacia anticonceptiva del parche APLEEK PATCH®. Un estudio que incluyó usuarias en la Unión Europea, América Latina y Australia tuvo como resultado una tasa de falla de aproximadamente 1% por año. Un segundo estudio de apoyo, que incluyó usuarias en Estados Unidos de América (EUA) tuvo como resultado una tasa de falla de aproximadamente 4% por año. La tasa de falla puede aumentar cuando se usa el parche APLEEK PATCH® de forma incorrecta.
Día de cambio del parche:
Se debe aplicar cada parche nuevo el mismo día de la semana. Este día se conoce como el “Día de cambio del parche”. Por ejemplo, si el primer parche se aplica un domingo, todos los parches subsiguientes deben aplicarse un domingo. Se debe usar solamente un parche por vez.
Primer parche
Día 1: Aplicación del primer parche (para mujeres que comienzan a usar el parche APLEEK PATCH®, véase “Cuándo comenzar con el parche APLEEK PATCH® por primera vez”).
Segundo parche:
Día 8: Retiro del primer parche y aplicación inmediata del segundo parche.
Tercer parche
Día 15: Retiro del segundo parche y aplicación inmediata del tercer parche.
Ningún parche
Día 22: Retiro del tercer parche (sin parche entre los días 22 a 28).
El retiro del parche se realiza el mismo día de la semana (“Día de cambio del parche”). Los cambios de parche pueden realizarse en cualquier momento del “Día de cambio del parche”. Los ciclos subsiguientes comienzan el mismo “Día de cambio del parche”, después del intervalo de 7 días sin parche (Días 22 a 28).
Días sin parche: No se usa ningún parche desde el día 22 (después del retiro del tercer parche) hasta el día 28 (“Semana cuatro”).
Bajo ninguna circunstancia debe haber un intervalo de más de 7 días sin parche entre ciclos.
Si pasan más de siete días sin parche, es probable que la mujer no esté protegida contra un embarazo. Tan pronto la mujer note que ha olvidado iniciar un nuevo ciclo con parches, debe aplicar un parche nuevo y debe usar anticoncepción de respaldo como condones, espermicida o diafragma durante los próximos siete días. Al igual que con los AOC, el riesgo de ovulación aumenta con cada día que alargue el periodo recomendado sin anticonceptivo.
Si ha tenido relaciones sexuales durante un intervalo prolongado sin parche, debe considerarse la posibilidad de embarazo.
Consulte además la sección “Manejo de los parches desprendidos, olvidados o no reemplazados”.
Método de administración:
Dónde aplicar el parche:
El parche únicamente debe aplicarse en alguno de los lugares de aplicación indicados (ver la figura a continuación): abdomen, nalgas, parte externa de la parte superior del brazo.
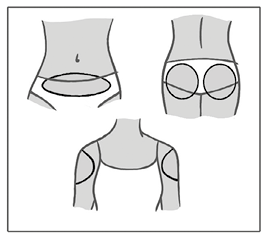
Se deben evitar las áreas de las que pueda desprenderse el parche (por ejemplo, cinturilla de las prendas de vestir).
El parche debe aplicarse sobre piel limpia, seca, intacta, saludable y preferentemente sin vello.
El parche APLEEK PATCH® no debe colocarse sobre piel grasosa, enrojecida, irritada, cortada o dañada.
Los parches no deben aplicarse en las mamas.
Para impedir la interferencia con las propiedades adhesivas del parche APLEEK PATCH®, no deben aplicarse cosméticos, cremas, lociones, polvos ni otros productos tópicos en el área de la piel en la que encuentra o se colocará el parche APLEEK PATCH®.
Los lugares de aplicación deben variar. Se puede hacer esto usando áreas diferentes en el mismo lugar de aplicación. Por ejemplo, una mujer puede pasar del lado izquierdo al lado derecho del abdomen o de la parte superior del brazo o de la nalga izquierda a la derecha.
La mujer también puede usar un lugar de aplicación diferente cada semana (por ejemplo, una semana la parte externa de la parte superior del brazo, a la semana siguiente el abdomen).
Tenga en cuenta lo siguiente:
• Se debe usar solamente un parche por vez.
• Si el parche se aplica correctamente, la mujer puede bañarse o ducharse como siempre.
• La mujer debe controlar visualmente el parche todos los días para verificar que continúe adherido de forma adecuada.
• El parche transparente tiene filtro UV/protección solar de modo que puede exponerse al sol y no es necesario que quede cubierto por las prendas de vestir.
En caso de irritación de la piel:
Si el uso del parche produce una irritación incómoda en el lugar de su aplicación, se debe retirar y colocar un parche nuevo en un lugar diferente. Ese parche nuevo se usa hasta el próximo “Día de cambio del parche” programado.
Cómo preparar el parche para su aplicación:
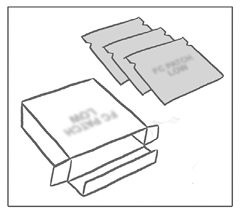
El parche APLEEK PATCH® viene en una caja que incluye: un folleto más 3 o 9 sobres sellados, cada uno de los cuales contiene un parche transdérmico APLEEK PATCH®.
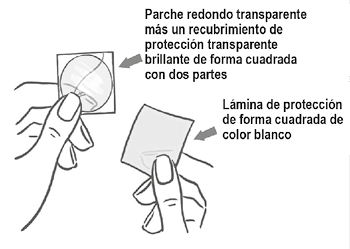
El parche es redondo y transparente:
Del lado adhesivo, el parche está cubierto por una capa de protección transparente brillante de forma cuadrada con dos partes. Este recubrimiento protege el lado adhesivo que contiene los componentes activos del parche. También asegura que se conserve la superficie adhesiva hasta la aplicación.
Del lado opuesto, el parche está cubierto por una lámina de protección de forma cuadrada de color blanco, que impide que el parche se pegue al sobre.
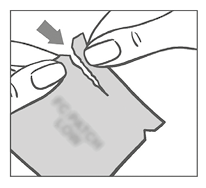
La mujer debe rasgar la parte superior del sobre con los dedos. Las muescas ayudarán a guiar el rasgado.
La mujer no debe usar tijeras ni tampoco cortar, dañar o alterar el parche de ninguna forma porque esto puede reducir el efecto anticonceptivo.
Como se describió inicialmente, el parche anticonceptivo circular viene entre un recubrimiento de protección transparente brillante de forma cuadrada con dos partes y una lámina de protección de forma cuadrada de color blanco. Es importante que retire el parche del sobre junto con el recubrimiento de protección transparente y la lámina de protección de color blanco. No se debe desechar el sobre. Debe conservarse para desechar el parche después del uso.
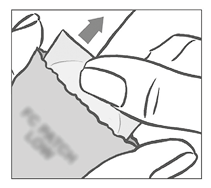
El parche debe aplicarse inmediatamente después de abrir el sobre de la siguiente manera:
Primero la mujer retira la lámina de protección de forma cuadrada de color blanco de una pieza de la parte superior del parche.
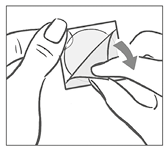
Después de retirarla del parche, debe desecharse la lámina de protección de forma cuadrada de color blanco que impide que el parche se pegue al sobre.
A continuación, la mujer retira la mitad del recubrimiento de protección transparente brillante de forma cuadrada con dos partes que cubre la parte inferior (adhesiva) del parche circular transparente. (El lado adhesivo contiene los fármacos activos). Debe evitar tocar la superficie adhesiva del parche de modo que se conserve su capacidad de adhesión.
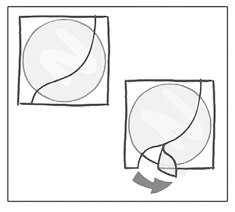
Mientras sostiene el parche por el borde que aún está cubierto por la otra mitad del recubrimiento de protección, debe colocar el parche sobre la piel en la zona donde lo usará.
Se debe retirar la segunda mitad del recubrimiento de protección con la otra mitad del parche suavemente adherida al lugar de aplicación.
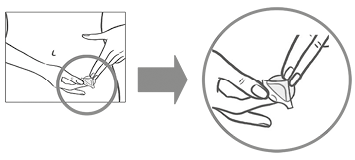
La mujer debe presionar el parche con firmeza con la palma de la mano durante 30 segundos y debe verificar que los bordes se hayan adherido bien.
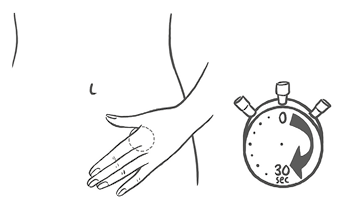
Nota: No debe desecharse el sobre, ya que será necesario para desechar el parche después de su uso.
Cómo desechar los parches:
Consulte la sección “Instrucciones de uso y manejo”.
Manejo de los parches desprendidos, olvidados o no reemplazados:
El manejo de las desviaciones de la aplicación del parche se basa en la regla siguiente:
Se necesitan al menos 7 días consecutivos con un parche aplicado de forma correcta para suprimir adecuadamente el eje hipotálamo-hipófisis-ovario como base para la eficacia anticonceptiva.
• Si un parche está desprendido parcial o completamente:
Durante menos de un día (hasta 24 horas):
La mujer debe intentar volver a aplicarlo en el mismo lugar o reemplazarlo con un parche nuevo de inmediato. No es necesario usar anticoncepción de respaldo. El “Día de cambio del parche” de la mujer seguirá siendo el mismo.
Durante más de un día (24 horas o más), o si la mujer no está segura de cuánto tiempo el parche ha estado desprendido:
Es probable que no esté protegida contra un embarazo. Debe detener el ciclo anticonceptivo actual e iniciar un ciclo nuevo de inmediato aplicando un parche nuevo. Ahora hay un “Día 1” nuevo y un “Día de cambio del parche” nuevo. Se debe usar anticoncepción de respaldo, como condones, espermicida o diafragma, durante la primera semana del nuevo ciclo.
No se debe volver a aplicar un parche si ya no se adhiere, si se ha doblado y pegado o se ha pegado a otra superficie, si tiene otro material pegado o si se ha desprendido o caído previamente. Si no se puede volver a aplicar el parche, se debe aplicar uno nuevo de inmediato. No se deben usar adhesivos complementarios ni envoltorios para mantener el parche APLEEK PATCH® en su lugar.
• Si la mujer se olvida de cambiar el parche:
Al inicio de cualquier ciclo de parches (semana uno/día 1):
Es probable que no esté protegida contra un embarazo. Debe aplicarse el primer parche del nuevo ciclo apenas lo recuerde. Ahora hay un nuevo “Día de cambio del parche” y un nuevo “Día 1” nuevo. La mujer debe usar anticoncepción de respaldo, como condones, espermicida o diafragma, durante la primera semana del ciclo nuevo.
En el medio del ciclo del parche (semana dos/día 8 o semana tres/día 15):
Durante uno o dos días (hasta 48 horas): Debe aplicarse un parche nuevo de inmediato. El parche siguiente debe aplicarse el “Día de cambio del parche” habitual. No es necesario usar anticoncepción de respaldo.
Durante más de dos días (48 horas o más):
es probable que no esté protegida contra un embarazo. Debe detener el ciclo anticonceptivo actual e iniciar un ciclo nuevo de cuatro semanas de inmediato aplicando un parche nuevo. Ahora hay un nuevo “Día de cambio del parche” y un nuevo “Día 1”. La mujer debe usar anticoncepción de respaldo durante una semana.
Al final del ciclo del parche (semana cuatro/día 22):
Semana cuatro (día 22): Si la mujer se olvida de retirar el parche el Día 22, debe quitárselo tan pronto lo recuerde (a más tardar el Día 28). Debe comenzarse el próximo ciclo con un parche nuevo (el día después del DÍA 28 - el “Día de cambio del parche” habitual), nunca más tarde. No es necesario usar anticoncepción de respaldo.
• Consecuencias de los parches desprendidos, olvidados o no reemplazados:
Tabla 1
|
Parches desprendidosa |
Plazo |
Consecuencias sobre la confiabilidad anticonceptiva a |
Acción necesaria a |
|
Parche desprendido |
< 24 horas |
Eficacia anticonceptiva asegurada |
- Aplicar un parche nuevo de inmediato. - No es necesario usar anticoncepción de respaldo. - No se modifica el “Día de cambio del parche”. |
|
> 24 horas |
Eficacia anticonceptiva comprometida |
- Comenzar un ciclo nuevo de cuatro semanas de inmediato aplicando un parche nuevo. - Usar anticoncepción de respaldo durante los próximos 7 díasb . - Anotar el nuevo “Día de cambio del parche”. |
|
|
Los parches no se reemplazan a tiempoa |
Plazo |
Consecuencias sobre la confiabilidad anticonceptiva a |
Acción necesaria a |
|
No se aplicó el primer parche (semana 1, día 1) en término |
Intervalo sin parche d > 7 días |
Eficacia anticonceptiva comprometida |
- Comenzar un ciclo nuevo de cuatro semanas de inmediato aplicando un parche nuevo. - Usar anticoncepción de respaldo durante los próximos 7 días b. - Anotar el nuevo “Día de cambio del parche”. |
|
El primer o el segundo parche (semana 1/2 o 2/3) no se reemplazó en término |
< 48 horas |
Eficacia anticonceptiva asegurada |
- Aplicar un parche nuevo de inmediato. - No es necesario usar anticoncepción de respaldo. - No se modifica el “Día de cambio del parche”. |
|
> 48 horas |
Eficacia anticonceptiva comprometida |
- Comenzar un ciclo nuevo de cuatro semanas de inmediato aplicando un parche nuevo. - Usar anticoncepción de respaldo durante los próximos 7 díasb. - Anotar el nuevo “Día de cambio del parche”. |
|
|
El tercer parche (semana 3/4) no se retiró en término |
Eficacia anticonceptiva asegurada c |
- Retirar el parche. - Iniciar el próximo ciclo de 4 semanas el “Día de cambio del parche” normal. |
a Se aplica a cada ciclo.
b La anticoncepción de respaldo es cualquier método anticonceptivo no hormonal adicional con excepción del método del calendario y el método de la temperatura.
c Siempre que el tercer parche haya sido reemplazado por uno nuevo a más tardar el Día 1 regular del ciclo del parche nuevo.
d Tiempo desde el retiro del último parche del ciclo anterior.
Ajuste del “Día de cambio del parche”: Si la mujer desea cambiar el “Día de cambio del parche”, debe completar el ciclo actual, retirando el tercer parche el día correcto. Durante la semana sin parche, puede seleccionar un “Día de cambio del parche” más temprano aplicando un parche nuevo el día deseado. En ningún caso deben pasar más de 7 días consecutivos sin parche.
Información adicional sobre poblaciones especiales:
Pacientes pediátricas:
El parche APLEEK PATCH® está indicado después de la menarca solamente. El parche APLEEK PATCH® no se ha estudiado en mujeres menores de 18 años de edad.
Pacientes geriátricas:
No aplica. El parche APLEEK PATCH® no está indicado después de la menopausia.
Pacientes con insuficiencia hepática:
El parche APLEEK PATCH® está contraindicado en mujeres con enfermedades hepáticas severas. Véase Contraindicaciones.
Pacientes con insuficiencia renal:
El parche APLEEK PATCH® no se ha estudiado en mujeres con insuficiencia renal. Debido a la metabolización completa del etinilestradiol y del gestodeno en metabolitos inactivos antes de la eliminación y debido a la disponibilidad de una segunda vía de eliminación a través del hígado1, no se espera que aumente el riesgo para las mujeres con insuficiencia renal.
Sexo: El parche APLEEK PATCH® está indicado en mujeres únicamente.
Diferencias étnicas: Se estudió la farmacocinética del etinilestradiol en combinación con otra progestina en mujeres caucásicas, chinas y japonesas, y no reveló ninguna diferencia clínicamente significativa.2 3 4 No se ha estudiado la farmacocinética del parche APLEEK PATCH® específicamente en mujeres de etnias diferentes. No se conocen enzimas polimórficas que contribuyan en una medida importante en la metabolización del gestodeno. De este modo, los datos disponibles no indican ninguna diferencia en la farmacocinética del parche APLEEK PATCH® entre mujeres de etnias diferentes.
MANIFESTACIONES Y MANEJO DE LA SOBREDOSIFICACIÓN O INGESTA ACCIDENTAL: La sobredosis es poco probable con este tipo de aplicación. No se han presentado informes de efectos nocivos serios por sobredosis de anticonceptivos orales combinados. Los síntomas que pueden producirse en este caso son: náuseas, vómitos y sangrado por privación, incluso en niñas antes de su menarca. No hay antídotos; se deben retirar los parches adicionales o usados de forma indebida de la piel y el tratamiento posterior debe ser sintomático.
PRESENTACIONES:
Caja con 3 o 9 parches transdérmicos de 11 cm2 cada uno, con gestodeno 2.1 mg y etinilestradiol 0.55 mg.
RECOMENDACIONES SOBRE ALMACENAMIENTO: Consérvese en el sobre original a temperatura ambiente a no más de 30°C y en lugar seco. No congelar.
LEYENDAS DE PROTECCIÓN:
Su venta requiere receta médica. No se deje a alcance de los niños. No se use durante el embarazo y lactancia.
Hecho en Alemania por:
Acino AG
Am Windfeld 35
83714 Miesbach
Alemania
Acondicionamiento secundario por:
Bayer Weimar GmbH und Co. KG
Döbereinerstraße 20
99427 Weimar
Thüringen
Alemania
Para:
Bayer Pharma AG
Müllerstraße 178
13353 Berlin
Alemania
Distribuido por:
BAYER DE MÉXICO, S. A. de C. V.
Carr. México-Toluca km 52.5
C.P. 52000, Lerma, México
Reg. Núm. 123M2016, SSA IV
153300CT050313


