
ADEMPAS - Comprimidos
Sustancia(s):
- Riociguat
Presentaciones:
- 1 Caja , 42 Comprimidos , 0.5 Miligramos
- 1 Caja , 84 Comprimidos , 0.5 Miligramos
- 1 Caja , 42 Comprimidos , 1 Miligramos
- 1 Caja , 84 Comprimidos , 1 Miligramos
- 1 Caja , 42 Comprimidos , 1.5 Miligramos
- 1 Caja , 84 Comprimidos , 1.5 Miligramos
- 1 Caja , 42 Comprimidos , 2 Miligramos
- 1 Caja , 84 Comprimidos , 2 Miligramos
- 1 Caja , 42 Comprimidos , 2.5 Miligramos
- 1 Caja , 84 Comprimidos , 2.5 Miligramos
FORMA FARMACÉUTICA Y FORMULACIÓN:
Cada COMPRIMIDO contiene:
|
Riociguat |
0.5 mg |
|
Excipiente, c.b.p. 1 comprimido. |
|
Cada comprimido recubierto de 0.5 mg
contiene 39.8 mg de lactosa monohidratada
(= 37.8 mg de lactosa).
|
Riociguat |
1 mg |
|
Excipiente, c.b.p. 1 comprimido. |
|
Cada comprimido recubierto de 1.0 mg
contiene 39.2 mg de lactosa monohidratada
(= 37.2 mg de lactosa).
|
Riociguat |
1.5 mg |
|
Excipiente, c.b.p. 1 comprimido. |
|
Cada comprimido recubierto de 1.5 mg
contiene 38.7 mg de lactosa monohidratada
(= 36.8 mg de lactosa).
|
Riociguat |
2 mg |
|
Excipiente, c.b.p. 1 comprimido. |
|
Cada comprimido recubierto de 2.0 mg
contiene 38.2 mg de lactosa monohidratada
(= 36.3 mg de lactosa).
|
Riociguat |
2.5 mg |
|
Excipiente, c.b.p. 1 comprimido. |
|
Cada comprimido recubierto de 2.5 mg
contiene 37.7 mg de lactosa monohidratada
(= 35.8 mg de lactosa).
INDICACIONES TERAPÉUTICAS:
Hipertensión pulmonar tromboembólica crónica (HPTEC, grupo 4 de la OMS):
ADEMPAS® está indicado para el tratamiento de pacientes adultos con:
• HPTEC inoperable.
• HPTEC persistente o recurrente tras el tratamiento quirúrgico para mejorar la capacidad de ejercicio y la clase funcional de la OMS.
Hipertensión arterial pulmonar (HAP, grupo 1 de la OMS):
ADEMPAS® está indicado para el tratamiento de pacientes adultos con HAP para mejorar la capacidad de ejercicio, y la clase funcional de la OMS y para retrasar el tiempo al deterioro clínico.
La eficacia se demostró en pacientes tratados con riociguat como monoterapia o en combinación con antagonistas de los receptores de la endotelina o prostanoides.
Los estudios que demostraron la eficacia incluían de forma predominante a pacientes con clases funcionales II-III de la OMS y etiologías de HAP idiopática o hereditaria, o HAP relacionada con enfermedad del tejido conjuntivo.
FARMACOCINÉTICA Y FARMACODINAMIA:
Propiedades farmacodinámicas.
Grupo farmacoterapéutico:
Código ATC: No ha sido asignado aún.
Mecanismo de acción/efectos farmacodinámicos.
Riociguat es un estimulador de la guanilato-ciclasa soluble (GCs), una enzima del sistema cardiopulmonar, y que es el receptor del óxido nítrico (NO).
Cuando el NO se une a la GCs, la enzima cataliza la síntesis de la molécula señalizadora guanosina-monofosfato cíclico (GMPc). El GMPc intracelular desempeña un papel importante en la regulación de procesos que influyen en el tono, la proliferación, la fibrosis y la inflamación vascular.
La hipertensión pulmonar está relacionada con la disfunción endotelial, el deterioro de la síntesis de óxido nítrico y la estimulación insuficiente de la vía NO-GCs-GMPc.
Riociguat tiene un mecanismo de acción doble. Sensibiliza la GCs frente al NO endógeno estabilizando la unión NO-GCs. Riociguat también estimula de forma directa la GCs a través de un sitio de unión distinto, independientemente del NO.
Riociguat restaura la vía NO-GCs-GMPc y provoca un aumento de la generación de GMPc.
Eficacia clínica:
Eficacia en pacientes con hipertensión pulmonar tromboembólica crónica (HPTEC):
Diseño del estudio: Se realizó un estudio de asignación aleatoria, doble ciego, multinacional, multicéntrico, controlado con placebo de fase III (CHEST-1) en pacientes con hipertensión pulmonar tromboembólica crónica (HPTEC). Se incluyeron pacientes con enfermedad inoperable (evaluada por un comité de adjudicación independiente) o que presentaban HPTEC recurrente o persistente tras la endarterectomía pulmonar (PEA).
La población de pacientes incluía hombres y mujeres de edades entre 18 y 80 años. 72% de los pacientes presentaban HPTEC inoperable, 28% presentaban HPTEC recurrente o persistente tras la PEA.
La mayoría de los pacientes estaban en las clases funcionales II (31%) o III (64%) de la Organización Mundial de la Salud (OMS) al inicio del estudio. La media basal de la prueba de caminata de seis minutos (6MWT) fue 347 m. Ninguno de los pacientes había recibido tratamiento previamente (se excluyó el tratamiento específico para la HAP).
El estudio CHEST-1 incluía 261 pacientes tratados y válidos para la evaluación de la seguridad, distribuidos de forma aleatoria a uno de dos grupos de tratamiento: titulación de la dosis de forma individual (TDI) de riociguat hasta 2.5 mg 3 veces al día (n = 173, denominado grupo de riociguat), o placebo (n = 88). Durante la fase de titulación de ocho semanas, se ajustó la dosis de riociguat cada 2 semanas teniendo en cuenta la presión arterial sistólica y los signos o síntomas de hipotensión del paciente. Se alcanzó una dosis individualizada al final de la titulación.
Criterios de valoración de la eficacia: Todos los valores de p se basan en la prueba de Wilcoxon estratificada (a no ser que se mencione una prueba distinta). Todos los IC de 95% y los efectos del tratamiento se basan en el análisis de la covarianza (ANCOVA).
Criterio principal de valoración: El criterio principal de valoración fue el cambio desde la prueba basal hasta la semana 16 (última visita) en la 6MWT en comparación con el placebo.
Se apreciaron mejorías en la distancia caminada a partir de la semana 2 en adelante, y en la semana 16 (n = 261) el aumento en la 6MWT en el grupo de riociguat fue de 46 metros (intervalo de confianza de 95% (IC): de 25 a 67 m; p < 0.0001) en comparación con el placebo (análisis por ITT, véase la tabla 2). Se observaron mejorías con riociguat en comparación con el placebo en todos los subgrupos evaluados. Los pacientes considerados inoperables (n = 189) presentaron un aumento en la 6MWT de 54 m (IC de 95%: de 29 a 79 m), y los pacientes con HPTEC recurrente o persistente tras la PEA (n = 72) presentaron un aumento en la 6MWT de 27 m (IC de 95%: de -10 a 63 m).
Tabla 2. Efectos del riociguat sobre la 6MWT en el estudio CHEST-1 en la semana 16 (última visita; conjunto de análisis por ITT).
|
Muestra total de pacientes |
Riociguat (TDI) (n = 173) |
Placebo (n = 88) |
|
Inicio (m) (SD ) |
342 (82) |
356 (75) |
|
Cambio desde la basal (m) (SD ) |
39 (79) |
-6 (84) |
|
Diferencia corregida con placebo (m) IC de 95%; [valor de p] |
46 25 a 67 m; (< 0.0001) |
|
|
Grupo de pacientes inoperables |
Riociguat (TDI) (n = 121) |
Placebo (n = 68) |
|
Inicio (m) (SD ) |
335 (83) |
351 (75) |
|
Cambio desde la basal (m) (SD ) |
44 (84) |
-8 (88) |
|
Diferencia corregida con placebo (m) IC de 95% |
54 29 a 79 m |
|
|
Grupo de pacientes con HPTEC tras la PEA |
Riociguat (TDI) (n = 52) |
Placebo (n = 20) |
|
Inicio (m) (SD ) |
360 (78) |
374 (72) |
|
Cambio desde la basal (m) (SD) |
27 (68) |
2 (73) |
|
Diferencia corregida con placebo (m) IC de 95% |
27 -10 a 63 m |
|
Criterios de valoración secundarios:
La mejoría en la distancia caminada se acompañó de las mejorías consistentes en criterios de valoración secundarios clínicamente relevantes.
Se demostró una mejoría estadísticamente significativa en el grupo de riociguat en comparación con el placebo en el caso de las siguientes variables secundarias de eficacia:
• Resistencia vascular pulmonar (RVP): RVP significativamente reducida (p < 0.0001, media del cambio desde la basal, corregida con placebo: -246 dyn*s*cm-5; IC de 95%: de -303 a -190; p < 0.0001; véase la tabla 3).
• NT-proBNP: niveles de NT-proBNP significativamente reducidos (media del cambio desde la basal, corregida con placebo: -444 ng/L, IC: -843 a -45; véase la tabla 3).
• Clase funcional de la OMS: se observó una mejoría significativa de por lo menos una clase funcional en el grupo de riociguat en la semana 16 (última visita) de 33%, en comparación con 15% en el grupo de placebo, y una disminución de por lo menos una clase funcional en 5% de los pacientes en el grupo de riociguat, en comparación con 7% en el grupo del placebo (p = 0.0026; véase la tabla 4). La clase funcional permaneció sin cambios en 62% de los pacientes en el grupo de riociguat, en comparación con 78% en el grupo de placebo.
Se demostró un efecto a favor del grupo de riociguat (por debajo del umbral de los análisis jerárquicos1) en el caso de:
• El tiempo hasta el deterioro clínico: los pacientes tratados con riociguat presentaron un retraso del tiempo hasta el deterioro clínico, en comparación con los pacientes tratados con placebo (p = 0.1724; prueba de rangos logarítmicos estratificada). Se observó una tendencia hacia una menor incidencia de episodios de deterioro clínico en la semana 16 (última visita) en los pacientes tratados con riociguat (2.3%) en comparación con el placebo (5.7%) (p = 0.2180, estimado de Mantel-Haenszel, véase la tabla 5, véase la figura 1).
• La escala CR 10 de Borg: mejoría en la escala CR 10 de Borg (-0.8 para el riociguat en comparación con +0.2 para el placebo, p = 0.0035).
• El cuestionario europeo de calidad de vida (EQ-5D): mejoría en el cuestionario EQ- 5D (cambio desde la basal: 0.13; IC de 95%: de 0.06 a 0.21; p < 0.0001).
• El cuestionario de calidad de vida Viviendo con Hipertensión Pulmonar (Living with Pulmonary Hypertension, LPH): mejoría en la escala del cuestionario LPH (cambio desde la basal: -5.8; p = 0.1220; IC de 95%: de -10.45 a -1.06).
Tabla 3. Efectos de riociguat sobre la RVP y el NT-proBNP en la semana 16 (última visita) en el estudio CHEST-1.
|
Muestra de población del estudio |
Inicio (SD) |
Cambio desde la basal (SD) |
Diferencia corregida con placebo |
IC de 95% |
Valor de p |
|
RVP (dyn*s*cm-5) Riociguat (TDI) (n = 151) |
791 (432) |
-226 (248) |
-246 |
de -303 a -190 |
< 0.0001 |
|
RVP (dyn*s*cm-5) Placebo (n = 82) |
779 (401) |
23 (274) |
- |
- |
- |
|
NT-proBNP (ng/L) Riociguat (TDI) (n = 150) |
1,508 (2,338) |
-291 (1,717) |
-444 |
de -843 a -45 |
< 0.0001 |
|
NT-proBNP (ng/L) Placebo (n = 73) |
1,706 (2,567) |
76 (1,447) |
- |
- |
- |
Tabla 4. Efectos de riociguat sobre el cambio en la clase funcional en la semana 16 (última visita; conjunto de análisis por ITT) en el estudio CHEST-1.
|
Cambio en la clase funcional |
Riociguat (n = 173) |
Placebo (n = 87) |
|
Mejoría |
57 (33%) |
13 (15%) |
|
Estable |
107 (62%) |
68 (78%) |
|
Deterioro |
9 (5%) |
6 (7%) |
|
Valor de p = 0.0026 |
||
Tabla 5. Efectos de riociguat sobre los episodios de deterioro clínico (conjunto de análisis por ITT) en el estudio CHEST-1.
|
Episodios de deterioro clínico |
Riociguat (TDI) (n = 173) |
Placebo (n = 88) |
|
Pacientes con cualquier deterioro clínico* |
4 (2.3%) |
5 (5.7%) |
|
Muerte |
2 (1.2%) |
3 (3.4%) |
|
Hospitalizaciones debidas a HP |
0 |
1 (1.1%) |
|
Descenso en el valor de la 6MWT debido a HP |
1 (0.6%) |
2 (2.3%) |
|
Deterioro persistente de la CF debido a HP |
0 |
1 (1.1%) |
|
Comienzo de un nuevo tratamiento para la HP |
2 (1.2%) |
1 (1.1%) |
* Valor de p = 0.2180 (estimado de Mantel-Haenszel).
Nota: Los pacientes pueden haber sufrido más de un evento de deterioro clínico.
Figura 1. Gráfica de Kaplan-Meier para tiempo hasta el deterioro clínico (Bay 63-2,521 = Riociguat; conjunto de análisis por ITT) en el estudio CHEST-1.
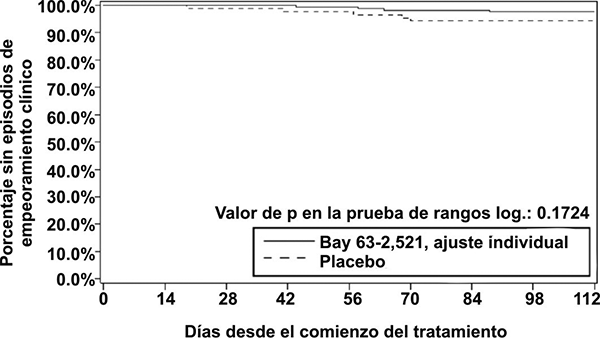
Parámetros hemodinámicos: Se realizó un cateterismo cardiaco derecho al comienzo y al final del periodo del estudio controlado con placebo en 233 pacientes, con el fin de obtener un conjunto amplio y comprensible de datos hemodinámicos cardiopulmonares (véase la tabla 6).
Se demostró una reducción estadísticamente significativa de la RVP (véase el apartado anterior) y de la presión media de la arteria pulmonar (PAPmedia ) (-5.0 mm Hg, p < 0.0001), y un aumento del índice cardiaco (0.47 L/min/m2; p < 0.0001) en el grupo de riociguat, en comparación con el placebo. La mejoría observada en el caso de las variables hemodinámicas descritas anteriormente también se observó en otros parámetros hemodinámicos relevantes.
Tabla 6. Estudio CHEST-1, cambio en los parámetros hemodinámicos desde la basal hasta la última visita: comparación entre riociguat (RIO), de 1.0-2.5 mg, y placebo (PBO) (conjunto de análisis por ITT).
|
Parámetro (unidades) |
Media del cambio |
Diferencia media de MC |
IC de 95% |
ANCOVA |
Prueba de Wilcoxon estratificada |
|
|
RIO |
PBO |
valor de p |
valor de p |
|||
|
PECP (mm Hg) |
0.59 |
0.18 |
0.58 |
de –0.36 a 1.53 |
0.2268 |
0.2285 |
|
PAD (mm Hg) |
–1.04 |
–0.55 |
–0.55 |
de –1.72 a 0.62 |
0.3566 |
0.3593 |
|
PSAP (mm Hg) |
–6.84 |
0.95 |
–7.52 |
de –10.88 a –4.16 |
< 0.0001 |
< 0.0001 |
|
PDAP (mm Hg) |
–3.05 |
0.67 |
–3.62 |
de –5.30 a –1.95 |
< 0.0001 |
0.0002 |
|
PMAP (mm Hg) |
–4.31 |
0.76 |
–4.96 |
de –6.75 a –3.16 |
< 0.0001 |
< 0.0001 |
|
TAM (mm Hg) |
–9.27 |
–0.29 |
–9.15 |
de –11.83 a –6.46 |
< 0.0001 |
< 0.0001 |
|
SvO2 (%) |
2.95 |
–0.44 |
3.85 |
de 1.46 a 6.25 |
0.0017 |
0.0010 |
|
GC (L/min) |
0.81 |
–0.03 |
0.86 |
de 0.59 a 1.12 |
< 0.0001 |
< 0.0001 |
|
IC (L/min/m2) |
0.45 |
–0.01 |
0.47 |
de 0.33 a 0.62 |
< 0.0001 |
< 0.0001 |
|
RVP* (dyn*s*cm-5) |
–226 |
23.1 |
–246.43 |
de –303.33 a –189.53 |
< 0.0001 |
< 0.0001 |
|
IRVP (dyn*s*cm-5*m2) |
–397 |
48.3 |
–448.95 |
de –553.62 a –344.27 |
< 0.0001 |
< 0.0001 |
|
RVS (dyn*s*cm-5) |
–445 |
16.6 |
–478.24 |
de –602.30 a –354.19 |
< 0.0001 |
< 0.0001 |
|
IRVS (dyn*s*cm-5*m2) |
–799 |
53.7 |
–914.16 |
de –1140.97 a –687.35 |
< 0.0001 |
< 0.0001 |
* La RVP fue un criterio de valoración secundario en este estudio.
El resto de los parámetros no se especificaron previamente como criterios de valoración.
Tratamiento prolongado de la HPTEC: Un estudio abierto, de extensión (CHEST-2) incluyó a 237 pacientes que habían completado el estudio CHEST-1. La duración media del tratamiento en la fecha de corte fueron 388 días, con una mediana de duración de 336 días (intervalo: de 15 a 989 días) y una exposición total al riociguat de 206 años-paciente.
En el estudio CHEST-2 también se observaron mejorías adicionales en la 6MWT y la clase funcional.
La probabilidad de supervivencia al cabo de un año fue de 98%.
Eficacia en pacientes con hipertensión arterial pulmonar (HAP).
Diseño del estudio: Se realizó un estudio aleatorizado, con doble ciego, multinacional, multicéntrico, controlado con placebo, de fase III (PATENT-1) en pacientes con hipertensión arterial pulmonar (HAP) que no habían recibido tratamiento previo o que habían sido tratados con un antagonista de los receptores de la endotelina (ERA) o con un análogo de la prostaciclina (administrado por inhalación, por vía oral o subcutánea).
La población total de pacientes incluía hombres y mujeres de 18 a 80 años con diagnóstico de HAP idiopática (61%), HAP familiar (2%), HAP relacionada con enfermedad del tejido conectivo (25%), enfermedad cardiaca congénita (8%), hipertensión portal (3%) y HAP relacionada con el uso de anorexígenos o anfetaminas (1%).
La mayoría de los pacientes se encontraban en las clases funcionales III (54%) o II (42%) de la Organización Mundial de la Salud (OMS) basal. La media global basal de la 6MWT fue 363 m. 50% de los pacientes no habían recibido tratamiento previo, 44% habían sido tratados con ERA, 6% sólo con análogos de la prostaciclina.
El estudio PATENT-1 incluía 443 pacientes tratados y válidos para la evaluación de la seguridad, asignados de forma aleatoria a uno de tres grupos de tratamiento: titulación de la dosis de forma individual de riociguat hasta 2.5 mg 3 veces día (n = 254); placebo (n = 126); y una titulación limitada de la dosis hasta 1.5 mg 3 veces día (n = 63; grupo de dosis exploratoria; no se realizaron pruebas estadísticas). Durante la fase de titulación de 8 semanas se ajustó la dosis de riociguat cada 2 semanas teniendo en cuenta la presión arterial sistólica y los signos o síntomas de hipotensión del paciente. Se alcanzó una dosis individualizada al final de la titulación.
Criterios de valoración de la eficacia: El análisis principal preespecificado se realiza con el grupo tratado con 2.5 mg de riociguat (denominado grupo de riociguat) en comparación con el placebo. Todos los valores de p se basan en la prueba de Wilcoxon estratificada (a no ser que se mencione una prueba distinta). Todos los IC de 95% y los efectos del tratamiento se basan en el análisis de la covarianza (ANCOVA).
Criterio principal de valoración: El criterio principal de valoración fue el cambio desde la basal hasta la semana 12 (última visita) en la prueba de caminata de seis minutos (6MWT) en comparación con el placebo.
Se apreciaron mejorías en la distancia recorrida a partir de la semana 2 en adelante, y en la semana 12 en el caso del grupo de riociguat el valor fue de 36 m (intervalo de confianza (IC) de 95%: de 20 a 52 m; p < 0.0001) en comparación con el placebo (análisis por ITT, véase la tabla 7). Se observaron mejorías con riociguat en comparación con el placebo en todos los subgrupos evaluados. Los pacientes que no habían recibido tratamiento previo (n = 189) demostraron un aumento en la 6MWT de 38 m (IC de 95%: de 14 a 62 m).
Los pacientes que habían sido previamente tratados (n = 191) presentaron un aumento en la 6MWT de 36 m (IC de 95%: de 15 a 56 m). El análisis adicional de subgrupos de pacientes previamente tratados con ERA (n = 167) demostró un efecto calculado del tratamiento de 26 m (IC de 95%: de 5 a 46 m). En pacientes previamente tratados con análogos de la prostaciclina (n = 272) el efecto calculado del tratamiento fue de 101 m (IC de 95%: de 27 a 176 m).
Tabla 7. Efectos de riociguat sobre la 6MWT en PATENT-1 en la semana 12 (última visita; conjunto de análisis por ITT).
|
Muestra total de pacientes |
Riociguat (TDI) (n = 254) |
Placebo (n = 126) |
|
Basal (m) (SD) |
361 (68) |
368 (75) |
|
Cambio desde la basal (m) (SD) |
30 (66) |
-6 (86) |
|
Diferencia corregida con placebo (m) IC de 95%; [valor de p] |
36 de 20 a 52 m (< 0.0001) |
|
|
Grupo de pacientes sin tratamiento previo |
Riociguat (TDI) (n = 123) |
Placebo (n = 66) |
|
Basal (m) (SD) |
370 (66) |
360 (80) |
|
Cambio desde la basal (m) (SD) |
32 (74) |
-6 (88) |
|
Diferencia corregida con placebo (m) IC de 95% |
38 de 14 a 62 m |
|
|
Grupo de pacientes con tratamiento previo |
Riociguat (TDI) (n = 131) |
Placebo (n = 60) |
|
Basal (m) (SD) |
353 (69) |
376 (68) |
|
Cambio desde la basal (m) (SD) |
27 (58) |
-5 (83) |
|
Diferencia corregida con placebo (m) IC de 95% |
36 de 15 a 56 m |
|
Criterios de valoración secundarios: La mejoría en la distancia de caminada se acompañó de mejorías consistentes en los criterios de valoración secundarios clínicamente relevantes.
Se demostró una mejoría estadísticamente significativa en el grupo de riociguat en comparación con placebo en el caso de las siguientes variables secundarias de eficacia:
• Resistencia vascular pulmonar (RVP): RVP significativamente reducida (p < 0.0001, media del cambio desde la basal, corregida con placebo: -226 dyn*s*cm-5 IC de 95%: de -281 a -170; p < 0.0001; véase la tabla 8).
• NT-proBNP: niveles de NT-proBNP significativamente reducidos (media de cambio desde la basal, corregida con placebo: -432 ng/L, IC de 95%: de -782 a -82; véase la tabla 8).
• Clase funcional de la OMS: se observó una mejoría significativa de por lo menos una clase funcional en el grupo de riociguat en la semana 12 (última visita) de 21%, en comparación con 14% en el grupo placebo, y una disminución de por lo menos una clase funcional en 4% de los pacientes en el grupo de riociguat, en comparación con 14% en el grupo placebo (p = 0.0033; véase la tabla 9). La clase funcional permaneció sin cambios en 76% de los pacientes en el grupo de riociguat, en comparación con 71% en el grupo placebo.
• Tiempo hasta el deterioro clínico: los pacientes tratados con riociguat presentaron un retraso significativo del tiempo hasta el deterioro clínico, en comparación con los pacientes tratados con placebo (p = 0.0046; prueba de rangos logarítmicos estratificada). Se observó un número significativamente menor de episodios de deterioro clínico hasta la semana 12 (última visita) en los pacientes tratados con riociguat (1.2%) en comparación con placebo (6.3%) (p = 0.0285, estimado de Mantel-Haenszel, véase la tabla 10, véase la figura 2).
• Escala CR 10 de Borg: mejoría significativa en la escala CR 10 de Borg (-0.4 para riociguat en comparación con + 0.1 para el placebo, p = 0.0022; véase la tabla 8).
Se demostró un efecto a favor del grupo de riociguat (por debajo del umbral de los análisis jerárquicos3) para el bienestar de los pacientes en lo que respecta al:
• Cuestionario europeo de calidad de vida (EQ-5D): cambio desde la basal: 0.06 (IC de 95%: de 0.01 a 0.11; p = 0.0663).
• Cuestionario de calidad de vida Viviendo con Hipertensión Pulmonar (LPH): mejoría en la escala del cuestionario LPH (cambio desde la basal: -6.2; p = 0.0019; IC de 95%: de -9.8 a -2.5).
Tabla 8. Efectos de riociguat sobre la RVP, el NT-proBNP y la escala CR 10 de Borg en la semana 12 (última visita) en el estudio PATENT-1.
|
Población del estudio |
Inicio (SD) |
Cambio desde la basal (SD ) |
Diferencia corregida con placebo |
IC de 95% |
Valor de p |
|
RVP (dyn*s*cm-5) Riociguat (TDI) (n = 232) |
791 (453) |
-223 (260) |
-226 |
de -281 a -170 |
< 0.0001 |
|
RVP (dyn*s*cm-5) Placebo (n = 107) |
834 (477) |
-9 (317) |
- |
- |
- |
|
NT-proBNP (ng/L) Riociguat (ADI) (n = 228) |
1,027 (1,799) |
-198 (1,721) |
-432 |
de -782 a -82 |
< 0.0001 |
|
NT-proBNP (ng/L) Placebo (n = 106) |
1,228 (1,775) |
232 (1,011) |
- |
- |
- |
|
Escala CR 10 de Borg Riociguat (TDI) (n = 254) |
3.9 (2.2) |
-0.4 (1.7) |
- |
- |
0.0022 |
|
Escala CR 10 de Borg Placebo (n = 126) |
3.9 (2.5) |
0.09 (2.1) |
- |
- |
- |
Tabla 9. Efectos de riociguat sobre el cambio en la clase funcional en la semana 12 (última visita; conjunto de análisis por ITT) en el PATENT-1.
|
Cambio en la clase funcional |
Riociguat (TDI) (n = 254) |
Placebo (n = 125) |
|
Mejoría |
53 (21%) |
18 (14%) |
|
Estable |
192 (76%) |
89 (71%) |
|
Deterioro |
9 (4%) |
18 (14%) |
|
Valor de p = 0.0033 |
||
Tabla 10. Efectos de riociguat sobre los episodios de deterioro clínico (conjunto de análisis por ITT) en el estudio PATENT-1.
|
Episodios de deterioro clínico |
Riociguat (TDI) (n = 254) |
Placebo (n = 126) |
|
Pacientes con cualquier deterioro clínico* |
3 (1.2%) |
8 (6.3%) |
|
Muerte |
2 (0.8%) |
3 (2.4%) |
|
Hospitalizaciones debidas a HP |
1 (0.4%) |
4 (3.2%) |
|
Descenso en el resultado de la 6MWT debido a HP |
1 (0.4%) |
2 (1.6%) |
|
Deterioro persistente de la CF debido a HP |
0 |
1 (0.8%) |
|
Comienzo de un nuevo tratamiento contra la HP |
1 (0.4%) |
5 (4.0%) |
* Valor de p = 0.0285 (estimado de Mantel-Haenszel).
Nota: Los pacientes pueden haber sufrido más de un episodio de deterioro clínico.
Figura 2. Gráfica de Kaplan-Meier para tiempo hasta el deterioro clínico (Bay 63- 2,521 = riociguat; conjunto de análisis por ITT) en el estudio PATENT-1.
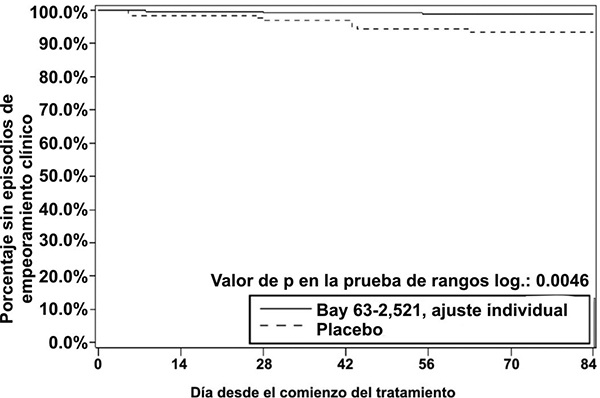
Parámetros hemodinámicos: Se realizó un cateterismo cardiaco derecho al comienzo y al final del periodo del estudio controlado con placebo en 339 pacientes, con el fin de obtener un conjunto amplio y comprensible de datos hemodinámicos cardiopulmonares (véase la tabla 11).
Se demostró una reducción estadísticamente significativa de la RVP (véase el apartado anterior) y de la presión media de la arteria pulmonar (PAPmedia ) (-3.8 mm Hg, p < 0.0001) y un aumento del índice cardiaco (0.56 L/min/m2; p < 0.0001) en el grupo de riociguat, en comparación con placebo. La mejoría observada en el caso de las variables hemodinámicas descritas anteriormente también se observó en otros parámetros hemodinámicos relevantes.
Tabla 11. Estudio PATENT-1; cambio en los parámetros hemodinámicos desde la basal hasta la última visita: comparación entre riociguat (RIO), de 1.0 a 2.5 mg, y placebo (PBO): conjunto de análisis por ITT.
|
Parámetro (unidades) |
Media del cambio |
Diferencia media de MC |
IC de 95% |
ANCOVA |
Prueba Wilcoxon estratifada |
|
|
RIO |
PBO |
valor de p |
valor de p |
|||
|
PECP (mm Hg) |
1.08 |
0.46 |
0.41 |
de –0.36 a 1.18 |
0.2972 |
0.0830 |
|
PAD (mm Hg) |
–0.20 |
0.97 |
–1.01 |
de –2.15 a 0.13 |
0.0832 |
0.0734 |
|
PSAP (mm Hg) |
–5.39 |
0.78 |
–6.73 |
de –9.43 a –4.04 |
< 0.0001 |
< 0.0001 |
|
PDAP (mm Hg) |
–3.19 |
–1.12 |
–2.41 |
de –4.15 a –0.68 |
0.0066 |
0.0110 |
|
PMAP (mm Hg) |
–3.93 |
–0.50 |
–3.83 |
de –5.61 a –2.06 |
< 0.0001 |
0.0002 |
|
TAM (mm Hg) |
–8.54 |
–1.40 |
–7.25 |
de –9.60 a –4.90 |
< 0.0001 |
< 0.0001 |
|
SvO2 (%) |
3.15 |
–2.33 |
5.02 |
de 3.20 a 6.84 |
< 0.0001 |
< 0.0001 |
|
GC (L/min) |
0.93 |
–0.01 |
0.93 |
de 0.70 a 1.15 |
< 0.0001 |
< 0.0001 |
|
IC (L/min/m2) |
0.54 |
–0.02 |
0.56 |
de 0.44 a 0.69 |
< 0.0001 |
< 0.0001 |
|
RVP* (dyn*s*cm-5) |
–223 |
–8.9 |
–225.72 |
de –281.37 a – 170.08 |
< 0.0001 |
< 0.0001 |
|
IRVP (dyn*s*cm-5*m2) |
–374 |
–22.4 |
–376.81 |
de –468.90 a – 284.72 |
< 0.0001 |
< 0.0001 |
|
RVS (dyn*s*cm-5) |
–448 |
–67.5 |
–394.57 |
de –472.95 a – 316.19 |
< 0.0001 |
< 0.0001 |
|
IRVS (dyn*s*cm-5*m2) |
–753 |
–130 |
–675.31 |
de –800.84 a – 549.79 |
< 0.0001 |
< 0.0001 |
* La RVP fue un criterio de valoración secundario en este estudio.
El resto de los parámetros no se especificaron previamente como criterios de valoración.
Tratamiento prolongado de la HAP: Un estudio abierto, ampliado (PATENT-2) incluyó a 363 pacientes que habían completado el estudio PATENT-1. La duración media del tratamiento en el estudio PATENT-2 en la fecha de corte fue de 438 días, con una mediana de duración de 441 días (intervalo de 1 a 1,078 días) y una exposición total al riociguat de 436 años-paciente.
En el PATENT-2 también se observaron mejorías adicionales en la 6MWT y la clase funcional. La probabilidad de supervivencia en un año fue de 96%.
Propiedades farmacocinéticas:
Absorción: La biodisponibilidad absoluta de riociguat es elevada (94%). Riociguat se absorbe rápidamente y alcanza concentraciones máximas (Cmáx) entre 1 y 1.5 horas después de la administración del comprimido.
La ingesta de alimentos no afecta al ABC de riociguat. La Cmáx se vio ligeramente reducida (disminución de 35%). Este efecto no se considera clínicamente relevante. Por consiguiente, riociguat pueda administrarse con o sin alimentos.
Distribución: La unión a las proteínas plasmáticas en los humanos es alta, de aproximadamente 95%, siendo la seroalbúmina y la glucoproteína ácida α1 los principales componentes a los que se une el fármaco.
El volumen de distribución es moderado, con un valor de distribución en estado de equilibrio de aproximadamente 30 L.
Metabolismo/biotransformación: La N-desmetilación, catalizada por CYP 1A1, CYP 3A4, CYP 2C8 y CYP 2J2, es la principal ruta de biotransformación de riociguat, que da lugar a su principal metabolito activo circulante (actividad farmacológica: de 1/10 a 1/3 de la de riociguat), que a su vez se metaboliza para formar el compuesto N-glucurónido, que es farmacológicamente inactivo.
El CYP1A1 cataliza la formación del principal metabolito de riociguat en el hígado y en los pulmones, y se sabe que es inducible por hidrocarburos aromáticos policíclicos, que están presentes, por ejemplo, en el humo de los cigarrillos.
Eliminación/excreción: El riociguat total (compuesto precursor y sus metabolitos) se excreta tanto por la vía renal (33-45%) como por las vías biliar/fecal (48-59%). Aproximadamente 4-19% de la dosis administrada se excreta en forma de riociguat sin cambios a través de los riñones. Aproximadamente 9-44% de la dosis administrada se encontró en forma de riociguat sin cambios en las heces.
Según los estudios in vitro, riociguat y su principal metabolito son sustratos de las proteínas transportadoras gp-P (glucoproteína P) y BCRP (proteína de resistencia del cáncer de mama).
Con una depuración sistémica de aproximadamente 3-6 L/h, riociguat puede clasificarse como un fármaco con bajo depuración. La vida media de eliminación es aproximadamente 7 horas en sujetos sanos, y aproximadamente 12 horas en pacientes.
Linealidad/no linealidad: La farmacocinética de riociguat es lineal desde 0.5 hasta 2.5 mg.
La variabilidad interindividual (% del CV) de la exposición al riociguat (ABC) de todas las dosis es aproximadamente 60%.
Información adicional sobre poblaciones especiales:
Pacientes ancianos: Los pacientes ancianos (≥ 65 años) mostraron mayores concentraciones plasmáticas que los pacientes más jóvenes, con medias del ABC siendo aproximadamente 40% mayores en los ancianos, debido principalmente a una disminución (aparente) en el depuración renal y total (véase Régimen de dosificación).
Pacientes con insuficiencia hepática: No hubo cambios clínicamente relevantes en la exposición en sujetos cirróticos con insuficiencia hepática leve (clasificado como Child-Pugh A).
En sujetos cirróticos con insuficiencia hepática moderada (clasificado como Child Pugh B), la media del ABC de riociguat aumentó en 50-70% en comparación con los controles sanos (véase Régimen de dosificación).
No existen datos en pacientes con insuficiencia hepática severa (clasificado como Child-Pugh C); por consiguiente, no se recomienda la administración de ADEMPAS® a estos pacientes (véase Régimen de dosificación, Advertencias y precauciones especiales de empleo).
Pacientes con insuficiencia renal: En general, los valores de exposición normalizados para la dosis media y peso, fueron mayores en sujetos con insuficiencia renal que en sujetos con función renal normal. Los valores correspondientes para el metabolito principal fueron mayores en sujetos con insuficiencia renal en comparación con los sujetos sanos. En los sujetos con insuficiencia renal leve (depuración de creatinina de 80-50 ml/min), moderada (depuración de creatinina < 50-30 ml/min) o severa (depuración de creatinina < 30 ml/min) las concentraciones plasmáticas de riociguat (ABC) aumentaron en 43, 104 y 44% respectivamente (véase Régimen de dosificación).
No existen datos en pacientes con depuración de creatinina < 15 ml/min o sometidos a diálisis. Por consiguiente, no se recomienda la administración a pacientes con depuración de creatinina < 15 ml/min o sometidos a diálisis (véanse Dosis y vía de administración y Advertencias y precauciones especiales de empleo).
Debido a la elevada unión de riociguat a las proteínas plasmáticas, no se espera que sea dializable.
Género, diferencias entre razas y categorías de peso: Los datos farmacocinéticos no muestran diferencias importantes en la exposición al riociguat debidas al género, raza ni peso.
Relación farmacocinética/farmacodinámica: Hay una relación directa entre la concentración plasmática de riociguat y los parámetros hemodinámicos como la resistencia vascular sistémica y pulmonar, la presión arterial sistólica y el gasto cardiaco.
1 Ninguno de los criterios de valoración subsecuentes puede considerarse estadísticamente significativo en un sentido formal, ya que no se alcanzó la importancia estadística en el caso del cuestionario del tiempo transcurrido hasta el deterioro clínico en los análisis jerárquicos de las variables secundarias de eficacia.
2 Tres pacientes habían sido tratados previamente con un ERA y un análogo de la prostaciclina al mismo tiempo.
3 Ninguno de los criterios de valoración posteriores puede considerarse estadísticamente significativo en un sentido formal, ya que no se alcanzó la importancia estadística en el caso del cuestionario EQ-5D en los análisis jerárquicos de las variables secundarias de eficacia.
CONTRAINDICACIONES: Hipersensibilidad al producto o alguno de los excipientes de la fórmula.
ADEMPAS® está contraindicado durante el embarazo y lactancia (véase Embarazo y lactancia) y para menores de 18 años.
La administración conjunta de ADEMPAS® con nitratos o con donadores de óxido nítrico (tal como nitrito de amilo) de cualquier forma está contraindicada (véase Interacciones medicamentosas y de otro género), así como inhibidores de la fosfodiesterasa.
RESTRICCIONES DE USO DURANTE EL EMBARAZO Y LA LACTANCIA:
Embarazo: No existen datos adecuados sobre el uso de riociguat en mujeres embarazadas. Los estudios en animales han demostrado toxicidad reproductiva (véase Datos preclínicos de seguridad).
Por consiguiente, ADEMPAS® está contraindicado durante el embarazo (véase Contraindicaciones).
Los estudios de toxicidad para el desarrollo realizados en ratas y conejos han demostrado la toxicidad del riociguat para la función reproductora. En ratas se observó un aumento de la tasa de malformaciones cardiacas, así como una disminución de la tasa de gestación debida a la reabsorción temprana con una exposición sistémica materna de aproximadamente siete veces la exposición en humanos (2.5 mg tres veces al día). En conejos se observaron abortos y toxicidad fetal a partir de una exposición sistémica de aproximadamente tres veces la exposición en humanos (2.5 mg tres veces al día).
En las mujeres en edad reproductiva se deberá descartar embarazo antes del inicio del tratamiento, mensualmente durante el tratamiento y después de suspender el tratamiento mediante prueba de embarazo.
Lactancia: No se dispone de datos sobre el uso de riociguat en mujeres en periodo de lactancia. Los datos procedentes de los estudios en animales indican que riociguat se excreta en la leche.
Debido a la posible aparición de eventos adversos graves en los lactantes, no debe utilizarse ADEMPAS® durante la lactancia. Es necesario decidir si conviene interrumpir la lactancia o discontinuar/abstenerse del tratamiento, teniendo en cuenta la importancia del fármaco para la madre.
REACCIONES SECUNDARIAS Y ADVERSAS:
Resumen del perfil de seguridad: La seguridad de ADEMPAS® ha sido evaluada en estudios de fase III con más de 650 pacientes con HPTEC o HAP que recibieron por lo menos una dosis de riociguat (véase Propiedades farmacodinámicas).
El perfil de seguridad de ADEMPAS® en ambas muestras de población parece ser similar; por consiguiente, las reacciones adversas al medicamento (ADRs) identificados en los ensayos clínicos controlados con placebo de 12 y 16 semanas se presentan como frecuencias agrupadas en la tabla (véase la tabla 10).
La mayoría de los eventos adversos están causados por la relajación de las células musculares lisas en la vasculatura o en el tracto gastrointestinal.
Los eventos adversos reportados con más frecuencia, que ocurrieron en ≥ 10% de los pacientes tratados con ADEMPAS® (hasta 2.5 mg 3 veces al día, fueron cefalea, mareo, dispepsia, edema periférico, náuseas, diarrea y vómitos.
Con la observación prolongada en los estudios de extensión a largo plazo, no controlados, el perfil de seguridad fue similar al observado en los estudios de fase III controlados con placebo.
Se han observado casos graves de hemoptisis y hemorragia pulmonar, incluidos casos con desenlace mortal en pacientes con HPTEC o HAP tratados con ADEMPAS® (véase Advertencias y precauciones especiales de empleo).
Las tasas generales de suspensión del tratamiento debido a un evento adverso (AE) en los estudios pivotes controlados con placebo fueron bajas en todos los grupos de tratamiento (datos agrupados: 2.9% en el grupo de ADEMPAS® y 5.1% en el grupo placebo).
Lista tabular de los eventos adversos: Los eventos adversos al medicamento observados con ADEMPAS® se presentan en la tabla siguiente.
Están ordenadas según la clase de sistema u órgano (MedDRA versión [15.0]). El término MedDRA más adecuado se utiliza para describir una determinada reacción y sus sinónimos y trastornos relacionados.
Los eventos adversos al medicamento observados en los ensayos clínicos se clasifican por orden de frecuencia. Los grupos de frecuencia se definen con arreglo al siguiente acuerdo:
Muy frecuentes (≥ 1/10), frecuentes (de ≥ 1/100 a < 1/10), poco frecuentes (de ≥ 1/1,000 a < 1/100), raras (≥ 1/10,000 a < 1/1,000), muy raras (< 1/10,000).
Tabla 10. Lista completa de reacciones adversas al medicamento surgidas durante el tratamiento y reportadas en pacientes en los estudios de fase III (datos agrupados de CHEST 1 y PATENT 1)
|
Clase de sistema u órgano (MedDRA) |
Muy frecuentes |
Frecuentes |
Poco frecuentes |
|
Infecciones e infestaciones |
Gastroenteritis |
||
|
Trastornos de la sangre y del sistema linfático |
Anemia (incl. los parámetros analíticos correspondientes) |
||
|
Trastornos del sistema nervioso |
Mareo Cefalea |
||
|
Trastornos cardiacos |
Palpitaciones |
||
|
Trastornos vasculares |
Hipotensión |
||
|
Trastornos respiratorios, torácicos y mediastínicos |
Hemoptisis Epistaxis Congestión nasal |
Hemorragia pulmonar* |
|
|
Trastornos gastrointestinales |
Dispepsia Diarrea Náuseas Vómitos |
Gastritis Enfermedad por reflujo gastroesofágico Disfagia Dolores gastrointestinales y abdominales Estreñimiento Distensión abdominal |
|
|
Trastornos generales y alteraciones en el lugar de la administración |
Edema periférico |
* Se reportó hemorragia pulmonar mortal en los estudios de extensión a largo plazo, no controlados.
PRECAUCIONES EN RELACIÓN CON EFECTOS DE CARCINOGÉNESIS, MUTAGÉNESIS, TERATOGÉNESIS Y SOBRE LA FERTILIDAD:
Datos preclínicos de seguridad: Los datos no clínicos no revelaron algún riesgo específico para los humanos en los estudios convencionales de farmacología de seguridad, toxicidad de las dosis únicas, fototoxicidad, genotoxicidad y carcinogenicidad.
Los efectos observados en los estudios de toxicidad con dosis repetidas se debían principalmente a la actividad farmacodinámica exagerada de riociguat (efectos hemodinámicos y relajantes del músculo liso).
En ratas adolescentes de crecimiento rápido se observaron efectos sobre la formación ósea (por ejemplo, un aumento de la masa ósea total). No se observaron dichos efectos tras la administración de riociguat a ratas adultas.
En ratas no se observaron efectos sobre la fertilidad de machos o hembras.
Los estudios de toxicidad para el desarrollo realizados en ratas y conejos han demostrado la toxicidad reproductiva de riociguat. En ratas se observó un aumento de la tasa de malformaciones cardiacas, así como una disminución de la tasa de gestación debida a la reabsorción temprana con una exposición sistémica materna de aproximadamente siete veces la exposición en humanos (2.5 mg tres veces al día). En conejos se observaron abortos y toxicidad fetal a partir de una exposición sistémica de aproximadamente tres veces la exposición en humanos (2.5 mg tres veces al día).
ADEMPAS® no resultó carcinógeno en ratas sometidas a una exposición sistémica equivalente a 7 veces la exposición en humanos.
En el estudio de carcinogenicidad en ratones, con niveles de exposición cercanos a los de la exposición terapéutica en humanos, se observó deterioro de la motilidad gastrointestinal, disbiosis e inflamación crónica, seguido por degeneración de la mucosa e hiperplasia reactiva, así como un aumento, estadísticamente no significativo, de los tumores intestinales. Esta secuencia de eventos es una reacción típica en ratones a un estímulo tal como la inflamación o la degeneración; por consiguiente, estos tumores no se consideran tan relevantes para los humanos.
Fertilidad: No se han realizado estudios específicos con riociguat en humanos para evaluar los efectos sobre la fertilidad. En un estudio sobre la fertilidad en ratas machos y hembras no se observaron efectos (véase Datos preclínicos sobre seguridad).
Mujeres en edad reproductiva/anticoncepción: Las mujeres en edad reproductiva tienen que utilizar medidas anticonceptivas eficaces durante el tratamiento con ADEMPAS®.
INTERACCIONES MEDICAMENTOSAS Y DE OTRO GÉNERO:
Interacciones farmacocinéticas:
Efectos de otras sustancias sobre riociguat:
Riociguat se elimina principalmente por metabolismo oxidativo mediado por el citocromo P450 (CYP1A1, CYP3A4, CYP2C8, CYP2J2), excreción directa por vía biliar/fecal del fármaco sin cambios y excreción renal del fármaco sin cambios mediante filtración glomerular. A partir de los estudios in vitro se demostró que riociguat era un sustrato para las proteínas de transporte de membranas gp-P/BCRP. Los inhibidores o inductores de estas enzimas o transportadores pueden afectar la exposición a riociguat.
In vitro, el ketoconazol, clasificado como un potente inhibidor del CYP3A4 y de la glucoproteína P (gp-P), ha demostrado ser un inhibidor de múltiples vías metabólicas del CYP y gp-P/’proteína de resistencia del cáncer de mama (BCRP) para el metabolismo y la excreción de riociguat (véase Propiedades farmacocinéticas). La administración concomitante de 400 mg de ketoconazol una vez al día llevó a un aumento de 150% (con un intervalo hasta 370%) de la media del ABC de riociguat y a un aumento de 46% de la media de la Cmáx La vida media terminal aumentó desde 7.3 hasta 9.2 horas, y el depuración corporal total disminuyó desde 6.1 hasta 2.4 L/h.
Por consiguiente, no se recomienda el uso concomitante con inhibidores potentes de rutas metabólicas múltiples de CYP y gp-P/BCRP, como los antimicóticos azólicos (por ejemplo, ketoconazol, itraconazol) o los inhibidores de la proteasa del VIH (por ejemplo, ritonavir) (véase Advertencias y precauciones especiales de empleo Propiedades farmacocinéticas).
Los fármacos que son potentes inhibidores de la gp-P/BCRP, como el fármaco inmunosupresor ciclosporina A, deben emplearse con precaución (véase Advertencias y precauciones especiales de empleo).
De las isoformas de CYP recombinantes investigadas in vitro, el CYP1A1 catalizaba de forma más eficaz la formación del metabolito principal de riociguat. La clase de compuestos inhibidores de la tirosina-cinasa se identificó como potentes inhibidores del CYP1A1, de los que erlotinib y gefitinib presentaban la máxima potencia inhibidora in vitro. Por consiguiente, las interacciones entre fármacos mediadas por inhibición del CYP1A1 (véase Propiedades farmacocinéticas) podrían llevar a un aumento de la exposición al riociguat, especialmente en fumadores. Por consiguiente, los inhibidores potentes del CYP1A1 deben emplearse con precaución (véase Advertencias y precauciones especiales de empleo).
La administración conjunta de claritromicina (500 mg dos veces al día), que está clasificada como un inhibidor potente y selectivo del CYP3A4, y que se ha descrito también que es un inhibidor débil a moderado de la gp-P, aumentó de forma moderada la media del ABC de riociguat en 41% sin cambios significativos en la Cmáx. Esto no se considera clínicamente relevante.
Riociguat presenta en pH neutro una menor solubilidad que en medio ácido. La administración conjunta de fármacos que aumentan el pH del tracto gastrointestinal superior puede disminuir la biodisponibilidad oral.
El tratamiento previo y tratamiento concomitante con el inhibidor de la bomba de protones omeprazol (40 mg una vez al día) redujo la media del ABC de riociguat en 26%, y la media de la Cmáx en 35%. Esto no se considera clínicamente relevante.
La administración conjunta del antiácido hidróxido de aluminio/hidróxido de magnesio redujo la media del ABC de riociguat en 34%, y la media de la Cmáx en 56% (véase Posología y forma de administración). Los antiácidos deben administrarse por lo menos una hora después de tomar ADEMPAS®.
Bosentán, del que se ha descrito ser un inductor moderado del CYP3A4, llevó a una disminución de las concentraciones plasmáticas en estado de equilibrio de riociguat en pacientes con HAP de 27% sin que se viese afectada la eficacia de la combinación (véase Eficacia y seguridad clínicas).
El uso concomitante de riociguat con inductores potentes del CYP3A4 (por ejemplo, fenitoína, carbamazepina, fenobarbital o hierba de San Juan) también puede llevar a una reducción de la concentración plasmática de riociguat.
Efectos de riociguat sobre otras sustancias: Ni riociguat ni su metabolito principal son inhibidores o inductores de las principales isoformas del CYP (incluido el CYP 3A4) o de transportadores (por ejemplo, gp-P/BCRP) in vitro a las concentraciones plasmáticas terapéuticas.
La falta de interacciones farmacocinéticas entre riociguat y el sustrato de prueba del CYP3A4 midazolam se demostró in vivo.
Riociguat y su principal metabolito demostraron ser potentes inhibidores del CYP1A1 in vitro. Por consiguiente, no se pueden descartar interacciones clínicamente relevantes medicamento- medicamento con la administración conjunta de medicamentos que se eliminan predominantemente por biotransformación mediada por CYP1A1, como el erlotinib o el granisetrón.
Interacciones farmacodinámicas.
Nitratos: Los comprimidos de 2.5 mg de ADEMPAS® potenciaron el efecto reductor de la presión arterial de la nitroglicerina sublingual (0.4 mg) administrada 4 y 8 horas después de la ingesta. Por consiguiente, la administración conjunta de ADEMPAS® con nitratos o donadores de óxido nítrico (como el nitrito de amilo) de cualquier forma está contraindicada (véase Contraindicaciones).
Inhibidores de la PDE-5: Los estudios preclínicos en modelos de animales demostraron un efecto aditivo de reducción de la presión arterial sistémica cuando se combinaba riociguat con sildenafil o vardenafil. Al aumentar las dosis se observaron efectos súper aditivos sobre la presión arterial sistémica en algunos casos.
En un estudio exploratorio de la interacción con 7 pacientes con HAP tratados de forma estable con sildenafil (20 mg tres veces al día), las dosis únicas de riociguat (0.5 y 1 mg de forma secuencial) demostraron tener efectos hemodinámicos aditivos. En este estudio no se investigaron dosis superiores a 1 mg de riociguat.
Se realizó un estudio de combinación de 12 semanas en 18 pacientes con HAP tratados de forma estable con sildenafil (20 mg tres veces al día) y riociguat (de 1.0 a 2.5 mg tres veces al día) en comparación con sildenafil solo. En la sección de extensión a largo plazo (no controlado), el uso concomitante de sildenafil y riociguat dio lugar a una elevada tasa de abandonos, debidos principalmente a hipotensión. No se demostró algún efecto clínico favorable de la combinación en la población estudiada.
La administración conjunta de riociguat con inhibidores de la PDE-5 (como sildenafil, tadalafil, vardenafil) está contraindicada (véase Contraindicaciones).
Warfarina/fenprocumon: El tratamiento concomitante de riociguat y warfarina no alteró el tiempo de protrombina inducido por el anticoagulante. No se espera que el uso concomitante de riociguat con otros derivados de la cumarina (por ejemplo, fenprocumon) altere el tiempo de protrombina.
La falta de interacciones farmacocinéticas entre riociguat y el sustrato del CYP2C9 warfarina se demostró in vivo.
Ácido acetilsalicílico: Riociguat no potenció el tiempo de sangrado causado por el ácido acetilsalicílico ni afectó a la agregación plaquetaria en los humanos.
Alimentos y productos lácteos: No se observaron interacciones clínicamente relevantes con los alimentos (véase Propiedades farmacocinéticas).
Información adicional sobre poblaciones especiales: La exposición a riociguat en los fumadores de cigarrillos se reduce en 50-60% (véase Propiedades farmacocinéticas). Por consiguiente, se recomienda a los pacientes que dejen de fumar (véase Posología y forma de administración).
ALTERACIONES EN LOS RESULTADOS DE PRUEBAS DE LABORATORIO: No existe evidencia.
PRECAUCIONES GENERALES: Este producto se deberá administrar bajo vigilancia médica, para monitorear la tensión arterial del paciente.
Enfermedad venooclusiva pulmonar: Los vasodilatadores pulmonares pueden empeorar significativamente el estado cardiovascular de los pacientes con enfermedad venooclusiva pulmonar (EVOP). Por consiguiente, no se recomienda la administración de ADEMPAS® a dichos pacientes. En el caso de que aparezcan signos de edema pulmonar, debe considerarse la posibilidad de que exista una EVOP concomitante, por lo que debe suspenderse el tratamiento con ADEMPAS®.
Hemorragia de las vías respiratorias: En los pacientes con hipertensión pulmonar existe una probabilidad mayor de que aparezcan hemorragias de las vías respiratorias, especialmente en aquellos que reciben tratamiento con anticoagulantes.
El riesgo de hemorragias graves o mortales de las vías respiratorias puede aumentar aún más con el tratamiento con ADEMPAS®, especialmente cuando hay factores de riesgo, como episodios recientes de hemoptisis grave, incluidos los episodios tratados con embolización de arterias bronquiales. El médico encargado debe valorar de forma periódica la relación riesgo-beneficio en cada paciente de forma individual.
Acción vasodilatadora: ADEMPAS® tiene propiedades vasodilatadoras, que pueden dar lugar a una disminución de la presión arterial. Antes de recetar ADEMPAS®, el médico debe sopesar cuidadosamente si los pacientes con ciertos trastornos subyacentes pueden verse perjudicados por dichos efectos vasodilatadores (por ejemplo, pacientes tratados con antihipertensivos o que presentan hipotensión en reposo, hipovolemia, obstrucción severa del tracto de salida del ventrículo izquierdo o disfunción autonómica).
Uso concomitante con otros medicamentos: No se recomienda el uso concomitante de riociguat con inhibidores potentes de rutas metabólicas múltiples de CYP y gp-P/BCRP, como los antimicóticos azólicos (por ejemplo, ketoconazol, itraconazol) o los inhibidores de la proteasa del VIH (por ejemplo, ritonavir), debido a un marcado aumento en la exposición a riociguat (véase Interacciones medicamentosas y de otro género).
El uso concomitante de riociguat con inhibidores potentes del CYP1A1, como el inhibidor de la tirosina-cinasa, erlotinib e inhibidores potentes de la gp-P/BCRP, como el inmunosupresor ciclosporina A, puede aumentar la exposición al riociguat (véase Interacciones medicamentosas y de otro género). Estos fármacos deben utilizarse con precaución. La presión arterial debe vigilarse, y debe considerarse una disminución de la dosis de riociguat.
Grupos de pacientes no estudiados: ADEMPAS® no ha sido estudiado con los siguientes grupos de pacientes, por lo que no se recomienda su administración a:
• Pacientes con presión arterial sistólica < 95 mm Hg al inicio del tratamiento.
• Pacientes con insuficiencia hepática severa (Child-Pugh C).
• Pacientes con depuración de creatinina < 15 ml/min o sometidos a diálisis.
Efectos sobre la capacidad para conducir y utilizar máquinas: Se han descrito mareos, que pueden afectar a la capacidad para conducir y utilizar máquinas (véase Eventos adversos). Los pacientes deben estar conscientes de la manera cómo reaccionan a ADEMPAS® antes de conducir o utilizar máquinas.
DOSIS Y VÍA DE ADMINISTRACIÓN: Uso oral.
Régimen de dosificación:
El tratamiento sólo se debe iniciar y controlar por un médico/profesional sanitario con experiencia en el tratamiento de la HPTEC o la HAP.
Adultos:
Inicio del tratamiento: La dosis inicial recomendada es 1.0 mg tres veces al día durante 2 semanas. Los comprimidos deben administrarse tres veces al día, con aproximadamente 6 a 8 horas de diferencia, con o sin alimentos.
La dosis debe aumentarse en intervalos de 2 semanas mediante incrementos de 0.5 mg hasta un máximo de 2.5 mg tres veces al día, si la presión arterial sistólica es ≥ 95 mm Hg y el paciente no presenta signos ni síntomas de hipotensión. Si la presión sistólica desciende por debajo de 95 mm Hg debe mantenerse la dosis, siempre que el paciente no presente signos ni síntomas de hipotensión. Si en algún momento durante la fase de titulación de la dosis la presión arterial sistólica desciende por debajo de 95 mm Hg y el paciente presenta signos o síntomas de hipotensión, la dosis en cuestión deberá reducirse por 0.5 mg 3 veces al día.
Dosis de mantenimiento: La dosis individual establecida debe mantenerse, a no ser que aparezcan signos o síntomas de hipotensión. La dosis diaria total máxima de ADEMPAS® es de 7.5 mg. En el caso que se omita una dosis, debe continuarse el tratamiento con la dosis siguiente, como estaba previsto.
Si la dosis no se tolera, puede considerarse una reducción de la misma en cualquier momento.
Interrupción del tratamiento: En caso de que tenga que interrumpirse el tratamiento durante 3 días o más, debe reiniciarse el tratamiento con 1 mg tres veces al día durante 2 semanas, y el tratamiento debe continuarse con el régimen de ajuste de la dosis descrito anteriormente.
Información adicional sobre poblaciones especiales: La titulación de la dosis individual al inicio del tratamiento permite adaptar la dosis a las necesidades del paciente.
Pediatría: No se ha estudiado la seguridad ni la eficacia de ADEMPAS® en pacientes menores de 18 años. No se dispone de datos. Por consiguiente, no se recomienda la administración de ADEMPAS® en pacientes pediátricos.
Pacientes ancianos: En el caso de los adultos mayores (≥ 65 años) debe tenerse especial precaución durante la titulación de la dosis individual (Véase Propiedades farmacocinéticas).
Pacientes con insuficiencia hepática: Los pacientes con insuficiencia hepática leve (clase A de Child-Pugh) presentaban concentraciones plasmáticas de riociguat similares a las de los testigos sanos.
Los pacientes con insuficiencia hepática moderada (clase B de Child-Pugh) presentaron una mayor exposición a ADEMPAS®. Debe tenerse precaución especial durante el ajuste de la dosis individual.
Los pacientes con insuficiencia hepática intensa (clase C de Child-Pugh) no han sido objeto de estudio, por lo que no se recomienda la administración de ADEMPAS® a estos pacientes (véase Propiedades farmacocinéticas).
Pacientes con insuficiencia renal: Los pacientes con insuficiencia renal leve, moderada o severa (depuración de creatinina entre 80-15 ml/min) demostraron una mayor exposición a ADEMPAS® (véase Propiedades farmacocinéticas). Debe tenerse precaución especial durante el ajuste de la dosis individual.
Tabaquismo: A los pacientes que sean fumadores se les debe recomendar que dejen de fumar. Las concentraciones plasmáticas de riociguat de los fumadores se encuentran reducidas en comparación con las de los no fumadores. Puede ser necesario un ajuste de la dosis de riociguat en pacientes que hayan dejado de fumar o que hayan comenzado a fumar durante el tratamiento (véase Interacciones medicamentosas y de otro género, Propiedades farmacocinéticas).
La exposición al riociguat está reducida entre 50 y 60% en los fumadores de cigarrillos. Por consiguiente, se recomienda a los pacientes que dejen de fumar.
MANIFESTACIONES Y MANEJO DE LA SOBREDOSIFICACIÓN O INGESTA ACCIDENTAL: Se han reportado sobredosis accidentales con dosis diarias totales de 9-25 mg de riociguat entre 2 y 32 días.
Los eventos adversos fueron similares a los observados con dosis menores (véase Eventos adversos). No se dispone de un antídoto específico.
En caso de sobredosis, se deben adoptar las medidas sintomáticas habituales, según sea necesario.
En caso de que aparezca una hipotensión marcada, puede ser necesario aplicar medidas de asistencia cardiovascular.
Debido a la elevada unión a las proteínas plasmáticas de riociguat, no se espera que sea dializable.
PRESENTACIONES:
ADEMPAS® 0.5 mg.
Caja con 42 u 84 comprimidos.
ADEMPAS® 1 mg.
Caja con 42 u 84 comprimidos.
ADEMPAS® 1.5 mg.
Caja con 42 u 84 comprimidos.
ADEMPAS® 2 mg.
Caja con 42 u 84 comprimidos.
ADEMPAS® 2.5 mg.
Caja con 42 u 84 comprimidos.
RECOMENDACIONES SOBRE ALMACENAMIENTO: Consérvese a no más de 30°C y en lugar seco.
LEYENDAS DE PROTECCIÓN:
Su venta requiere receta médica. No se deje a alcance de los niños. No se use en pacientes menores de 18 años. Este medicamento contiene lactosa. No se use durante el embarazo y lactancia. Literatura exclusiva para el médico.
Reporte las sospechas de reacción adversa al correo:
farmacovigilancia@cofepris.gob.mx
Hecho en Alemania por:
Bayer Pharma AG
Kaiser-Wilhelm-Allee
51368 Leverkusen, Alemania
Distribuido por:
BAYER DE MÉXICO, S.A. de C.V.
Carr. México-Toluca Km 52.5
C.P. 52000, Lerma, México
Reg. Núm. 233M2014, SSA IV
143300415D0312
®Marca registrada

