

REGIVAS
DRONEDARONA
Tabletas
1 Caja,1 Envase(s) de burbuja,20 Tabletas,400 mg
1 Caja,1 Envase(s) de burbuja,60 Tabletas,400 mg
FORMA FARMACÉUTICA Y FORMULACIÓN:
Cada TABLETA contiene:
Clorhidrato de dronedarona equivalente a 400 mg de dronedarona
Excipiente cbp 1 tableta
INDICACIONES TERAPÉUTICAS: REGIVAS® está indicado en pacientes con fibrilación auricular (FA) paroxística o persistente, o flútter auricular (FLA), que se encuentren en ritmo sinusal o que van a ser cardiovertidos:
• para el mantenimiento del ritmo sinusal y/o
• para la reducción del riesgo de hospitalización cardiovascular o muerte.
FARMACOCINÉTICA Y FARMACODINAMIA:
Propiedades farmacocinéticas:
Absorción: Después de la administración oral con alimentos, dronedarona es bien absorbido (por lo menos en un 70%). Sin embargo, debido a un metabolismo pre-sistémico de primer paso, la biodisponibilidad absoluta de dronedarona (administrada con alimentos) es del 15%. La administración concomitante de alimentos incrementa la biodisponibilidad de dronedarona en promedio de 2 a 4 veces. Después de la administración oral con alimentos en el estómago, las concentraciones plasmáticas pico de dronedarona y del principal metabolito activo circulante (metabolito N-debutilo) se alcanzan entre 3 y 6 horas. Después de la administración de dosis repetidas de 400 mg, 2 veces al día, se logra el estado estable entre los 4 y 8 días de tratamiento, y el radio promedio de acumulación de dronedarona se encuentra en el rango de 2.6 a 4.5.
La Cmax de dronedarona en estado estable es de 84-147 ng/ml y la exposición del principal metabolito N-debutilo es similar a la del compuesto padre. La farmacocinética de dronedarona y de su metabolito N-debutilo, se modifican moderadamente en proporción con la dosis: un incremento del doble en la dosis resulta en aproximadamente de 2.5 a 3 veces un incremento en la Cmax y en el área bajo la curva (ABC).
Distribución: La unión de dronedarona in vitro a las proteínas plasmáticas y de su metabolito N-debutilo es > 98% y no es saturable. Ambos compuestos se unen principalmente a la albúmina. Después de su administración intravenosa (IV), el volumen de distribución en estado estable (Vss) se encuentra en el rango de 1,200 a 1,400 l.
Metabolismo: Dronedarona se metaboliza ampliamente, principalmente por CYP 3A4 (Ver Interacciones medicamentosas y de otro género). La principal vía metabólica incluye N-debutilación para formar el principal metabolito activo circulante seguido por oxidación, y por oxidación directa. El metabolito N-debutilo muestra actividad farmacodinámica, pero es de 3 a 10 veces menos potente que la dronedarona.
Eliminación: Después de su administración por vía oral, aproximadamente el 6% del fármaco marcado es excretado a través de la orina principalmente en forma de metabolitos (en la orina se elimina medicamento sin cambios), y el 84% se elimina a través de las heces principalmente en forma de metabolitos. Después de su administración IV, el aclaramiento plasmático de dronedarona es de 130 a 150 l/h. La vida media de eliminación terminal de dronedarona es de alrededor de 25-30 horas y la del metabolito N-debutilo es de alrededor de 20-25 horas. En los pacientes, dronedarona y su metabolito se eliminan completamente del plasma dentro de las dos primeras semanas después de completar un tratamiento con una dosis de 400 mg 2 veces al día.
Poblaciones especiales: La farmacocinética de dronedarona en pacientes con fibrilación auricular es consistente con la de sujetos sanos. Las principales fuentes de variabilidad en la exposición a dronedarona (edad, género, peso corporal, tratamiento concomitante, con inhibidores débiles a moderados de CYP 3A4) tienen una magnitud modesta (menos del doble).
Género: En pacientes del género femenino, la exposición a dronedarona es en promedio 30% mayor en comparación con pacientes del género masculino.
Vejez: Del total de sujetos incluidos en estudios clínicos con dronedarona, el 73% fue mayor de 65 años, y el 34% fue mayor de 75 años. En los pacientes de 65 años o mayores, la exposición a dronedarona fue 23% mayor en comparación con pacientes menores de 65 años.
Insuficiencia hepática: En sujetos con insuficiencia hepática moderada, la exposición a dronedarona en sus fracciones total y no conjugada se incrementan 1.3 y 2 veces, respectivamente. Las del metabolito activo, disminuyen 1.6 a 1.9 veces (Ver Dosis y vía de administración). No se ha estudiado el efecto de la insuficiencia hepática severa sobre la farmacocinética de dronedarona (Ver Contraindicaciones).
Insuficiencia renal: En los estudios clínicos se incluyó a pacientes con insuficiencia renal. En consistencia con la muy débil excreción renal de dronedarona, no se han observado modificaciones en la farmacocinética en los pacientes con insuficiencia renal, en particular en aquellos con insuficiencia renal severa (Ver Dosis y vía de administración).
PROPIEDADES FARMACODINÁMICAS:
Mecanismo de acción: En animales, dronedarona evita la fibrilación auricular o restablece el ritmo sinusal normal dependiendo del modelo empleado. Asimismo previene la taquicardia ventricular y la fibrilación ventricular en muchos modelos animales. Estos efectos son más probablemente el resultado de sus propiedades electrofisiológicas pertenecientes a las cuatro clases de Vaughan-Williams. Dronedarona es un bloqueador multicanal que inhibe la corriente de potasio (inclusive IK [Ach], IKur, IKr, IKs), prolongando de esta manera el potencial de acción cardiaco y los periodos refractarios (clase III). También inhibe la corriente de sodio (Clase Ib) y de Calcio (Clase IV). De forma no competitiva antagoniza las actividades adrenérgicas (Clase II). En modelos animales, dronedarona reduce la frecuencia cardiaca. Prolonga la duración del ciclo de Wenckebach y los intervalos AH-, PQ-, QT; sin un efecto marcado o con un débil incremento en los intervalos QTc, HV- y QRS. Incrementa el periodo refractario efectivo en la aurícula, en el nodo atrio-ventricular y en el ventrículo con un grado mínimo de dependencia de uso reverso.
Dronedarona disminuye la presión arterial sanguínea y la contractilidad del miocardio (dP/dt max) sin cambios en la fracción de eyección del ventrículo izquierdo y reduce el consumo de oxígeno del miocardio.
Dronedarona tiene propiedades vasodilatadoras, más acentuadas en las arterias coronarias (relacionadas con la activación de la vía del oxido nítrico) que en las arterias periféricas.
Dronedarona tiene efectos antiadrenérgicos indirectos; reduce la respuesta alfa-adrenérgica de la presión arterial sanguínea ante la adrenalina, y las respuestas beta 1 y beta 2 ante isoproterenol.
INFORMACIÓN CLÍNICA:
Reducción del riesgo cardiovascular de hospitalización o muerte: En el estudio ATHENA, multicéntrico, multinacional, doble ciego, aleatorizado y controlado con placebo, se demostró la eficacia de dronedarona en la reducción del riesgo de hospitalización cardiovascular o muerte por cualquier causa en pacientes con FA o flútter auricular, o con antecedentes de FA/flútter auricular y factores de riesgo adicionales.
Los pacientes debieron haber tenido al menos un factor de riesgo (incluyendo edad, hipertensión, diabetes, evento cerebrovascular previo, diámetro auricular ≥ 50 mm o FEVI < 0.40) junto con FA/flútter auricular y ritmo sinusal, ambos documentados dentro de los últimos 6 meses. Los pacientes se pudieron haber encontrado en FA/flútter auricular, o en ritmo sinusal después de conversión espontánea, o después de intervención.
Se aleatorizó a cuatro mil seiscientos veintiocho pacientes, y se les dio tratamiento hasta por un máximo de 30 meses (mediana de seguimiento: 22 meses) ya sea con 400 mg de dronedarona 2 veces al día (2,301 pacientes), o placebo (2,327 pacientes), además de una terapia convencional, entre la que se incluye betabloqueadores (71%), IECA o ARAII (69%), digital (14%), calcio antagonistas (14%), estatinas (39%), anticoagulantes orales (60%), terapia antiplaquetaria crónica (5%) y/o diuréticos (54%).
El punto final primario del estudio fue el tiempo transcurrido para la primera hospitalización por causas cardiovasculares o muerte por cualquier causa. Los puntos finales secundarios evaluados consistieron en muerte por cualquier causa, tiempo transcurrido para la primera hospitalización por causas cardiovasculares, tiempo transcurrido para muerte por causa cardiovascular. Además, también se evaluó el tiempo que transcurrió para la muerte súbita.
Los pacientes se encontraban en un rango de edad de 23 a 97 años, y el 42% eran mayores de 75 años. El 47% de los pacientes fueron del sexo femenino y en su mayoría caucásicos (89%).
La mayoría de la población tenía hipertensión (86%) y cardiopatía estructural (60%) (incluyendo enfermedad arterial coronaria: 30%; insuficiencia cardiaca congestiva [ICC]): 30%; disfunción ventricular izquierda < 45%: 12%). El 25% tenían FA en el periodo basal.
Dronedarona disminuyó la incidencia de hospitalización por causa cardiovascular o de muerte por cualquier causa en un 24%, comparativamente con placebo (p = 2 x 10-8).
En la Figura 1 se muestra la incidencia global de eventos. Las curvas por evento se separaron tempranamente y continuaron separándose a lo largo de los 30 meses de seguimiento.
Figura 1. Curvas de Kaplan-Meier de incidencia acumulada desde la aleatorización a la primera hospitalización por causa cardiovascular o muerte por cualquier causa
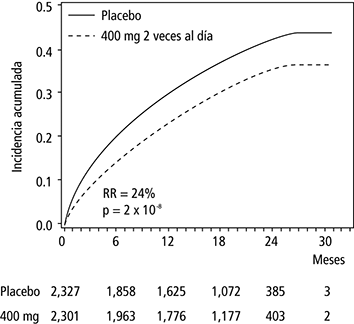
2 veces al día.
La reducción de hospitalización por causa cardiovascular o de muerte por cualquier causa fue consistente en todos los subgrupos, independientemente de las características basales o de los tratamientos (IECA, ARA II, betabloqueadores, digital, estatinas, calcioantagonistas, diuréticos) (Ver Figura 2).
Figura 2. Riesgo relativo (dronedarona 400 mg 2 veces al día versus placebo) estimado con un intervalo de confianza del 95%, de acuerdo con características basales seleccionadas –primera hospitalización por causa cardiovascular o muerte por cualquier causa.
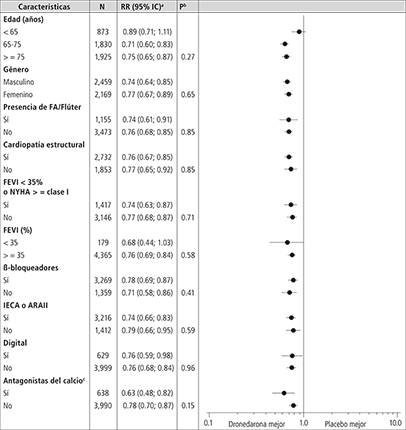
a Determinado por Modelo de Regresión de Cox.
b Valor de P, de interacción entre las características basales y el tratamiento, en base a un Modelo de Regresión de Cox.
c Calcioantagonistas con efectos reductores de la frecuencia cardiaca restringidos a diltiazem, verapamilo y bepridilo.
Se obtuvieron resultados similares en la incidencia de hospitalización por causa cardiovascular con una reducción de riesgo de 25.5% (p = 9 x 10-9).
Aunque una reducción en la hospitalización por FA fue dominante, el riesgo de una primera hospitalización no determinada por FA/flútter auricular disminuyó significativamente en un 14.5% en el grupo con dronedarona comparativamente con placebo (p = 0.0162). También hubo menos hospitalizaciones por empeoramiento de ICC (3.4% vs. 4.0% [placebo]), infarto del miocardio o angina inestable (2.1 vs. 2.6% [placebo]), y AIT/ECV (1.2 vs. 1.5% [placebo]) en el grupo con dronedarona.
Las hospitalizaciones por sangrado mayor (0.9% vs. 1% [placebo]), síncope (0.9% vs. 1% [placebo]) o arritmia ventricular (incluyendo extrasístoles ventriculares, taquicardia ventricular, fibrilación ventricular y otras arritmias ventriculares) (0.4% vs. 0.3% [placebo]) fueron similares en ambos grupos.
Además, la duración total de hospitalización fue menor en los pacientes con dronedarona en comparación con aquellos con placebo (9,995 noches vs 13,986 noches [placebo]); con una reducción mayor (47%) en el número de noches de hospitalización cardiovascular en la Unidad de Cuidados Intensivos/Unidad de Terapia de Cardiología.
El número de fallecimientos fue menor en el grupo que recibió dronedarona 400 mg 2 veces al día (n = 116 vs 139 [placebo], con reducción de riesgo de 15.6%, p = 0.176), con una marcada reducción del 30.2% en el riesgo de muerte cardiovascular (p = 0.025; 2.8% vs 4.0% [placebo]). Esto se debió a una reducción del 59.5% en el riesgo de muerte súbita (p = 0.0031; 0.6% vs. 1.5% [placebo]), y por una reducción del 38.3% en el riesgo de muerte por evento cerebrovascular (p = 0.2021; 0.5% vs. 0.8% [placebo]).
Curvas acumulativas de incidencia de Kaplan Meier (Ver Figuras 3 y 4) desde la aleatorización a la muerte por causa cardiovascular, que señalan un efecto temprano de dronedarona para evitar fallecimientos y que se mantuvo a lo largo del tiempo.
Figura 3. Curvas de incidencia acumulada de Kaplan Meier desde la aleatorización hasta muerte por causa cardiovascular durante el estudio
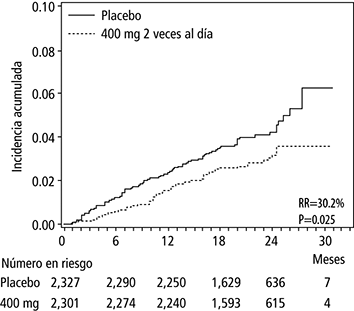
2 veces al día.
Figura 4. Curvas de Kaplan-Meier de incidencia acumulada de muerte súbita durante el estudio
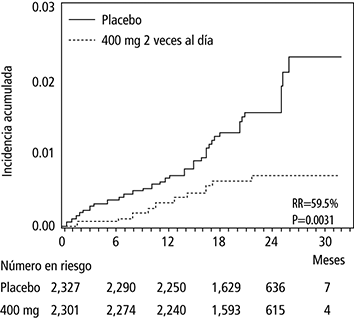
2 veces al día.
Mantenimiento del ritmo sinusal: En los estudios EURIDIS y ADONIS, se aleatorizaron en forma ambulatoria a un total de 1,237 pacientes con un episodio previo de FA o de flútter auricular y se les inició dronedarona a la dosis de 400 mg, 2 veces al día (n = 828) o placebo (n = 409) sobre su tratamiento de base (incluyendo anticoagulantes orales, betabloqueadores, IECAs, ARAII, antiagregantes plaquetarios de uso crónico, diuréticos, estatinas, digital, y calcioantagonistas). Los pacientes tenían documentado con ECG al menos un episodio de FA/flútter auricular durante los últimos 3 meses y se mantenían en ritmo sinusal durante al menos una hora, con seguimiento durante 12 meses.
Los pacientes se encontraban en rangos de edad de 20 a 88 años, la mayoría caucásicos (97%) y de género masculino (69%). Las comorbilidades más comunes fueron hipertensión arterial (56.8%), cardiopatía estructural (41.5%), incluyendo enfermedad arterial coronaria (21.8%).
En las bases de datos de EURIDIS y ADONIS, así como en estudios individuales, dronedarona consistentemente retardó el tiempo para la aparición del primer evento de FA/flútter auricular (punto final primario).
En comparación con placebo, dronedarona disminuyó el riesgo de la primera recurrencia de FA/flútter auricular durante los 12 meses del estudio en un 25% (p = 0.00007). La mediana del tiempo desde la aleatorización a la primera recurrencia en el grupo con dronedarona fue de 116 días, esto es, 22 veces más que en el grupo con placebo (53 días). La mayoría (60%) de las primeras recurrencias fueron sintomáticas. Dronedarona también retardó el tiempo para la aparición de la primera recurrencia sintomática de FA/flútter auricular en ambos estudios. Con dronedarona 400 mg, 2 veces al día, el porcentaje de pacientes con una primera recurrencia de FA/flútter auricular asintomático a un año fue de 62.3%.
En el estudio DAFNE, en el cual se inició la administración de dronedarona antes de la conversión, la mediana del tiempo de la recurrencia de FA, medida por telemetría y por ECG de 12 derivaciones, fue de 60 días en el grupo al que se le administró dronedarona a la dosis de 400 mg, 2 veces al día, comparativamente con 5 días al grupo con placebo. Dronedarona a la dosis de 400 mg, 2 veces al día disminuyo en 55% (p = 0.001) el riesgo de la primera recurrencia de FA comparativamente con placebo durante el periodo de 6 meses del estudio.
Control de la frecuencia ventricular: En el estudio ERATO, doble ciego, controlado con placebo, con duración de 6 meses, participaron 174 pacientes con FA sintomática permanente (con duración de 6 meses) y se les aleatorizó con dronedarona 400 mg 2 veces al día (85 pacientes) o placebo (89 pacientes), además de su tratamiento de base. La edad de los pacientes estaba en el rango de los 31 a los 86 años, la mayoría de ellos caucásicos (99%), del género masculino (70%). Las comorbilidades más comunes fueron hipertensión arterial (49%) y cardiopatía estructural (39%).
Al día 14 del estudio, dronedarona disminuyó el promedio de la frecuencia ventricular comparativamente con placebo. Este efecto fue independiente del control logrado con el tratamiento de base, y se conservó 4 meses después de iniciado el tratamiento con una disminución promedio comparada con el estado basal de 8.8 latidos por minuto (p < 0.0001). Bajó el consumo de betabloqueadores, digital y calcioantagonistas bradicardizantes, la disminución promedio de la frecuencia cardiaca ventricular, y con un IC 95%, fue de 14.9 latidos por minuto (-20; -10), 11.5 latidos por minuto (-17; -6.4) y 5.05 (-11; 0.92), respectivamente.
También se observó una disminución de la frecuencia ventricular durante un ejercicio máximo al día 14 (-24.5 latidos por minuto, p < 0.0001).
En las bases de datos de EURIDIS y ADONIS, los pacientes que fueron tratados con 400 mg de dronedarona 2 veces al día, tuvieron promedios más bajos de frecuencias ventriculares al tiempo de la primera recurrencia (103.4 latidos por minuto) comparativamente con los pacientes con placebo (117.1 latidos por minuto) determinado por telemetría, p < 0.0001).
Estudio ANDROMEDA: En este estudio, los pacientes recientemente hospitalizados con insuficiencia cardiaca sintomática y disfunción sistólica ventricular izquierda severa (índice de movimiento de pared ≤ 1.2) fueron aleatorizados a recibir REGIVAS® 400 mg dos veces al día o placebo, con un punto final primario compuesto de mortalidad por todas las causas u hospitalización por insuficiencia cardiaca. Después del enrolamiento de 627 de 1000 pacientes planeados (310 y 317 en los grupos de dronedarona y placebo respectivamente) y un seguimiento medio de 63 días. El estudio se interrumpió prematuramente debido a que se observó una desproporción de muertes en el grupo con dronedarona [n= 25 versus 12 (placebo), p = 0.027]. La razón principal de muerte fue el empeoramiento de la insuficiencia cardiaca: existió también un exceso en hospitalización por causas cardiovasculares en el grupo de dronedarona (71) vs placebo (51). (Ver Contraindicaciones).
Las poblaciones enroladas en los estudios ANDROMEDA y ATHENA fueron significativamente diferentes. Los pacientes enrolados en ANDROMEDA tenían insuficiencia cardiaca relativamente severa , habían sido hospitalizados o referidos a una clínica especializada, por el empeoramiento de los síntomas de insuficiencia cardiaca o disnea significativa. Cabe señalar que estos pacientes habían mejorado clínicamente al momento del enrolamiento y es el historial de descompensación era lo que los caracterizaba. Los pacientes enrolados en ANDROMEDA fueron predominantemente de una Clase funcional II NYHA (40%) y III NYHA (57%), solamente el 38% tenían un historial de fibrilación o flútter auricular (25% tenían fibrilación auricular al momento de la aleatorización). En contraste en ATHENA, 71% de los pacientes no tenían falla cardiaca, el 25% tenía una clase funcional I o II NYHA y sólo el 4% estaban en clase funcional III. Todos los pacientes tenían historial de fibrilación o flútter auricular.
CONTRAINDICACIONES:
Pacientes con intolerancia a la galactosa: Debido a la presencia de lactosa en los excipientes, los pacientes con los raros problemas hereditarios de intolerancia a la galactosa, deficiencia de lactasa Lapp o malabsorción de glucosa-galactosa, no deben tomar este medicamento.
• Hipersensibilidad al ingrediente activo o a los excipientes contenidos en su formulación.
• Bloqueo AV de segundo o de tercer grado, bloqueo de rama completo, bloqueo distal, disfunción del nodo sinusal, defectos de conducción auricular o síndrome del seno enfermo (excepto cuando se usa en combinación con un marcapasos).
• Bradicardia < 50 latidos por minuto.
• Intervalo QTc Bazzet ≥ 500 mseg.
• Pacientes con FA permanente con una duración de la FA ≥ 6 meses (o duración desconocida) y cuando un intento de restaurar el ritmo sinusal no es considerado por el médico tratante.
• Condiciones hemodinámicas inestables.
• Historia de, o padecimiento actual de insuficiencia cardiaca o disfunción sistólica ventricular izquierda.
• Administración simultánea con inhibidores potentes del CYP 3A4, tales como ketoconazol, itraconazol, voriconazol, telitromicina, claritromicina, nefazodona, ciclosporina y ritonavir (Ver Interacciones medicamentosas y de otro género).
• Productos que puedan provocar torsades de pointes, tales como fenotiazinas, cisaprida, bepridilo, antidepresivos tricílicos, terfenadina y ciertos macrólidos orales, antiarrítmicos Clase I y III (Ver Interacciones medicamentosas y de otro género).
• Insuficiencia hepática severa.
• Pacientes con toxicidad hepática o pulmonar relacionada al uso previo de amiodarona.
• Embarazo (Ver Restricciones de uso durante el embarazo y lactancia).
• Lactancia (Ver Restricciones de uso durante el embarazo y lactancia).
• Pacientes alérgicos a las sulfas.
RESTRICCIONES DE USO DURANTE EL EMBARAZO Y LA LACTANCIA:
Embarazo: No se dispone de información sobre el uso de dronedarona en mujeres embarazadas. Estudios en animales han demostrado toxicidad sobre la reproducción (teratogenicidad en ratas) (Ver Precauciones en relación con efectos de carcinogénesis, mutagénesis, teratogénesis y sobre la fertilidad). Dronedarona está contraindicada en mujeres embarazadas (Ver Contraindicaciones). Las mujeres en edad reproductiva deben emplear métodos adecuados de control de la fertilidad durante el tratamiento con REGIVAS®.
Lactancia: No se sabe si dronedarona se excreta a través de la leche materna. Estudios en animales han demostrado la excreción de dronedarona y sus metabolitos en la leche. Las mujeres no deben dar lactancia al seno materno mientras se encuentren en tratamiento con REGIVAS® (Ver Precauciones en relación con efectos de carcinogénesis, mutagénesis, teratogénesis y sobre la fertilidad). Por lo tanto, cuando dronedarona está indicada, la madre debe ser advertida a descontinuar.
REACCIONES SECUNDARIAS Y ADVERSAS: El perfil de seguridad de 400 mg de dronedarona 2 veces al día en pacientes con FA o con flútter auricular se sustenta en 5 estudios controlados con placebo: ATHENA, EURIDIS, ADONIS, ERATO y DAFNE. En estos estudios se aleatorizaron y dio tratamiento a un total de 6,285 pacientes. De éstos, 3,282 pacientes fueron tratados con 400 mg de dronedarona 2 veces al día, y 2,785 con placebo. El promedio de la exposición a dronedarona entre los estudios fue de 13 meses. En ATHENA, el máximo de seguimiento fue de 30 meses.
La evaluación de factores intrínsecos, tales como género o edad, sobre la incidencia de cualquier tratamiento emergente como consecuencia de eventos adversos, no es sugestivo de un exceso de eventos adversos en particular en ninguno de los subgrupos.
En los estudios clínicos, la interrupción prematura del tratamiento por reacciones adversas ocurrió en 11.8% en el grupo tratado con dronedarona y 7.7% en el grupo con placebo. Las razones mas comunes para interrumpir el tratamiento con REGIVAS® fueron por alteraciones gastrointestinales (3.2% de los pacientes vs 1.8% en el grupo con placebo).
Las reacciones secundarias más frecuentemente observadas en los pacientes con dronedarona 400 mg, 2 veces al día en los 5 estudios fueron diarrea, náuseas y vómitos, fatiga y astenia.
La Tabla 1 muestra las reacciones adversas asociadas con la administración de 400 mg de dronedarona 2 veces al día en pacientes con FA y flútter auricular, presentados por clase por órganos y sistemas, y por orden decreciente de frecuencia.
Las reacciones adversas incluidas en la clase por órganos y sistemas en "Investigación" se muestran por separado.
Las frecuencias se definen como: muy común (≥ 1/10); común (≥ 1/100, < 1/10); poco común (≥ 1/1,000), < 1/100); raro (≥ 1/10000, < 1/1000); muy raro (< 1/10000).
Dentro de cada grupo de frecuencia, las reacciones indeseables se muestran en orden decreciente de severidad.
Tabla 1. Reacciones Adversas medicamentosas
|
Clase por órgano y sistema |
Común (≥ 1/100, < 1/10) |
Poco común (≥ 1/1000, <1/100) |
Raro (≥ 1/10 000, < 1/1000) |
|
Alteraciones cardiacas |
Bradicardia |
||
|
Trastornos del sistema nervioso |
Ageusia, disgeusia |
||
|
Alteraciones gastrointestinales |
Diarrea |
||
|
Vómitos |
|||
|
Náuseas |
|||
|
Dolor abdominal |
|||
|
Dispepsia |
|||
|
Piel y tejido celular subcutáneo |
Erupciones cutáneas (generalizadas, maculares, maculo-papulares) Prurito |
Eritemas (inclusive eritema y erupciones cutáneas eritematosas) |
|
|
Eczema |
|||
|
Reacciones de fotosensibilidad |
|||
|
Dermatitis alérgica |
|||
|
Dermatitis |
|||
|
Trastornos generales y en el sitio de administración |
Fatiga Astenia |
Además, también se ha informado de las siguientes alteraciones en resultados de laboratorio/parámetros de ECG con frecuencia muy común (> 1/10) con la administración de 400 mg de dronedarona 2 veces al día:
|
Placebo (N = 2875) |
Dronedarona 400 mg 2 veces al día (N = 3282) |
|
|
Elevación de la creatinina sanguínea ≥ 10% cinco días después de iniciado el tratamiento |
20.6% |
50.9% |
|
(N = 2237) |
(N = 2701) |
|
|
Prolongación del QTc Bazzet (> 450 mseg en hombres y > 470 mseg en mujeres) |
18.7% |
27.6% |
Experiencia post-comercialización: Las siguientes reacciones adversas han sido identificadas durante el uso de REGIVAS®. Estas reacciones adversas derivan de reportes espontáneos y por ello, la frecuencia no se conoce (no puede ser estimada con los datos disponibles).
Trastornos cardiacos:
Insuficiencia cardiaca congestiva*.
Algunos casos de flútter auricular 1:1 con conducción atrioventricular ha sido reportado.
Trastornos hepatobiliares:
Anormalidades en pruebas de función hepáticas.
Daño hepatocelular, que incluye falla hepática que amenaza la vida.
Alteraciones vasculares:
Vasculitis, incluyendo vasculitis leucocitoclástica.
Trastornos respiratorios:
Enfermedad pulmonar intersticial, incluyendo neumonitis y fibrosis pulmonar**.
Alteraciones del sistema inmune:
Reacciones anafilácticas, incluyendo angioedema.
* La insuficiencia cardiaca congestiva es una complicación de diversas patologías cardiacas, incluyendo fibrilacion y flútter auricular; sin embargo, la posibilidad de estar relacionada al medicamento no puede ser excluida.
** Algunos pacientes fueron expuestos previamente a amiodarona.
PRECAUCIONES EN RELACIÓN CON EFECTOS DE CARCINOGÉNESIS, MUTAGÉNESIS, TERATOGÉNESIS Y SOBRE LA FERTILIDAD: En estudios de carcinogenicidad de 2 años, la dosis más elevada de dronedarona administrada durante los 24 meses fue de 70 mg/kg/día en rata y de 300 mg/kg/día en ratón. Se observaron un aumento en la incidencia de tumores de las glándulas mamarias en ratones hembras, sarcomas histiocíticos en ratones y hemangiomas de los ganglios mesentéricos en ratas, sólo en los casos en los que se administraron las dosis más elevadas (correspondientes a una exposición de 5 a 10 veces la dosis terapéutica para humanos). Los hemangiomas no son precancerosos y no se transforman en hemangiosarcomas malignos en los animales ni en los humanos. Ninguna de estas observaciones se considera que sea de importancia para los seres humanos.
En estudios de toxicidad crónica, se observó fosfolipidosis leve y reversible (acumulación de macrófagos espumosos) en los ganglios mesentéricos, principalmente en la rata. Este efecto se considera específico de estas especies, y sin importancia para los humanos.
Dronedarona ocasionó marcados efectos sobre el desarrollo embrio-fetal en dosis elevadas en ratas, tales como un incremento en las pérdidas post-implantación del cigoto, reducción de peso fetal y placentario, y malformaciones externas, viscerales y esqueléticas.
Dronedarona no tiene efectos genotóxicos, basados en una prueba in vivo de micronúcleo en ratón y en 4 pruebas in vitro: Prueba de Ames con y sin activación metabólica, una prueba de reparación de DNA en hepatocitos de rata, un ensayo de mutación de genes en fibroblastos de hámsters y un estudio de citogenética de linfocitos humanos.
No se ha demostrado que dronedarona afecte la fertilidad en estudios en animales.
INTERACCIONES MEDICAMENTOSAS Y DE OTRO GÉNERO: Dronedarona es metabolizado principalmente por el CYP 3A4 (Ver Propiedades farmacocinéticas) y es un inhibidor moderado del CYP 3A4, e inhibidor leve de CYP 2D6. Por lo tanto, los inhibidores e inductores de CYP 3A4 tienen el potencial de interactuar con dronedarona, así como dronedarona tiene también el potencial de interactuar con los medicamentos cuyos substratos sean CYP 3A4 y CYP 2D6. También tiene el potencial de inhibir a los transportadores de la glucoproteína P (P-gP). Dronedarona no tiene potencial significativo para inhibir a CYP 1A2, CYP 2C9, CYP 2C19, CYP 2C8 y CYP 2B6.
Es de esperarse una potencial interacción farmacodinámica con los betabloqueadores, calcioantagonistas y digital.
En los estudios clínicos, los pacientes tratados con dronedarona recibieron una variedad de medicamentos concomitantemente, entre los que se incluyen betabloqueadores, digital, calcioantagonistas (inclusive aquellos con efectos bradicardizantes), estatinas y anticoagulantes orales.
Efecto de otros medicamentos sobre REGIVAS®:
Medicamentos que provocan torsades de pointes: Los medicamentos que provocan torsades de pointes tales como las fenotiazinas, cisaprida, bepridil, antidepresivos tricíclicos, ciertos macrólidos orales, terfenadina y antiarrítmicos Clase I y III están contraindicados debido al potencial riesgo pro-arrítmico.
Inhibidores potentes del CYP 3A4: La administración repetida de dosis de 200 mg diarios de ketoconazol resultó en un incremento de 17 veces en la exposición a dronedarona. Por lo tanto, está contraindicado el uso concomitante de ketoconazol así como de otros inhibidores potentes del CYP 3A4, tales como itraconazol, voriconazol, ritonavir, telitromicina, claritromicina, ciclosporina, nefazodona (Ver Contraindicaciones).
Inhibidores débiles/moderados del CYP 3A4:
Calcioantagonistas: Los calcioantagonistas son substrato y/o inhibidores moderados del CYP 3A4. Además, los calcioantagonistas con propiedades bradicardizantes tienen el potencial de interactuar con dronedarona desde un punto de vista farmacodinámico.
La administración de dosis repetidas de diltiazem (240 mg 2 veces al día), verapamilo (240 mg una vez al día) y nifedipino (20 mg 2 veces al día), resultó en un incremento en la exposición a dronedarona de 1.7, 1.4 y 1.2 veces, respectivamente. Los calcioantagonistas también ven incrementada su exposición por acción de dronedarona (400 mg 2 veces al día) (verapamilo 1.4 veces y nisoldipino 1.5 veces). En estudios clínicos, no hubo problemas de seguridad cuando se administraron concomitantemente dronedarona con calcioantagonistas con propiedades bradicardizantes.
Además, debido a la interacción farmacocinética y a la posible interacción farmacodinámica, los calcioantagonistas con efectos depresores sobre el seno y sobre el nodo atrio-ventricular, tales como verapamilo y diltiazem deben emplearse con precaución en combinación con dronedarona. Estos medicamentos deben ser iniciados a dosis bajas y una titulación a la alza debe ser efectuada únicamente después de una evaluación por electrocardiograma. En pacientes que ya toman calcioantagonistas al momento del inicio de dronedarona, un electrocardiograma debe ser realizado y la dosis del calcioantagonista debe ser ajustada si se requiere.
Eritromicina: Las dosis repetidas de eritromicina (500 mg tres veces al día por 10 días) resultaron en un incremento en la exposición del estado estable de dronedarona en 3.8 veces.
Otros inhibidores del CYP3A4 son también responsables del incremento de exposición de la dronedarona.
Inductores del CYP 3A4: Rifampicina (600 mg 1 vez al día) disminuyó la exposición a dronedarona 5 veces sin modificaciones mayores en la exposición a sus metabolitos activos. Por lo tanto, la administración conjunta de rifampicina y otros inductores potentes de CYP 3A4 como fenobarbital, carbamazepina, fenitoína, hierba de San Juan, no son recomendables ya que estos incrementan la exposición a dronedarona.
Efectos de REGIVAS® sobre otros medicamentos:
Interacción con medicamentos metabolizados por CYP 3A4:
• Estatinas substratos de CYP 3A4 y/o substratos de glucoproteína P:
Dronedarona puede incrementar la exposición a las estatinas, las cuales son substratos de CYP 3A4. Dronedarona (400 mg 2 veces al día) incrementó la exposición a simvastatina y simvastatina ácida en 4 y 2 veces, respectivamente. Se estima que dronedarona también pueda incrementar la exposición de lovastatina, atorvastatina y pravastatina dentro del mismo grado que simvastatina ácida. En estudios clínicos, no hubo evidencia de problemas de seguridad cuando se administró dronedarona con estatinas metabolizadas por CYP 3A4.
Como las dosis elevadas de estatinas incrementan el riesgo de miopatía, la administración simultánea de estatinas que tienen como substrato a CYP 3A4 y/o substrato de P-gP (simvastatina, lovastatina, atorvastatina y pravastatina) deben usarse con precaución y vigilar a los pacientes ante la manifestación clínica de toxicidad muscular.
Es poco probable una interacción significativa entre dronedarona y estatinas que no tienen como substrato a CYP 3A4/p-gP, como son fluvastatina y rosuvastatina.
• Calcioantagonistas: La interacción de dronedarona con calcioantagonistas se describe más adelante.
• Sirolimus, tacrolimus: Dronedarona podría incrementar las concentraciones plasmáticas de tacrolimus y sirolimus. Es recomendable el monitoreo de las concentraciones plasmáticas de los anteriores, y el ajuste de su dosis en caso de administrarse concomitantemente con dronedarona.
• Anticonceptivos orales: No se observaron disminuciones en las concentraciones de etinilestradiol ni de levonorgestrel en sujetos sanos a los que se les administró dronedarona (800 mg 2 veces al día) conjuntamente con contraceptivos orales.
Interacciones con medicamentos metabolizados por CYP 2D6: betabloqueadores, antidepresivos:
• Betabloqueadores: Los betabloqueadores que son metabolizados por CYP 2D6 pueden ver incrementada su exposición por dronedarona. Además, los betabloqueadores tienen el potencial de interactuar con dronedarona desde un punto de vista farmacodinámico. Droneradona 800 mg al día incrementó la exposición a metoprolol 1.6 veces y a propranolol 1.3 veces (esto es, muy por debajo de las 6 veces las diferencias observadas entre los metabolizadores pobres y extensos de CYP 2D6). En estudios clínicos, se observó con más frecuencia bradicardia cuando se administró dronedarona en combinación con betabloqueadores. Debido a la interacción farmacocinética y a una posible interacción farmacodinámica, los betabloqueadores deben utilizarse con precaución simultáneamente con dronedarona. Estos medicamentos deben ser iniciados a dosis bajas efectuando una titulación a la alza únicamente después de una evaluación electrocardiográfica. En pacientes que ya toman beta-bloqueadores al momento de iniciar dronedarona, un electrocardiograma debe ser realizado y ajustar la dosis de los betabloqueadores si es necesario.
• Antidepresivos: Dado que dronedarona es un inhibidor débil de CYP 2D6 en humanos, se espera que tenga una limitada interacción con los antidepresivos metabolizados por CYP 2D6.
Interacción de substrato P-gP:
• Digoxina: Dronedarona (400 mg 2 veces al día) incrementa la exposición a digoxina 2.5 veces mediante inhibición del transportador P-gP. Además, digital tiene el potencial de interactuar con dronedarona desde un punto de vista farmacodinámico. En estudios clínicos, se observaron elevaciones en los niveles de digital y/o trastornos gastrointestinales cuando se administró dronedarona concomitantemente con digital. Debido a una interacción farmacocinética y a una posible interacción farmacodinámica, debe administrarse digoxina con precaución simultáneamente con dronedarona y monitorear cuidadosamente los niveles de digoxina, especialmente durante la primera semana de tratamiento conjunto. Si el tratamiento con digoxina continua, se recomienda reducir la dosis de digoxina en un 50% monitorizar niveles séricos de digoxina estrechamente y observar datos clínicos de toxicidad en el paciente.
• Dabigatrán: Cuando dabigatrán etexilato fue coadministrado con una dosis repetida de 400 mg de dronedarona 2 veces al día, el ABC 0-24 y la Cmax se incrementaron un 100% y 70% respectivamente: No se observó efecto de dronedarona en la depuración renal de dabigatrán.
• Otros substratos de P-gP: Dronedarona inhibe P-gP y por lo tanto, puede haber interacciones con doxorrubicina, fexofenadina y talinolol.
Interacción con warfarina y losartán (substratos de CYP 2C9):
• Warfarina y otros antagonistas de vitamina K: Dronedarona (600 mg 2 veces al día) incrementó 1.2 veces la exposición a S-warfarina sin cambios en R warfarina y con sólo un incremento de 1.07 en el INR.
Una mayor cantidad de pacientes presentaron elevaciones clínicamente significativas del INR (≥ 5), usualmente 1 semana después del inicio de dronedarona vs. el grupo placebo en pacientes quienes tomaban anticoagulantes orales en el estudio ATHENA. Sin embargo, no se observó un exceso de riesgo de sangrado en el grupo de dronedarona.
En la etapa de comercialización del producto han sido reportados casos de incremento del INR con y sin sangrado en pacientes que toman antagonistas de vitamina K por vía oral y que son tratados con dronedarona. El INR debe ser monitoreado después del inicio de dronedarona en pacientes que toman antagonistas de vitamina K según su indicación.
• Losartán y otros ARA II: No se observaron interacciones entre dronedarona y losartán, y no son de esperarse interacciones entre dronedarona y otros ARAII.
Interacción con teofilina (substrato de CYP 1A2): Dronedarona 400 mg 2 veces al día no incrementa la exposición de teofilina en estado estable.
Interacción con metformina (substrato de OCT1 y OCT2): No se ha observado interacción entre dronedarona y metformina, un substrato de OCT1 y OCT2.
Interacción con omeprazol (substrato de CYP2C19): No se ha observado interacción entre dronedarona y omeprazol, un substrato de CYP2C19.
Interacción con clopidogrel: No se ha observado interacción entre dronedarona y clopidogrel.
Otra información: Pantoprazol (40 mg 1 vez al día), un medicamento que eleva el pH gástrico sin ningún efecto sobre el citocromo P450, no interactúa significativamente sobre la farmacocinética de dronedarona.
Jugo de toronja (inhibidor de CYP 3A4): El consumo de dosis repetidas de 300 ml de jugo de toronja 3 veces al día, dio lugar a una exposición del triple de dronedarona. Por lo tanto, debe advertirse a los pacientes que eviten bebidas que contengan jugo de toronja mientras se encuentren bajo tratamiento con dronedarona.
ALTERACIONES EN LOS RESULTADOS DE PRUEBAS DE LABORATORIO: Se ha observado un incremento en la creatinina plasmática con la administración de 400 mg de dronedarona 2 veces al día en voluntarios sanos y en pacientes. Este incremento tiene lugar en forma temprana después del inicio del tratamiento y alcanza una meseta después de los 7 días. El promedio de la elevación en los pacientes con FA y con flútter auricular es de aproximadamente 10 μmol/l. Los niveles regresan a los valores basales a la semana de haber interrumpido el tratamiento.
PRECAUCIONES GENERALES:
Niños: La eficacia y seguridad en personas menores de 18 años no ha sido establecida, el uso de REGIVAS® en estos pacientes no se recomienda.
Pacientes que desarrollan FA permanente durante el tratamiento: Un estudio clínico en pacientes con FA permanente (FA con duración al menos de 6 meses) y factores de riesgo cardiovascular fue suspendido prematuramente debido a un exceso en la muerte cardiovascular, evento vascular cerebral y hospitalización cardiovascular no planeada. Se recomienda realizar un electrocardiograma al menos cada 6 meses mientras los pacientes se encuentren bajo tratamiento con REGIVAS®.Si estos pacientes desarrollan FA permanente el tratamiento con REGIVAS® debe ser descontinuado.
Pacientes con Insuficiencia Cardiaca Nueva o disfunción sistólica ventricular izquierda durante el tratamiento: REGIVAS® está contraindicado en pacientes en condiciones hemodinámicas inestables, historia de, o padecimiento actual de insuficiencia cardiaca o disfunción sistólica ventricular izquierda. Se debe recomendar a los pacientes que consulten a su médico si llegan a desarrollar signos o síntomas de insuficiencia cardiaca, tales como aumento de peso dependiente de edema, o incremento en la dificultad respiratoria. Si el paciente desarrolla insuficiencia cardiaca, el tratamiento con REGIVAS® debe ser descontinuado.
Los pacientes deben tener un seguimiento relativo al desarrollo de disfunción sistólica ventricular izquierda, durante el tratamiento. Si se desarrolla disfunción sistólica ventricular izquierda, el tratamiento con REGIVAS® debe ser descontinuado.
Daño hepático: Daño hepático de tipo hepatocelular incluyendo insuficiencia hepática aguda que pone en riesgo la vida, ha sido reportado en pocos pacientes tratados con REGIVAS® en su etapa de post-comercialización. Pruebas de función hepática deben de ser obtenidas previo al tratamiento con REGIVAS® y ser monitorizadas después de una semana y al mes de iniciarse el tratamiento, posteriormente de forma mensual, durante 6, 9 y 12 meses y periódicamente en lo sucesivo.
Si los niveles de alanina transferasa (ALT) se elevan > 3 x arriba del límite normal (ULN), los niveles se volverán a medir. Si se confirmaron niveles > 3 x ULN, retirar el tratamiento. Continuar con una observación cercana hasta normalización de SLT e investigar la posible causa. REGIVAS® no debe ser reiniciada en pacientes sin otra explicación de daño hepático observada.
Asesorar a pacientes para el reporte inmediato de cualquier síntoma potencial de daño hepático (como anorexia, náusea, vómito, fiebre, malestar general, fatiga, malestar en el cuadrante superior derecho del abdomen, ictericia, prurito, coluria) a los médicos.
Alteraciones respiratorias: Han sido reportados casos de enfermedad pulmonar intersticial, incluyendo neumonitis y fibrosis pulmonar en la experiencia de post-comercialización (ver Reacciones secundarias y adversas): la aparición de disnea o tos no productiva puede estar relacionada a toxicidad pulmonar y los pacientes deben ser evaluados clínicamente en forma cautelosa. Si se confirma toxicidad pulmonar el tratamiento debe ser descontinuado.
Desequilibrios electrolíticos: Dado que los medicamentos antiarrítmicos pueden ser inefectivos o arritmogénicos en pacientes con hipokalemia, es necesario corregir las deficiencias de potasio o de magnesio antes de iniciar y durante el tratamiento con dronedarona.
PRECAUCIONES:
Tratamiento con anticoagulantes: Los pacientes deben ser apropiadamente anticoagulados. Donde sea aplicable el Índice Internacional Normalizado (INR), debe ser monitoreado estrechamente después del inicio de dronedarona en pacientes que toman antagonistas de la vitamina K según su indicación terapéutica.
Incremento en la creatinina sérica: Los niveles de creatinina sérica se incrementan cerca de 0.1 mg/dl posteriores al inicio de dronedarona. Dicha elevación es de inicio súbito, alcanza una meseta después de 7 días y es reversible posterior a la descontinuación. Si el incremento de la creatinina sérica ocurre dentro de este periodo de tiempo y meseta, este incremento en el valor, debe ser usado en los pacientes como un nuevo valor basal. Ha sido demostrado que el cambio en estos niveles, es resultado de una inhibición de la secreción tubular de creatinina, sin efecto sobre la taza de filtración glomerular.
Incrementos mayores en la creatinina, incluyendo casos de azoemia pre-renal secundaria a ICC, hipo-perfusión o hipovolemia, después del inicio de dronedarona han sido reportados en la fase de post-comercialización. En algunos casos también se reportó incremento en los niveles de nitrógeno ureico en sangre. En la mayoría de los casos estos efectos aparentan ser reversibles después de la descontinuación del fármaco. Se recomienda monitorear la función renal periódicamente y considerar una investigación posterior según se requiera.
Prolongación del QT: La acción farmacológica de dronedarona puede inducir una prolongación moderada (aproximadamente de 10 mseg) del QTc Bazett, relacionado con una prolongación de la re-polarización. Estos cambios están vinculados con el efecto terapéutico de dronedarona y no son un reflejo de toxicidad. Se recomienda un seguimiento adecuado de los pacientes durante el tratamiento, entre lo que se incluye ECG. Si el intervalo del QT es ≥ 500 mseg, se debe interrumpir la administración de dronedarona (Ver Contraindicaciones).
Con base en la experiencia clínica, dronedarona tiene muy baja acción pro-arrítmica. Se observó una disminución en la muerte por arritmias en el estudio ATHENA (Ver Propiedades farmacodinámicas). Sin embargo, los efectos pro-arrítmicos pueden ocurrir en condiciones particulares, tales como el uso concomitante de medicamentos que favorecen las arritmias y/o alteraciones electrolíticas (Ver Precauciones generales e Interacciones medicamentosas y de otro género).
Pacientes con enfermedad arterial coronaria: Se recomienda precaución en este tipo de pacientes.
Pacientes ancianos: Se recomienda precaución en pacientes ancianos ≥ 75 años con múltiples comorbilidades.
Efectos sobre la capacidad de manejar vehículos y uso de maquinaria: No se han llevado a cabo estudios sobre la capacidad de manejar vehículos y uso de maquinaria.
DOSIS Y VÍA DE ADMINISTRACIÓN: El tratamiento con REGIVAS® se puede iniciar en forma ambulatoria.
El tratamiento con los antiarrítmicos Clase I o III (como flecainida, propafenona, quinidina, disopiramida, dofetilida, sotalol, amiodarona), debe interrumpirse antes de iniciar el tratamiento con REGIVAS® (Ver Contraindicaciones).
Existe información limitada relativa al momento óptimo para hacer el cambio de amiodarona a REGIVAS®. Debe considerarse que amiodarona puede tener una larga duración de acción, debido a su vida media larga. Si se prevee un cambio, este debe ser realizado con cautela y bajo la supervisión de un especialista.
Adultos: La dosis recomendada es de 400 mg 2 veces al día; se debe tomar:
• Una tableta con el desayuno.
• Una tableta con la cena.
Niños y adolescentes: No existe experiencia en niños ni en adolescentes.
Ancianos: Un número importante de pacientes ancianos con FA /flútter auricular han participado en el programa de estudios clínicos con REGIVAS® (más de 4,500 pacientes de 65 años o mayores, de los cuales, más de 2,000 tenían 75 años o más). La eficacia y la seguridad son comparables en ancianos y en pacientes jóvenes. Se recomienda precaución en pacientes ≥ 75 años con múltiples comorbilidades.
Insuficiencia hepática: No es necesario ajustar la dosis en pacientes con insuficiencia hepática de leve a moderada (Ver Propiedades farmacocinéticas). REGIVAS® está contraindicado en pacientes con insuficiencia hepática severa debido a la ausencia de información relacionada al respecto (Ver Contraindicaciones).
Insuficiencia renal: No es necesario ajustar la dosis (Ver Propiedades farmacocinéticas).
MANIFESTACIONES Y MANEJO DE LA SOBREDOSIFICACIÓN O INGESTA ACCIDENTAL: No se sabe si dronedarona y/o sus metabolitos pueden ser removidos mediante diálisis (hemodiálisis, diálisis peritoneal, o hemofiltración).
No existe un antídoto específico disponible. En el caso de sobredosis, se debe dar tratamiento de apoyo orientado al manejo de los síntomas.
PRESENTACIONES: Caja con 20 y con 60 tabletas de 400 mg en envase de burbuja e instructivo anexo.
RECOMENDACIONES SOBRE ALMACENAMIENTO: Consérvese a no más de 30°C.
LEYENDAS DE PROTECCIÓN:
Este medicamento contiene lactosa. Literatura exclusiva para médicos. Su venta requiere receta médica. No se deje al alcance de los niños. No se use durante el embarazo o lactancia. No se recomienda su uso en menores de 18 años.
Reporte las sospechas de reacción adversa al correo: farmacovigilancia@cofepris.gob.mx
SANOFI-AVENTIS DE MÉXICO, S.A. de C.V.
Acueducto del Alto Lerma No. 2
Zona Industrial Ocoyoacac
C.P. 52740, Ocoyoacac, México, México
Reg. Núm. 278M2009, SSA IV
® Marca Registrada
Clave interna IPP: MX-(IPPA_REGIVAS / V4) - (Dronedarona CCDS /V 7, 8, 9)


