
PILOVAIT - Tabletas
Sustancia(s):
- Finasterida
Presentaciones:
- 1 Caja,100 Tabletas,
- 1 Caja,28 Tabletas,
- 1 Caja,56 Tabletas,
FORMA FARMACÉUTICA Y FORMULACIÓN:
Cada TABLETA contiene:
Finasterida 1 mg
Excipiente, c.b.p. 1 tableta.
PILOVAIT® es Finasterida, un compuesto 4-azasteroide sintético que inhibe de forma específica la enzima 5-a reductasa tipo II. Su fórmula es C23H36B2O2. Su peso molecular es de 372.549 g/mol. La siguiente es su fórmula estructural:
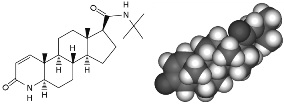
Mecanismo de acción: PILOVAIT® es Finasterida, un inhibidor competitivo de la enzima 5-alfa-reductasa que transforma la testosterona en dihidrotestosterona, su molécula activa. A dosis de 1 mg es útil en el tratamiento de la alopecia androgenética.
INDICACIONES TERAPÉUTICAS:
PILOVAIT® está indicado en el tratamiento de: Alopecia androgenética en hombres, para incrementar el crecimiento de cabello y evitar que la densidad de pelo disminuya, evitando la caída del mismo.
FARMACOCINÉTICA Y FARMACODINAMIA:
Farmacocinética:
En comparación con una dosis intravenosa de referencia la biodisponibilidad de la Finasterida administrada por vía oral es de 80% aproximadamente y no se afecta por la presencia de alimentos. La Finasterida alcanza concentraciones plasmáticas máximas aproximadamente dos horas después de la administración y su absorción es completa en seis a ocho horas. La unión de Finasterida con las proteínas plasmáticas es de 93% y su volumen de distribución es de 76 litros.
Después de la administración de dosis múltiples de Finasterida, se presenta una mínima acumulación plasmática. Una vez alcanzado el estado de equilibrio, con dosis de 1 mg diario, el promedio de concentración plasmática máxima de Finasterida fue de 9.2 ng/ml y se alcanzó después de una a dos horas; el AUC (0-24 h) fue de 53 ng/h/ml. Se ha detectado Finasterida en el líquido cefalorraquídeo, y mínima cantidad en el semen de los hombres tratados con este fármaco. Se metaboliza principalmente por la subfamilia 3A4 del citocromo P-450. En el hombre después de una dosis oral de 14C-Finasterida se identificaron dos metabolitos de la Finasterida. El 39% de la dosis total fue excretada con la orina en forma de metabolitos y 57% fue eliminado en la materia fecal.
La depuración plasmática de la Finasterida es de aproximadamente 165 ml/min.
La rapidez de eliminación de la Finasterida es inversamente proporcional a la edad. Sin embargo, no tiene ninguna importancia clínica, por tanto, no es necesario reducir la dosis en los pacientes de edad avanzada.
Pacientes con insuficiencia renal: En pacientes con insuficiencia renal crónica y depuración de creatinina de 9 a 55 mg/min, la eliminación de una dosis única de 14C-Finasterida no se modificó con respecto a la de los voluntarios sanos, y tampoco en su unión con las proteínas plasmáticas. Una porción de los metabolitos se eliminan por vía renal y por materia fecal. No se requiere ajustar la dosis en los pacientes con insuficiencia renal que no están sometidos a diálisis.
Farmacodinamia: No se observó efecto alguno de la Finasterida en las concentraciones circulantes de cortisol, de la hormona estimulante de la tiroides y la tiroxina, y del perfil de lípidos, ni la densidad mineral ósea. No se detectaron, en los estudios con Finasterida ningún cambio clínicamente significativo en las concentraciones de hormona luteinizante (HL) hormona foliculoestimulante (HFE), estradiol o prolactina. Las concentraciones de HL y HFE estimuladas por la hormona liberadora de gonadotropinas no se modificaron.
Las concentraciones circulantes de testosterona aumentaron 10-15% aproximadamente en comparación con el placebo, pero se mantuvieron dentro de los límites fisiológicos. No hubo ningún efecto sobre los parámetros del semen en hombres tratados con 1 mg diario de Finasterida durante 48 semanas.
La Finasterida inhibió el metabolismo de los esteroides C19 y de los C21, lo cual indicó un efecto inhibidor de la actividad de la 5a-reductasa de tipo II, hepática y periférica. También disminuyeron significativamente las concentraciones séricas de los metabolitos de la DHT glucurónido de androstenediol y glucurónido de androsterona.
Estudios clínicos:
Estudios en hombres: La eficacia de Finasterida se demostró en tres estudios en 1,879 hombres de 18 a 41 años con pérdida de cabello leve a moderada, pero no completa, del vértex y de las regiones frontal y parietales. En estos estudios el crecimiento del cabello se evaluó utilizando cuatro mediciones por separado, la cuenta de cabellos, evaluación fotográfica de la cabeza por un panel de expertos en dermatología, evaluación del investigador y evaluación del paciente.
En los dos estudios en hombres con pérdida de cabello en el vértex el tratamiento con Finasterida se realizó a cinco años, tiempo durante el cual los pacientes mejoraron respecto al inicio (comparado con el placebo) desde el tercer mes. El tratamiento con Finasterida durante cinco años resultó en una estabilización de la pérdida de cabello en 90% de los hombres según la evaluación fotográfica y en 93% según la evaluación del investigador. Además se reportó incremento de cabello en 65% de los hombres tratados con Finasterida, de acuerdo a la cuenta de cabello; en 48% según la evaluación fotográfica y, en 77% según la evaluación del investigador. En cambio, en el grupo con placebo se observó pérdida de cabello en forma gradual en el tiempo en 100% de los hombres según la cuenta de cabello (vs. 35% en el grupo de los hombres tratados con Finasterida); en 75% según la evaluación fotográfica (vs. 10% en el grupo de los hombres tratados con Finasterida) y en 38% según la evaluación del investigador (vs. 7% en el grupo de los hombres tratados con Finasterida). Además durante los cinco años de tratamiento con Finasterida, la autoevaluación del paciente demostró aumento significativo en la densidad del cabello, disminución de la pérdida de cabello, y mejoría en la apariencia del cabello. Mientras que las mediciones de mejoría del cabello respecto al inicio, fueron mayores en los hombres tratados con Finasterida a los dos años y gradualmente disminuyeron a partir de entonces. Así con base en esas cuatro mediciones las diferencias entre los grupos de tratamiento continuaron aumentando durante los cinco años del estudio.
El estudio de 12 meses en hombres con pérdida de cabello en las regiones frontal y parietal también demostró mejoría significativa en el crecimiento del cabello y en su apariencia, de acuerdo con las mismas evaluaciones mencionadas anteriormente.
Un estudio de 48 semanas controlado con placebo diseñado para evaluar los efectos de Finasterida en las fases del ciclo de crecimiento del pelo (fase anágena y fase de telógena) del vértex incluyó 212 hombres con alopecia androgenética. Al inicio y a las 48 semanas la cuenta de cabellos totales en telógeno y en anágeno fue obtenida de un área objetivo de 1 cm2. El tratamiento con Finasterida ocasionó un aumento en la cuenta de cabello en fase anágena, mientras que los hombres con placebo presentaron pérdida. A las 48 semanas respecto a placebo, los hombres tratados con Finasterida presentaron un aumento neto en la cuenta total de cabellos y en la cuenta en fase anágena de 17 y 27 cabellos respectivamente. El aumento en la cuenta de cabellos en fase anágena respecto a la cuenta de cabello total produjo un aumento neto en la fase anágeno a telógeno en una proporción de 47% a las 48 semanas en los hombres tratados con Finasterida comparados con el placebo.
Estos datos proporcionan evidencia directa de que el tratamiento con Finasterida proporciona una conversión de los folículos de los cabellos en una fase activa de crecimiento. En resumen, estos estudios demostraron que el tratamiento con Finasterida aumenta el crecimiento de cabello y evita que continúe la pérdida asociada con la alopecia androgenética.
CONTRAINDICACIONES: PILOVAIT* está contraindicado en mujeres embarazadas o que deseen embarazarse. Hipersensibilidad a cualquier componente del producto.
RESTRICCIONES DE USO DURANTE EL EMBARAZO Y LA LACTANCIA:
Embarazo: Finasterida está contraindicado en mujeres embarazadas o que deseen embarazarse.
Debido a la propiedad de los inhibidores de la 5a-reductasa de inhibir la conversión de la testosterona en dehidrotestosterona (DHT), como lo es la Finasterida, puede ocasionar alteraciones en los órganos genitales externos de los fetos de sexo masculino, si se administra a mujeres embarazadas. Las mujeres embarazadas o con riesgo de embarazo, no deben manipular tabletas rotas de Finasterida, debido a la posibilidad de absorción de Finasterida, y el consecuente riesgo potencial para los fetos de sexo masculino.
Lactancia: Se desconoce si la Finasterida se excreta en la leche materna.
REACCIONES SECUNDARIAS Y ADVERSAS: PILOVAIT* es generalmente bien tolerado. Usualmente los efectos han sido leves y transitorios, y no ameritaron la suspensión del tratamiento.
En los estudios realizados, se han reportado las siguientes reacciones adversas relacionadas con Finasterida, en ³ 1% de los hombres: disminución de la libido y disfunción de la eréctil (1.3%), disminución del volumen de la eyaculación. Estos efectos colaterales remitieron en su totalidad, en los hombres que suspendieron el tratamiento, y en otro estudio no se observaron cambios en el volumen de la eyaculación. En el quinto año de tratamiento con Finasterida, la incidencia de cada una de las reacciones adversas, anteriormente mencionadas disminuyeron a ? 0.3%.
Las siguientes experiencias adversas se han reportado con el uso del medicamento, trastornos de la eyaculación, hiperestesia y ginecomastia; reacciones de hipersensibilidad, como erupción, prurito, urticaria y edema de los labios y cara; y dolor testicular.
PRECAUCIONES EN RELACIÓN CON EFECTOS DE CARCINOGÉNESIS, MUTAGÉNESIS, TERATOGÉNESIS Y SOBRE LA FERTILIDAD:
Carcinogénesis: No se observó ningún indicio de efecto carcinogénico en un estudio de 24 meses en ratas que recibieron hasta 320 mg/kg/día de Finasterida (16,000 veces más que la dosis de 1 mg diario recomendada en humanos). En un estudio de carcinogenicidad de 19 meses en ratones se observó un aumento estadísticamente significativo (p ? 0.05) en la incidencia de adenomas testiculares de las células de Leydig con una dosis de 250 mg/kg/día (12,500 veces mayor que la de 1 mg diario recomendada en humanos); no se observaron adenomas en los ratones que recibieron 2.5 o 25 mg/kg/día (125 y 1,250 veces más que la dosis de 1 mg diario recomendada en humanos).
Se observó un aumento de la incidencia de la hiperplasia de las células de Leydig con una dosis de 25 mg/kg/día en los ratones y de 40 mg/kg/día o más en las ratas (1,250 y 2,000 o más veces mayores que la de 1 mg diario recomendada en humanos, respectivamente). Se corroboró una correlación positiva entre los cambios proliferativos de las células de Leydig y el aumento de las concentraciones séricas de la hormona luteinizante (HL), al doble o al triple que los controle, en ambas especies de roedores tratadas con dosis elevadas de Finasterida. Esto sugiere que los cambios en las células de Leydig son secundarios al aumento de las concentraciones séricas de HL y no debidos a un efecto directo de la Finasterida. No se observaron cambios de las células de Leydig relacionados con el medicamento en ratas o en perros tratados con Finasterida durante un año a dosis de 20 mg/kg/día y 45 mg/kg/día (1,000 y 2,250 veces mayores que la de 1 mg diario recomendada en los humanos, respectivamente) ni en ratones tratados durante 19 meses con 2.5 mg/kg/día (125 veces más que la dosis de 1 mg diario recomendada en humanos).
Mutagénesis: No se observó ningún indicio de mutagenicidad en un ensayo de mutagénesis bacteriana in vitro, en un ensayo de mutagénesis en células de mamífero, ni en un ensayo de elución alcalina in vitro. En un ensayo de aberración cromosómica in vitro cuando se trataron células de ovario de hámster chino con concentraciones altas (450-550 µmol) de Finasterida hubo un incremento leve de las aberraciones cromosómicas. Esas concentraciones son 18,000-22,000 veces mayores que las concentraciones plasmáticas máximas obtenidas en el hombre con una dosis total de 1 mg. En un ensayo de aberración cromosómica in vivo en ratones no se observó ningún aumento de las aberraciones cromosómicas relacionado con el tratamiento administrando Finasterida a la dosis máxima tolerada (250 mg/kg/día 12,500 veces mayor que la de 1 mg diario recomendada en los seres humanos).
Reproducción: En conejos machos sexualmente maduros tratados con 80 mg/kg/día de Finasterida (4,000 veces más que la dosis de 1 mg diario recomendada en humanos) hasta por 12 semanas no se observó ningún efecto sobre la fertilidad, el número de espermatozoides y el volumen de la eyaculación. En ratas machos sexualmente maduras tratadas con 80 mg/kg/día de Finasterida, no se observó ningún efecto significativo sobre la fertilidad después de 6 a 12 semanas de tratamiento; sin embargo, cuando se prolongó éste hasta 24 o 30 semanas, se observó una disminución de la fertilidad y la fecundidad acompañada de una reducción significativa del peso de las vesículas seminales y de la próstata. Todos estos efectos se revirtieron a las seis semanas siguientes, a la conclusión del tratamiento.
La disminución de la fertilidad en las ratas tratadas con Finasterida es secundaria al efecto de éste sobre los órganos sexuales accesorios (próstata y vesículas seminales) que evita la formación del tapón seminal indispensable para la fertilidad normal de las ratas. Este efecto carece de importancia en el hombre. No se ha observado en las ratas ni en los conejos ningún efecto relacionado con el medicamento sobre los testículos o el apareamiento.
Desarrollo: Se observó hipospadias dependiente de la dosificación con una frecuencia de 3.6 a 100% en los fetos machos de ratas embarazadas que recibieron Finasterida a dosis que variaron entre 100 µg/kg/día y 100 mg/kg/día (5 a 5,000 veces más que la dosis de 1 mg diario recomendada en los humanos). Cuando se administró Finasterida a dosis ³ 30 µg/kg/día (³ 1.5 veces mayores que la dosis de 1 mg diario recomendada en los seres humanos), las ratas embarazadas, dieron fetos machos, con menor peso de la próstata y de las vesículas seminales, retraso de la separación del prepucio y crecimiento temporal de los pezones y cuando se les suministraron dosis ³ 3 µg/kg/día (aproximadamente la quinta parte de la dosis de 1 mg diario recomendada en los humanos), se observó disminución de la distancia anogenital. Se ha determinado que el periodo crítico para la inducción de estos efectos en las ratas son los días 16-17 de la gestación.
Los cambios anteriormente descritos, son los efectos farmacológicos esperados de los inhibidores de la 5 a-reductasa de tipo II.
Muchos de los cambios observados en ratas machos expuestas in utero a la Finasterida como hipospadias, son similares a los encontrados en niños del sexo masculino, con deficiencia genética de 5 a-reductasa de tipo II. No se observó ningún efecto en las hembras expuestas in utero a la Finasterida, con las dosis utilizadas.
La administración de Finasterida a ratas (3 mg/kg/día 150 veces más que la dosis de 1 mg diario recomendada en humanos) durante la última etapa de la gestación y el periodo de lactancia, ocasionó una disminución leve de la fertilidad en los machos de la primera generación. No se observó ninguna alteración del desarrollo en las crías machos o hembras de la primera generación como resultado del apareamiento de ratas machos tratadas con Finasterida (80 mg/kg/día 4,000 veces más que la dosis de 1 mg diario recomendada en los seres humanos) con hembras no tratadas.
No se ha observado ningún indicio de malformaciones en fetos de conejo expuestos a Finasterida in utero del día 6 al día 18 de la gestación a dosis de hasta 100 mg/kg/día (5,000 veces más que la dosis de 1 mg diario recomendado en los seres humanos).
Los efectos de la exposición a la Finasterida in utero durante el periodo de desarrollo embrionario y fetal fueron evaluados en monos Rhesus (del día 20 al día 100 de la gestación), una especie más predictiva del desarrollo humano. La administración intravenosa de Finasterida a monas embarazadas a dosis de 800 ng/día (por lo menos 750 veces mayores que la exposición más alta estimada de mujeres embarazadas a la Finasterida existente en el semen de los hombres tratados con 1 mg diario) no produjo ninguna malformación en los fetos masculinos.
En confirmación de la significancia del modelo de monos Rhesus respecto al desarrollo del feto humano, la administración oral de dosis muy altas de Finasterida (2 mg/kg/día; 100 veces la dosis de 1 mg diario recomendada en los seres humanos o 12 millones de veces mayor que la exposición más alta estimada a la Finasterida existente en el semen de los hombres tratados con 1 mg diario) a monas embarazadas produjo malformaciones de los genitales externos en los fetos masculinos. No se observó ninguna otra alteración en los fetos masculinos, ni anormalidades relacionadas con la Finasterida en los fetos femeninos.
INTERACCIONES MEDICAMENTOSAS Y DE OTRO GÉNERO: No se han identificado interacciones medicamentosas, hasta el momento.
ALTERACIONES EN LOS RESULTADOS DE PRUEBAS DE LABORATORIO: Cuando se utilice PILOVAIT* como tratamiento contra la pérdida de cabello de patrón masculino en hombres de edad avanzada que además tienen hiperplasia prostática benigna (HPB), se debe considerar que en estos pacientes, las concentraciones de APE están disminuidas aproximadamente en un 50% aproximadamente.
PRECAUCIONES GENERALES: En los estudios clínicos con Finasterida en hombres de 18 a 41 años, el promedio de concentración de antígeno prostático específico (APE) sérico disminuyó de 0.7 ng/ml inicial a 0.5 ng/ml a los 12 meses. Cuando se utilice para el tratamiento de alopecia androgenética en hombres de edad avanzada que además presentan hiperplasia prostática benigna (HPB), se debe considerar que en estos pacientes las concentraciones de APE están disminuidas 50% en niños.
Finasterida no está indicado en niños.
DOSIS Y VÍA DE ADMINISTRACIÓN: PILOVAIT* se recomienda exclusivamente en hombres. Finasterida no se recomienda en mujeres ni en niños. Se administra por vía oral.
La dosis recomendada de PILOVAIT* es de una tableta de 1 mg al día, de forma indistinta con los alimentos.
En general, es necesario tomarlo diariamente durante tres meses o más para comenzar a percibir un aumento de la cantidad de cabello y/o para evitar su pérdida. Se recomienda tomarlo de forma continua para obtener el máximo beneficio. Se ha observado, que el efecto obtenido pierde, 12 meses después de suspender Finasterida.
MANIFESTACIONES Y MANEJO DE LA SOBREDOSIFICACIÓN O INGESTA ACCIDENTAL: En los estudios clínicos, las dosis únicas de Finasterida de hasta 400 mg y las dosis múltiples de hasta 80 mg.
PRESENTACIONES: Cajas con 28, 56 y 100 tabletas.
RECOMENDACIONES SOBRE ALMACENAMIENTO: Protéjase de la luz y la humedad. Consérvese a una temperatura ambiente no mayor de 30ºC y en un lugar seco.
LEYENDAS DE PROTECCIÓN:
Literatura exclusiva para médicos. Su venta requiere receta médica. No se deje al alcance de los niños.
Hecho en México por:
LABORATORIOS SERRAL, S. A. de C. V.
Adolfo Prieto No. 1009, Col. Del Valle
03100, México, D.F.
Núm. Reg. 149M2008, SSA IV

