
REBIF - Solución inyectable
Sustancia(s):
- Interferón Beta 1a
Presentaciones:
- 1 Caja , 1 Jeringa(s) prellenada(s) , 22/0.5 µg/ml
- 1 Caja , 3 Jeringa(s) prellenada(s) , 22/0.5 µg/ml
- 1 Caja , 6 Jeringa(s) prellenada(s) , 22/0.5 µg/ml
- 1 Caja , 12 Jeringa(s) prellenada(s) , 22/0.5 µg/ml
- 1 Caja , 1 Jeringa(s) prellenada(s) , 44/0.5 µg/ml
- 1 Caja , 3 Jeringa(s) prellenada(s) , 44/0.5 µg/ml
- 1 Caja , 6 Jeringa(s) prellenada(s) , 44/0.5 µg/ml
- 1 Caja , 12 Jeringa(s) prellenada(s) , 44/0.5 µg/ml
- 1 Caja , 12 Jeringa(s) prellenada(s) , 22/0.5 µg/ml
- 1 Caja , 12 Jeringa(s) prellenada(s) , 44/0.5 µg/ml
FORMA FARMACÉUTICA Y FORMULACIÓN:
SOLUCIÓN INYECTABLE:
Cada jeringa prellenada contiene:
** Interferón beta-1a (r IFN-ß 1a) 22 µg (6 MUI)* o 44 µg (12 MUI)*
Vehículo, c.b.p. 0.5 ml.
* Millón de unidades internacionales, valorado mediante el bioensayo del efecto citopático (CPE) frente al patrón interno de IFN beta-1a, que está calibrado frente al patrón internacional del NIH (GB-23-902-531).
** Producido por tecnología del ADN recombinante en células de ovario de hámster chino (CHO-K1).
INDICACIONES TERAPÉUTICAS: REBIF® está indicado para el tratamiento de pacientes con esclerosis múltiple.
FARMACOCINÉTICA Y FARMACODINAMIA:
Propiedades farmacodinámicas:
Grupo farmacoterapéutico: lnmunoestimulantes, interferones, código ATC: L03AB07.
Los interferones (IFNs) son un grupo de glicoproteínas endógenas, dotadas de propiedades inmunomoduladoras, antivirales y antiproliferativas.
REBIF® (interferón beta-1a) comparte la misma secuencia de aminoácidos con el interferón beta humano endógeno. Se produce en células de mamífero (ovario de hámster chino), por lo que está glicosilado, como la proteína natural.
REBIF® (interferón beta-1) comparte la misma secuencia de aminoácidos con el interferón beta humano endógeno. Se producen células de mamífero (ovario de hámster chino), por lo que está glicosilado, como la proteína natural.
El mecanismo de acción preciso de REBIF® en la esclerosis múltiple todavía se está investigando.
Esclerosis múltiple remitente-recurrente: Se ha evaluado la seguridad y eficacia de REBIF® en pacientes con esclerosis múltiple remitente-recurrente, a dosis comprendidas entre 11 y 44 µg (3-12 millones de U.I.), administradas por vía subcutánea, tres veces por semana. Se ha demostrado que, a la dosis autorizada, REBIF® 44 µg disminuye la incidencia (aproximadamente 30% en 2 años) y la gravedad de los brotes clínicos en pacientes con al menos 2 brotes en los dos años previos y con una escala de discapacidad ampliada de Kurtzke (EDSS) de 0-5.0 al inicio. La proporción de pacientes con progresión de la discapacidad, definida por un incremento de al menos un punto en la EDSS, confirmado tres meses más tarde, se redujo desde 39% (placebo) hasta 30% (REBIF® 22 µg) y 27% (REBIF® 44 µg). A lo largo de 4 años, la reducción en la tasa media de brotes fue de 22% en los pacientes tratados con REBIF® 22 µg y de 29% en los pacientes tratados con REBIF® 44 µg, en comparación con un grupo de pacientes tratados con placebo durante 2 años y posteriormente con REBIF® 22 o REBIF® 44 µg durante otros 2 años.
Esclerosis múltiple secundaria progresiva: En un estudio a 3 años en pacientes con esclerosis múltiple secundaria progresiva (EDSS 3-6.5) con evidencia de progresión clínica en los dos años previos y sin brotes en las 8 semanas anteriores, REBIF® no tuvo efecto significativo sobre la progresión de la discapacidad, pero redujo la tasa de brotes 30% aproximadamente. Al dividir la población de pacientes en 2 subgrupos (según hubieran presentado brotes o no en los 2 años previos a la entrada en el estudio), no se observó efecto sobre la discapacidad en los pacientes sin brotes; sin embargo, en aquellos que habían tenido brotes, la proporción de pacientes con progresión de la discapacidad al final del estudio se redujo desde 70% (placebo) hasta 57% (REBIF® 22 y 44 µg combinados). Estos resultados obtenidos en un subgrupo de pacientes a posteriori deben interpretarse con cautela.
Esclerosis múltiple primaria progresiva: REBIF® no se ha investigado todavía en pacientes con esclerosis múltiple primaria/progresiva y no debe utilizarse en estos pacientes.
Propiedades farmacocinéticas: Tras la administración intravenosa en voluntarios sanos, el interferón beta-1a muestra un pronunciado descenso multi-exponencial, y los niveles séricos son proporcionales a la dosis. La vida media de distribución es del orden de minutos y la vida media de eliminación es de varias horas, con la posible existencia de un compartimiento profundo. Cuando se administra por vía subcutánea o intramuscular, los niveles séricos de interferón beta permanecen bajos, pero siguen siendo detectables hasta 12-24 horas posinyección. La administración subcutánea o intramuscular de REBIF® produce una exposición equivalente al interferón beta. Tras una dosis única de 60 µg, la concentración máxima, determinada por inmunoensayo, es de alrededor de 6-10 U.I./ml, y se alcanza, como promedio, unas 3 horas después de la inyección. Tras la administración subcutánea de la misma dosis, repetida cada 48 horas hasta un total de 4 dosis, se produce una acumulación moderada (aproximadamente 2.5 veces para el ABC).
La administración de REBIF® se asocia a cambios farmacodinámicos pronunciados, independientemente de la vía utilizada. Tras una dosis única, la actividad intracelular y sérica de la 2-5A sintetasa y las concentraciones séricas de beta-2 microglobulina y neopterina aumentan en las primeras 24 horas, y comienzan a descender a los 2 días. La administración intramuscular y subcutánea da lugar a una respuesta totalmente superponible. Tras la administración subcutánea repetida cada 48 horas, hasta un total de 4 dosis, estas respuestas biológicas permanecen elevadas, sin signos de tolerancia. El interferón beta-1a se metaboliza y excreta principalmente por el hígado y los riñones.
CONTRAINDICACIONES:
• Inicio del tratamiento en el embarazo (véase Precauciones generales).
• Hipersensibilidad conocida al interferón beta natural o recombinante, o a alguno de los excipientes.
• Depresión grave activa y/o ideación suicida (véase Precauciones generales).
RESTRICCIONES DE USO DURANTE EL EMBARAZO Y LA LACTANCIA:
Embarazo: Existe una información limitada sobre el uso de REBIF® en el embarazo. Los datos disponibles indican que puede haber un incremento del riesgo de aborto espontáneo. Por lo tanto, durante el embarazo está contraindicado el inicio del tratamiento (véase Contraindicaciones).
Mujeres en edad fértil: Las mujeres en edad fértil deben utilizar medidas anticonceptivas apropiadas. Se debe informar a la paciente que quede embarazada o que esté planificando un embarazo, mientras está en tratamiento con REBIF®, de los riesgos potenciales y debe considerarse la posibilidad de interrumpir el tratamiento (véase Precauciones en relación con efectos de carcinogénesis, mutagénesis, teratogénesis y sobre la fertilidad).
En pacientes embarazadas que presentan una alta tasa de brotes antes de iniciar el tratamiento, el riesgo de la aparición de un brote grave tras la interrupción del tratamiento con REBIF® debe tenerse en cuenta frente al posible riesgo de un aborto espontáneo.
Lactancia: Se desconoce si REBIF® se excreta en la leche, pero por la posibilidad de aparición de reacciones adversas graves en los lactantes se debe decidir si interrumpir la lactancia o el tratamiento con REBIF®.
REACCIONES SECUNDARIAS Y ADVERSAS:
Resumen del perfil de seguridad: La incidencia más alta de reacciones adversas asociadas a la terapia con REBIF® está relacionada con el síndrome pseudogripal. Los síntomas pseudogripales tienden a ser más acusados al inicio de la terapia y disminuyen en frecuencia al continuar el tratamiento. Aproximadamente 70% de los pacientes tratados con REBIF® pueden presentar el típico síndrome pseudogripal durante los primeros seis meses tras iniciar el tratamiento. Aproximadamente 30% de los pacientes también presentarán reacciones en la zona de inyección, principalmente inflamación leve o eritema. También son frecuentes los incrementos asintomáticos de los parámetros analíticos de función hepática y los descensos de los leucocitos.
La mayoría de las reacciones adversas observadas con IFN-beta-1a suelen ser leves y reversibles, y responden bien a la disminución de la dosis. En caso de presentar efectos adversos graves o persistentes, la dosis de REBIF® puede disminuir o interrumpirse temporalmente, a juicio del facultativo .
Lista de reacciones adversas: Las reacciones adversas presentadas se han identificado en los ensayos clínicos y en los informes después de la comercialización (un asterisco [*] indica reacciones adversas identificadas durante la vigilancia después de la comercialización). Las siguientes definiciones corresponden a la terminología de frecuencia utilizada de aquí en adelante:
Muy frecuentes: ³ 1/10.
Frecuentes: ³ 1/100 < 1/10.
Poco frecuentes: ³ 1/1,000 < 1/100.
Raras: ³ 1/10,000 < 1/1,000.
Muy raras: <1/10,000.
Frecuencia no conocida: no puede estimarse a partir de los datos disponibles.
Trastornos de la sangre y del sistema linfático:
Muy frecuentes: neutropenia, linfopenia, leucopenia, trombocitopenia, anemia.
Raras: púrpura trombocitopénica trombótica/síndrome hemolítico urémico*, pancitopenia*.
Trastornos endocrinos:
Poco frecuentes: disfunción tiroidea, que a menudo se presenta como hipotiroidismo o hipertiroidismo.
Trastornos del sistema inmunológico:
Raros: reacciones anafilácticas*.
Trastornos hepatobiliares:
Muy frecuentes: elevación asintomática de las transaminasas.
Frecuentes: elevaciones graves de las transaminasas.
Poco frecuentes: hepatitis con o sin ictericia*.
Raros: fallo hepático* (véase Precauciones generales), hepatitis autoinmune*.
Trastornos psiquiátricos:
Frecuentes: depresión, insomnio.
Raros: intento de suicidio*.
Trastornos del sistema nervioso:
Muy frecuentes: cefalea.
Poco frecuentes: convulsiones*.
Frecuencia no conocida: síntomas neurológicos transitorios (por ejemplo, hipoestesia, espasmo muscular, parestesia, dificultad para caminar, rigidez musculoesquelética) que pueden imitar exacerbaciones de esclerosis múltiple*.
Trastornos oculares:
Poco frecuentes: trastornos vasculares retinales (por ejemplo, retinopatía, exudados algodonosos y obstrucción de la arteria o vena retiniana)*.
Trastornos vasculares:
Poco frecuentes: eventos tromboembólicos*.
Trastornos respiratorios, torácicos y mediastínicos:
Poco frecuentes : disnea*.
Trastornos gastrointestinales:
Frecuentes: diarrea, vómito, náuseas.
Trastornos de la piel y del tejido subcutáneo:
Frecuentes: prurito, erupción, erupción eritematosa, exantema maculopapular, alopecia*.
Poco frecuentes: urticaria*.
Raros: edema de Quincke (angioedema)*, eritema multiforme*, reacciones cutáneas tipo eritema multiforme*, síndrome de Stevens-Johnson*.
Trastornos musculoesqueléticos y del tejido conjuntivo:
Frecuentes: mialgias, artralgias.
Raros: lupus eritematoso inducido por fármacos*.
Trastornos generales y alteraciones en el lugar de administración:
Muy frecuentes: inflamación en la zona de inyección, reacción en el punto de inyección, síntomas pseudogripales.
Frecuentes: Dolor en la zona de inyección, fatiga, escalofríos, fiebre.
Poco frecuentes: necrosis en la zona de inyección, masa en la zona de inyección, absceso en la zona de inyección, infecciones en la zona de inyección*, aumento de la sudación*.
Raros: celulitis en la zona de la inyección*.
Efectos de clases: La administración de interferones se ha asociado a anorexia, vértigo, ansiedad, arritmias, vasodilatación y palpitaciones, menorragia y metrorragia.
Puede producirse un aumento de la formación de autoanticuerpos durante el tratamiento con interferón beta.
PRECAUCIONES EN RELACIÓN CON EFECTOS DE CARCINOGÉNESIS, MUTAGÉNESIS, TERATOGÉNESIS Y SOBRE LA FERTILIDAD: Los datos de los estudios no clínicos no muestran riesgos especiales para los humanos según los estudios convencionales de farmacología de seguridad, toxicidad a dosis repetidas y genotoxicidad.
No se han realizado estudios de carcinogénesis con REBIF®.
Un estudio de toxicidad embriofetal en monos no mostró ninguna evidencia de trastornos de la reproducción. Según las observaciones realizadas con otros interferones alfa y beta, no puede descartarse un aumento del riesgo de aborto. No se dispone de información sobre los efectos del interferón beta-1a sobre la fertilidad-masculina.
INTERACCIONES MEDICAMENTOSAS Y DE OTRO GÉNERO: No se han realizado estudios de interacción farmacológica con interferón beta-1a en humanos.
Se ha notificado que los interferones disminuyen la actividad de las enzimas hepáticas dependientes del citocromo P-450, en humanos y en animales. Debe tenerse precaución cuando se administre REBIF® asociado a medicamentos con un estrecho índice terapéutico y cuyo aclaramiento dependa en gran manera del sistema hepático del citocromo P-450, por ejemplo, los antiepilépticos y algunas clases de antidepresivos.
No se ha estudiado sistemáticamente la interacción de REBIF® con los corticoides o la hormona adrenocorticotropa o corticotropina (ACTH). Los ensayos clínicos indican que los pacientes con esclerosis múltiple pueden recibir REBIF® y corticoides o ACTH durante los brotes.
ALTERACIONES EN LOS RESULTADOS DE PRUEBAS DE LABORATORIO:
Alteraciones analíticas: El empleo de interferones puede acompañarse de alteraciones analíticas. La incidencia global de las mismas es ligeramente superior con REBIF® 44 que con REBIF® 22 µg. Por lo tanto, además de las pruebas de laboratorio requeridas normalmente para controlar a los pacientes con esclerosis múltiple tras el inicio del tratamiento con REBIF® y en ausencia de síntomas clínicos, se recomienda realizar un control de las enzimas hepáticas y un recuento celular, fórmula leucocitaria y determinación de plaquetas a intervalos regulares (1, 3 y 6 meses) y luego periódicamente.
Estas determinaciones deben ser más frecuentes cuando se inicie el tratamiento con REBIF® 44 µg.
PRECAUCIONES GENERALES:
General: Se debe informar a los pacientes de las reacciones adversas más frecuentes asociadas a la administración de interferón beta, incluyendo los síntomas del síndrome pseudogripal (véase Reacciones secundarias y adversas). Estos síntomas tienden a ser más intensos al comenzar el tratamiento, y disminuyen en frecuencia y gravedad al continuar el tratamiento.
Depresión e ideación suicida: REBIF® debe administrase con precaución en pacientes que presentan trastornos depresivos previos o activos, en particular en aquellos con antecedentes de ideación suicida (véase Contraindicaciones). Se sabe que existe una mayor frecuencia de depresión e ideación suicida en la población con esclerosis múltiple y en asociación con el uso de interferón. Se debe aconsejar a los pacientes tratados con REBIF® que notifiquen inmediatamente a su médico cualquier síntoma de depresión y/o ideación suicida. Los pacientes que presenten depresión deben controlarse estrechamente durante el tratamiento con REBIF® y tratarse de forma adecuada. Debe considerarse la posibilidad de interrumpir el tratamiento con REBIF® (véase Contraindicaciones y Reacciones secundarias y adversas).
Trastornos convulsivos: REBIF® debe administrarse con precaución en pacientes con historia previa de crisis epilépticas, en aquellos que reciben tratamiento con antiepilépticos, en especial si su epilepsia no está convenientemente controlada con antiepilépticos (véase Contraindicaciones y Reacciones secundarias y adversas).
Cardiopatías: Los pacientes que presentan cardiopatías, como angina, insuficiencia cardiaca congestiva o arritmias, deben vigilarse estrechamente para descartar un empeoramiento de su situación clínica al inicio del tratamiento con interferón beta-1a. Los síntomas del síndrome pseudogripal asociados al tratamiento con interferón beta-1a pueden alterar a los pacientes que presenten cardiopatías.
Necrosis en la zona de inyección: En pacientes que utilizan REBIF® se ha notificado necrosis en la zona de inyección (véase Reacciones secundarias y adversas). Para minimizar el riesgo de necrosis en la zona de inyección se debe recomendar a los pacientes que:
• Utilicen una técnica de inyección aséptica.
• Alternen las zonas de inyección cada vez que se inyecten.
Se debe revisar periódicamente el procedimiento de auto-administración por el propio paciente, especialmente si se han producido reacciones en el lugar de inyección.
Si el paciente presenta cualquier solución de continuidad, que pueda estar asociada a hinchazón o drenaje de líquido desde la zona de inyección, se le debe recomendar que consulte a su médico antes de continuar con las inyecciones de REBIF®. Si los pacientes presentan múltiples lesiones, se debe suspender el tratamiento con REBIF® hasta que se hayan curado. Los pacientes con una única lesión pueden continuar el tratamiento, siempre que la necrosis no sea demasiado extensa.
Disfunción hepática: En los ensayos clínicos con REBIF®, fue frecuente el aumento asintomático de las transaminasas hepáticas (especialmente la alanina aminotransferasa (ALAT)) y 1-3% de los pacientes presentaron elevación de dichas transaminasas por encima de 5 veces el límite superior de la normalidad (ULN).
En ausencia de síntomas clínicos, deben controlarse los niveles de ALAT antes de iniciar el tratamiento, al cabo de 1, 3 y 6 meses de tratamiento y luego periódicamente. Si la ALAT aumenta más de 5 veces el ULN debe considerarse una reducción de la dosis de REBIF® para volver a aumentarla gradualmente cuando se hayan normalizado los niveles enzimáticos. El tratamiento con REBIF® debe iniciarse con precaución en pacientes con historia de hepatopatía significativa, evidencia clínica de hepatopatía activa, abuso de alcohol o ALAT sérica elevada (> 2.5 veces el ULN). El tratamiento con REBIF® debe interrumpirse si aparece ictericia u otros síntomas clínicos de disfunción hepática (véase Reacciones secundarias y adversas) .
REBIF®, como los otros interferones beta, tiene cierto potencial para causar daño hepático grave incluyendo insuficiencia hepática aguda. El mecanismo de los casos raros de disfunción hepática sintomática no se conoce. No se han identificado factores de riesgo específicos.
Trastornos tiroideos: Los pacientes tratados con REBIF® pueden presentar ocasionalmente alteraciones en la función tiroidea de nuevo diagnóstico o un empeoramiento de las ya existentes. Se recomienda practicar pruebas de función tiroidea en situación basal y, si son anormales, cada 6-12 meses tras el comienzo del tratamiento. Si las pruebas basales son normales, no es necesario repetirlas de forma sistemática, pero deben realizarse si aparecen signos clínicos de disfunción tiroidea (véase Reacciones secundarias y adversas).
Insuficiencia renal o hepática graves y mielosupresión grave: Se deberá tener precaución y considerar una estrecha monitorización cuando se administre interferón beta-1a en pacientes con insuficiencia renal y hepática graves y en pacientes con mielosupresión grave.
Anticuerpos neutralizantes: Pueden aparecer en el suero anticuerpos neutralizantes (Nab) frente al interferón beta-1a. La incidencia exacta de la formación de anticuerpos todavía no está clara. Los datos clínicos sugieren que, después de 24 a 48 meses de tratamiento con REBIF® 22 µg, aproximadamente 24% y con REBIF® 44 µg 13-14% de los pacientes presentan anticuerpos en suero frente al interferón beta-1a, de forma persistente. La presencia de anticuerpos atenúa la respuesta farmacodinámica al interferón beta-1a (beta-2 microglobulina y neopterina). Aunque el significado clínico de la inducción de anticuerpos no se ha dilucidado totalmente, el desarrollo de anticuerpos neutralizantes se asocia a una reducción de la eficacia sobre los parámetros clínicos y de resonancia magnética. La terapia con interferón deberá ser inmediatamente reconsiderada si se observa una mala evolución clínica asociada a la presencia de anticuerpos neutralizantes persistentes.
El empleo de diversos análisis para detectar los anticuerpos en suero y las diferentes definiciones de "anticuerpos positivos", limitan la capacidad para comparar la antigenicidad entre distintos productos.
Otras formas de esclerosis múltiple: Se dispone de escasos datos de eficacia y seguridad en pacientes con esclerosis múltiple sin capacidad ambulatoria. REBIF® no ha sido investigado aún en pacientes con esclerosis múltiple primaria progresiva y no se debe utilizar en dichos pacientes.
Efectos sobre la capacidad para conducir y utilizar máquinas: Las reacciones adversas que afectan al sistema nervioso central, relacionadas con uso de interferón beta (por ejemplo, mareos), podrían influir sobre la capacidad del paciente para conducir u operar maquinaria (véase Reacciones secundarias y adversas).
DOSIS Y VÍA DE ADMINISTRACIÓN: El tratamiento debe instaurarse bajo la supervisión de un médico con experiencia en el tratamiento de la enfermedad.
Posología:
Inicio del tratamiento: Cuando se inicia por primera vez el tratamiento con REBIF®, la dosis debe irse aumentando gradualmente para lograr que aparezca la taquifilaxia (tolerancia) y que así disminuyan las reacciones adversas.
Por lo tanto, es recomendado que:
• 8.8 µg deben ser administrados tres veces por semana durante las dos primeras semanas de tratamiento, los que corresponden a 0.1 ml de la jeringa prellenada de 44 µg (o a 0.2 ml de la jeringa de 22 µg). Para la administración de 0.1 ml de la jeringa de 44 µg, mover el émbolo hasta la marca de 0.1 ml en la escala de la jeringa, descartando el líquido. El volumen de solución que queda en la jeringa corresponde a la dosis que debe administrarse.
• 22 µg deben ser administrados tres veces por semana, administrados durante la tercera y cuarta semanas de tratamiento, los cuales corresponden a 0.25 ml de la jeringa de 44 µg o al volumen total de la jeringa de 22 µg. Para la administración de 0.25 ml de la jeringa de 44 µg, mover el émbolo hasta la marca de 0.25 ml en la escala de la jeringa, descartando el líquido. El volumen de solución que queda en la jeringa corresponde a la dosis que debe administrarse.
• 44 µg sean administrados tres veces por semana, desde la quinta semana en adelante, que corresponde con el volumen total de la jeringa de 44 µg.
La posología recomendada de REBIF® es de 44 µg, administrados tres veces por semana por inyección subcutánea. Una dosis más baja de 22 µg, administrada también tres veces por semana por inyección subcutánea, se recomienda para los pacientes que no toleran la dosis más alta, según considere el especialista que los trate.
Uso pediátrico: No se han llevado a cabo ensayos clínicos formales ni estudios farmacocinéticos en niños ni adolescentes. Sin embargo, los datos publicados limitados sugieren que el perfil de seguridad en adolescentes de 12 a 16 años tratados con REBIF® 22 µg, por vía subcutánea, tres veces por semana, es similar al observado en adultos. Se dispone de una información limitada sobre el uso de REBIF® en niños menores de 12 años y, por lo tanto, REBIF® no debe utilizarse en esta población.
Forma de administración: REBIF® es administrado por inyección subcutánea. Antes de la inyección y durante 24 horas después de cada inyección, se recomienda la administración de un analgésico antipirético para reducir los síntomas pseudogripales asociados con la administración de REBIF®.
Actualmente, se desconoce el tiempo durante el cual se debe tratar a los pacientes. La seguridad y eficacia de REBIF® no se han demostrado durante el periodo posterior a 4 años de tratamiento. Se recomienda evaluar a los pacientes al menos cada dos años en el periodo de los 4 años siguientes al comienzo del tratamiento con REBIF® y que el médico decida entonces de forma individualizada si conviene prolongar el tratamiento durante más tiempo.
Instrucciones de uso:
Forma de administración: REBIF® debe administrarse por vía subcutánea (bajo la piel).
La(s) primera(s) inyección(es) deben administrarse bajo la supervisión de un profesional de la salud adecuadamente calificado. Tras recibir el entrenamiento adecuado, el paciente, un miembro de su familia, amigo o persona encargada de su cuidado puede usar jeringas de REBIF® para administrar el medicamento en el domicilio del paciente (véase Inicio del tratamiento).
Dónde inyectar REBIF®:
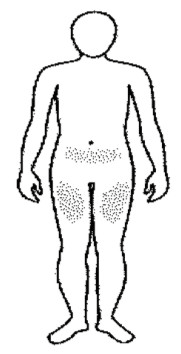
• Elegir un lugar para la inyección. El médico debe indicar dónde puede ponerse la inyección (son zonas adecuadas la parte superior de los muslos y la parte inferior del vientre).
• Se recomienda que se tome nota de las zonas de inyección y las vaya alternando de manera que no se inyecte con demasiada frecuencia en una zona determinada, a fin de reducir al mínimo el riesgo de necrosis en el lugar de inyección.
Nota: no utilizar ninguna zona con hinchazón, bultos duros o dolor; debe recomendarse al paciente que notifique a su médico o profesional de la salud sobre cualquier cosa que observe.
Como inyectar REBIF® jeringas prellenadas:
• Lavar las manos concienzudamente con agua y jabón.
• Sacar la jeringa de REBIF® de su envoltorio, quitando la cubierta de plástico.
• Antes de realizar las inyecciones debe usarse una gasa con alcohol para limpiar la piel en el lugar de inyección. Dejar secar la piel. Si queda algo de alcohol en la piel, el paciente puede notar una sensación de escozor.
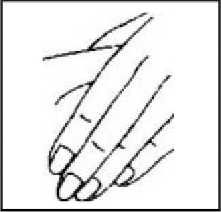
• Pellizcar suavemente la piel alrededor de la zona de inyección (para levantarla un poco).
• Apoyando la muñeca en la piel próxima a la zona, introducir la aguja directamente en la piel en ángulo recto, con un movimiento rápido y firme.
• Inyectar el medicamento presionando de forma lenta y sostenida (empujar el émbolo hasta el final, hasta que la jeringa esté vacía).
• Mantener una torunda en el lugar de inyección. Retirar la aguja de la piel.
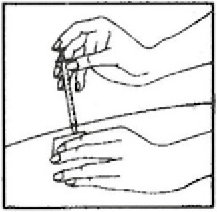
• Masajear suavemente la zona de inyección con una bola de algodón o gasa seca.
• Una vez que la inyección ha finalizado, la jeringa debe ser desechada inmediatamente en un recipiente apropiado para ello.
Precauciones especiales de eliminación y otras manipulaciones:
• La solución inyectable en jeringas prellenadas está lista para su uso. También puede administrarse con un autoinyector adecuado.
• Para un solo uso. Sólo deben utilizarse soluciones de claras a opalescentes, sin partículas y sin signos visibles de deterioro.
• La eliminación del medicamento no utilizado y de todos los materiales que hayan estado en contacto con él, se realizará de acuerdo con la normativa local.
MANIFESTACIONES Y MANEJO DE LA SOBREDOSIFICACIÓN O INGESTA ACCIDENTAL: En caso de sobredosis, se debe hospitalizar a los pacientes para observación e instaurar el tratamiento de soporte adecuado.
PRESENTACIONES:
Caja con 1, 3, 6 o 12 jeringas prellenadas de 22 µg (6 MUI)/0.5 ml en estuche individual e instructivo anexo.
Caja con 1, 3, 6 o 12 jeringas prellenadas de 44 µg (12 MUI)/0.5 ml en estuche individual e instructivo anexo.
Caja con 12 jeringas prellenadas de 22 µg (6 MUI)/0.5 ml en estuche individual e instructivo anexo, con un dispositivo médico Rebiject.
Caja con 12 jeringas prellenadas de 44 µg (12 MUI)/0.5 ml en estuche individual e instructivo anexo, con un dispositivo médico Rebiject.
RECOMENDACIONES SOBRE ALMACENAMIENTO: Conservar en refrigeración (entre 2 y 8°C). No congelar. Conservar en el embalaje original para protegerlo de la luz.
Periodo de validez: No utilice REBIF® después de la fecha de caducidad que aparece en el envase.
Precauciones especiales de conservación: Conservar entre 2 y 8ºC en el embalaje original. No congelar. Si temporalmente no se dispone de refrigeración, REBIF® puede ser conservado por el usuario, a una temperatura inferior a 25ºC durante un máximo de 30 días; luego debe ponerse de nuevo en refrigeración y utilizarse antes de la fecha de caducidad.
LEYENDAS DE PROTECCIÓN:
Literatura exclusiva para médicos. Su venta requiere receta médica. No se deje al alcance de los niños. No utilizar después de la fecha de caducidad indicada. No se administre si el contenedor está abierto. Almacenar en refrigeración (2º-8ºC). No congelar. No se use en el embarazo ni en la lactancia. Usar sólo por indicación y bajo supervisión médica. No use el medicamento si se distingue algún signo de deterioro. No se administre si la solución no es transparente, si contiene partículas en suspensión o sedimentos. Si no se administra todo el producto deséchese el sobrante.
Reporte las sospechas de reacción adversa al correo:
farmacovigilancia@cofepris.gob.mx
Hecho en Italia por:
Merck Serono S. p. A.
Vía Delle Magnolie 15
(loc. Frazione Zona Industriale)
70026 Modugno, (BA), Italia
Acondicionado en Uruguay por:
Ares Trading Uruguay, S. A. (A.T.U.S.A.)
Ruta 8, Km 17.500 Zona América
Montevideo, Uruguay
Distribuido por:
MERCK, S. A. de C. V.
Calle 5 No. 7
Fracc. Industrial Alce Blanco, C.P. 53370
Naucalpan de Juárez, Edo. de México
Reg. Núm. 109M97, SSA IV
®Marca registrada

