
IMNOVID
POMALIDOMIDA
Cápsulas
1 Caja, 21 Cápsulas, 1 Miligramos
1 Caja, 21 Cápsulas, 2 Miligramos
1 Caja, 21 Cápsulas, 3 Miligramos
1 Caja, 21 Cápsulas, 4 Miligramos
FORMA FARMACÉUTICA Y FORMULACIÓN:
Cada CÁPSULA contiene:
Pomalidomida 1 mg, 2 mg, 3 mg, 4 mg
Excipiente cbp 1 cápsula
INDICACIONES TERAPÉUTICAS:
En combinación con dexametasona-pacientes con mieloma múltiple en recaída y refractario: Pomalidomida en combinación con dexametasona está indicada para el tratamiento de mieloma múltiple en recaída y refractario en pacientes que han recibido lenalidomida y un inhibidor del proteosoma.
FARMACOCINÉTICA Y FARMACODINAMIA:
Farmacocinética:
Absorción: Pomalidomida es absorbida con una Cmáx entre 2 y 3 horas y es > 70% absorbida después de la administración de una sola dosis oral. La exposición sistémica (AUC) de pomalidomida aumenta de una manera aproximadamente proporcional a la dosis. Después de dosis múltiples, pomalidomida tiene una proporción de acumulación de 27 a 31 por ciento.
La coadministración con un alimento alto en grasas y alto en calorías disminuye el índice de absorción, disminuyendo la Cmáx media en plasma en ~27%, pero tiene un efecto mínimo sobre el grado de absorción global con una disminución del 8% en la media del AUC. Por lo tanto, pomalidomida se puede administrar independientemente de la ingesta de alimentos.
Distribución: Pomalidomida tiene una media del volumen aparente de distribución (Vd/F) entre 68.1 y 140 L. Pomalidomida se distribuye en el semen de sujetos sanos a una concentración de aproximadamente 67% del nivel de plasma a las 4 horas post-dosis (~Tmáx), después de 4 días de una dosis diaria de 4 mg. La unión in vitro de los enatiómeros de pomalidomida a las proteínas en el plasma humano está en un rango de 12% a 44% y no es dependiente de la dosis.
Metabolismo: Pomalidomida es el componente circulante principal (aproximadamente 70% de radioactividad en plasma) in vivo en sujetos sanos que recibieron una sola dosis oral de pomalidomida marcada con C14 (2 mg). No estuvieron presentes metabolitos a > 10% respecto al producto original o a la radiactividad total en plasma.
Pomalidomida se elimina en humanos a través de rutas múltiples incluyendo metabolismo mediado por CYP, hidrólisis no dependiente de CYP y excreción del fármaco intacto. Las rutas metabólicas predominantes de la radioactividad excretada son hidroxilación con glucuronidación o hidrólisis subsecuente. CYP1A2 y CYP3A4, in vitro, se identificaron como las enzimas primarias involucradas en la hidroxilación mediada por CYP de pomalidomida con contribuciones menores adicionales de CYP2C19 y CYP2D6.
La coadministración de pomalidomida con ketoconazol, fuerte inhibidor de CYP3A4/5 y glicoproteína P, o carbamazepina, fuerte inductor de CYP3A4/5, no tuvieron efecto clínicamente relevantes sobre la exposición a pomalidomida.
La coadministración de fluvoxamina, fuerte inhibidor de CYP1A2, con pomalidomida en presencia de ketoconazol, incrementó la exposición media a pomalidomida en 107% con un intervalo de confianza del 90% [91% a 124%] en comparación con pomalidomida más ketoconazol. En un estudio secundario para evaluar la contribución de un inhibidor de la CYP1A2 sobre los cambios metabólicos, la co-administración de la fluvoxamina sola en combinación con pomalidomida incrementó la exposición media a pomalidomida en 125% con un intervalo de confianza del 90% [98% a 156%] en comparación con pomalidomida sola. Reduzca en un 50% la dosis de pomalidomida si son co-administrados inhibidores fuertes de la CYP1A2 con la pomalidomida.
La coadministración de múltiples dosis de pomalidomida 4 mg con dexametasona 20 mg a 40 mg (un inductor débil a moderado de varias enzimas CYP, incluyendo CYP3A) a pacientes con mieloma múltiple, no tuvo efectos sobre la farmacocinética de pomalidomida en comparación con pomalidomida administrada sola.
Pomalidomida es un sustrato de la glicoproteína P in vitro, pero aparentemente esto no limita su absorción en humanos, en donde al menos 73% del fármaco fue absorbido. La coadministración de pomalidomida con ketoconazol, inhibidor de glicoproteína P, no tuvo efecto clínicamente relevante con la exposición a pomalidomida, por lo tanto, basado en lo anterior, no se anticipan interacciones medicamentosas clínicamente relevantes cuando se coadministra pomalidomida con inhibidores de la glucoproteína P.
Con base en los datos in vitro, la pomalidomida no es un inhibidor o inductor de isoenzimas del citocromo P-450 y no inhibe la glicoproteína P u otros transportadores estudiados. No se anticipan interacciones medicamentosas clínicamente relevantes cuando se coadministra pomalidomida con sustratos de estas rutas.
La administración de pomalidomida en fumadores, en el conocimiento de que el tabaco es un inductor de la CYP1A2, no tuvo un efecto clínico relevante en la exposición a la pomalidomida en relación a la exposición a pomalidomida observada en los no fumadores.
Excreción: El promedio de vida media de eliminación en plasma de Pomalidomida se encuentra entre 5.67 y 10.8 horas en sujetos sanos y entre 6.45 y 8.92 horas en pacientes con mieloma múltiple. Pomalidomida tiene una depuración corporal total promedio (CL/F) entre 6.78 y 10.74 L/hr para sujetos sanos y entre 3.9 y 9.96 L/hr para pacientes con mieloma múltiple.
Después de una sola administración oral de pomalidomida marcada con C14 (2 mg) a sujetos sanos, aproximadamente 73% y 15% de la dosis radiactiva fue eliminada en orina y heces respectivamente, con aproximadamente 2% y 8% del radiocarburo dosificado eliminado como pomalidomida en la orina y heces.
Pomalidomida es metabolizada extensamente antes de su excreción, eliminando los metabolitos resultante principalmente en la orina. Los 3 metabolitos predominantes en orina (que se forma mediante hidrólisis o hidroxilación con glucuronidación subsecuente) son responsables de aproximadamente 23%, 17% y 12%, respectivamente, de la dosis en la orina.
Los metabolitos dependientes de CYP son responsables de aproximadamente el 43% del total de radioactividad excretada, mientras que los metabolitos hidrolíticos no dependientes de CYP representan el 25% y la excreción de pomalidomida intacta representó el 10% (2% en orina y 8% en heces).
Farmacocinética en Niños: No existen datos disponibles sobre la administración de pomalidomida a sujetos pediátricos o adolescentes (< 18 años de edad).
Farmacocinética en pacientes Geriátricos: En sujetos de entre 61 a 82 años de edad, las medias de los parámetros farmacocinéticos de AUC (0-∞) y Cmax fueron generalmente similares a los sujetos más jóvenes.
Farmacocinética en Deterioro Renal: Los análisis de la farmacocinética poblacional mostraron que los parámetros farmacocinéticos de la pomalidomida no fueron marcadamente afectados en los pacientes con deterioro renal (definido por la eliminación de la creatinina o la tasa de filtración glomerular [eGFR, por sus siglas en inglés]) en relación a los pacientes con función renal normal (CrCl 60 mL/minuto).
La exposición media normalizada de la AUC para pomalidomida fue 98.2% con un intervalo de confianza del 90% [77.4% a 120.6%] en pacientes con deterioro renal moderado (eGFR ≥ 30 a ≤ 45 mL/minuto/1.73 m²) en relación a pacientes con función renal normal.
La exposición media normalizada de la AUC para pomalidomida fue de 100.2% con un intervalo de confianza del 90% [79.7% a 127.0%] en pacientes con deterioro renal grave que no requieren diálisis (CrCl < 30 o eGFR < 30 mL/minuto/1.73 m²) en relación a los pacientes con función renal normal.
La exposición media normalizada de la AUC a pomalidomida incrementó en un 35.8% con un intervalo de confianza de 90% [7.5% a 70.0%] en pacientes con deterioro renal grave que sí requieren diálisis (CrCl < 30 mL/minuto requiriendo diálisis) en relación a los pacientes con función renal normal. La media de los cambios en la exposición a pomalidomida en cada uno de estos grupos de pacientes con deterioro renal no son de una magnitud tal que requieran ajustes de la dosis.
Farmacocinética en Deterioro Hepático: Los parámetros farmacocinéticos fueron cambiados modestamente en pacientes con deterioro hepático (definido por el criterio de Child-Pugh) en relación a sujetos sanos. La exposición media a pomalidomida incremento en 51% con un intervalo de confianza del 90% [9% a 110%] en pacientes con deterioro hepático leve en comparación a sujetos sanos. La exposición media a pomalidomida incrementó en 58% con un intervalo de confianza del 90% [13% a 119%] en pacientes con deterioro hepático moderado en relación a sujetos sanos. La exposición media a pomalidomida incrementó en un 72% con un intervalo de confianza de 90% [24% a 138%] en pacientes con deterioro hepático grave en relación a sujetos sanos. Los incrementos medios en la exposición a pomalidomida en cada uno de estos grupos de pacientes con deterioro hepático no son de una magnitud tal que se requieran ajustes en la dosis o en el régimen de dosificación.
Farmacodinamia:
Mecanismo de acción:
Grupo Farmacoterapéutico: Agente inmunomodulador.
Pomalidomida tiene actividad directa tumoricida antimieloma, actividades inmunomoduladoras e inhibe el soporte de células estromales para el crecimiento de células tumorales de mieloma múltiple. Específicamente, pomalidomida inhibe la proliferación e induce apoptosis de las células hematopoyéticas tumorales. Adicionalmente, pomalidomida inhibe la proliferación de líneas celulares de mieloma múltiple resistentes a lenalidomida y tiene acción sinérgica con dexametasona tanto en líneas celulares sensibles a lenalidomida como resistentes a lenalidomida para inducir apoptosis de las células tumorales. Pomalidomida mejora la inmunidad mediada por células T y células asesinas naturales (NK) e inhibe la producción de citocinas proinflamatorias (por ejemplo, TNF-α y IL-6) mediante monocitos. Pomalidomida también inhibe la angiogénesis bloqueando la migración y adhesión de células endoteliales.
Pomalidomida se une directamente a la proteína cereblon (CRBN), que es parte de un complejo de la E3 ligasa que incluye la proteína 1 (DDB1), de unión al ácido desoxirribonucleico (DNA) dañado, culina 4 (CUL4) y Roc1 y puede inhibir la auto-ubiquitinación de CRBN dentro del complejo. Las E3 ubiquitin ligasas son responsables de la poliubiquitinación de una variedad de proteínas sustrato y puede explicar parcialmente los efectos celulares pleiotrópicos observados en el tratamiento con pomalidomida.
Se demostraron las actividades proeritropoyéticas de pomalidomida en células madre hematopoyéticas CD34+ inducidas para diferenciar el fenotipo eritroide. Estas actividades se manifestaron como una maduración eritroide retardada, incremento de la proliferación de células eritroides inmaduras e inducción de producción de hemoglobina fetal (HbF).
Estudios Clínicos:
Mieloma múltiple en recaída y refractario:
Resumen de eficacia: Se evaluó la eficacia y seguridad de pomalidomida en combinación con dexametasona en un estudio Fase III multicéntrico, aleatorizado, abierto (CC-4047-MM-003), en el que se comparó la terapia de pomalidomida más dexametasona en dosis baja (Pom + LD-dex) con dexametasona en dosis alta (HD-dex) sola en pacientes adultos previamente tratados con mieloma múltiple en recaída y refractario, que habían recibido por lo menos dos esquemas de tratamiento previos y habían presentado falla tanto con lenalidomida como con bortezomib y habían demostrado progresión de la enfermedad durante la última terapia. Un total de 455 sujetos fueron incluidos en el estudio: 302 en el grupo Pom + LD-dex y 153 en el grupo HD-dex. La mayoría de los sujetos eran hombres (59%) y de raza blanca (79%); la mediana de la edad para la población global fue 64 años (mín., máx.: 35, 87 años).
A los pacientes del grupo Pom + LD-dex se les administraron 4 mg de pomalidomida por vía oral los Días 1 a 21 de cada ciclo de 28 días. Se administró LD-dex (40 mg) una vez al día los Días 1, 8, 15 y 22 de un ciclo de 28 días. Los sujetos > 75 años de edad iniciaron tratamiento con 20 mg de dexametasona utilizando el mismo esquema. Para el grupo HD-dex, se administró dexametasona (40 mg) una vez al día los Días 1 a 4, 9 a 12 y 17 a 20 de un ciclo de 28 días. Los sujetos > 75 años de edad iniciaron tratamiento con 20 mg de dexametasona utilizando el mismo esquema. El tratamiento se continuó hasta que los sujetos presentaron progresión de la enfermedad. Los sujetos del grupo HD-dex después de la progresión de la enfermedad tenían la opción de recibir Pomalidomida sola en un estudio asociado (CC-4047-MM-003/C).
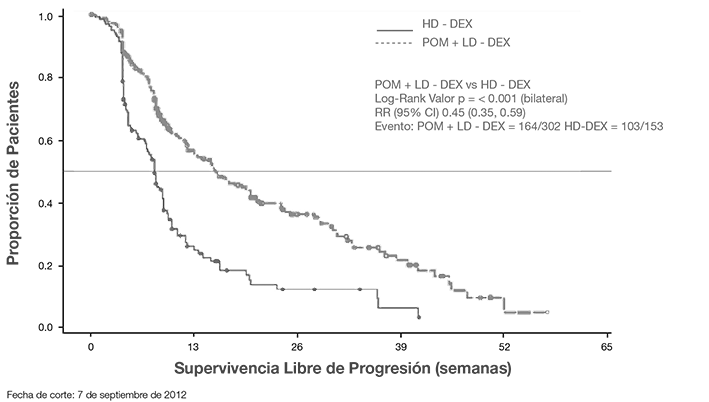
El punto final primario para evaluar eficacia fue supervivencia libre de progresión (PFS, por sus siglas en inglés) de conformidad con los criterios del Grupo de Trabajo Internacional de Mieloma (IMWG, por sus siglas en inglés). Para la población con Intención de Tratar (ITT, por sus siglas en inglés), la mediana del tiempo durante PFS mediante la revisión del Comité de Adjudicación y Revisión Independiente (IRAC) fue de 15.7 semanas (IC 95%: 13.0, 20.1) en el grupo Pom + LD-dex; el índice estimado de supervivencia libre de eventos a 26 semanas fue 35.99% ± 3.46. En el grupo HD-dex, la mediana del tiempo PFS fue 8.0 semanas (IC 95%: 7.0, 9.0); el índice estimado de supervivencia libre de eventos a 26 semanas fue 12.15% ± 3.63%. Se obtuvieron resultados idénticos en la revisión de IRAC con base en los criterios del Grupo Europeo para Sangre y Trasplante de Médula Ósea (EBMT). Los resultados del análisis de la población evaluable para eficacia se basaron en los criterios del IMWG, así como en los criterios EBMT, fueron consistentes con los observados en la población ITT. Independientemente del subgrupo evaluado, la PFS generalmente fue consistente con la observada en la población ITT para ambos grupos de tratamiento.
La Supervivencia Libre de Progresión se resume en la Tabla 1 para la población ITT. En la Figura 1 se presenta la curva de Kaplan-Meier para PFS en la población ITT.
Tabla 1. Tiempo de Supervivencia Libre de Progresión por Revisión IRAC con Base en Criterios IMWG
(Prueba de LogRank Estratificada) (Población ITT)
|
Pom + LD-Dex (N = 302) |
HD-Dex (N =153) |
Global (N =455) |
|
|
Tiempo de Supervivencia Libre de Progresión (semanas) |
|||
|
Mediana (IC 95% Bilateralb) |
15.7 [13.0, 20.1] |
8.0 [7.0, 9.0] |
11.9 [9.7, 13.6] |
|
Razón de Riesgo (Pom+LD Dex:HD -Dex) 2- IC 95% Bilateralc |
0.45 [0.35, 0.59] |
||
|
Valor-P con Prueba de Rango Logarítmico bilaterald |
< 0.001 |
||
IC = Intervalo de confianza; IRAC = Comité de Adjudicación de Revisión Independiente; NE = No Estimable.
a. La mediana se basa en el estimado de Kaplan-Meier.
b.Intervalo de confianza al 95% alrededor de la mediana del tiempo de supervivencia libre de progresión.
c.Basado en el modelo de riesgos proporcionales de Cox comparando las funciones de riesgo asociadas con grupos de tratamiento, estratificados por edad (≤ 75 vs > 75), población de enfermedades (refractario tanto a Lenalidomida como a Bortezomib vs no refractario a ambos fármacos) y número previo de terapia antimieloma (= 2 vs >2).
d. El valor -p se basa en la prueba de Log-Rank estratificada con los mismos factores de estratificación que el modelo de Cox anterior.
Fecha de corte: 7 de septiembre de 2012.
Figura 1. Supervivencia Libre de Progresión Basada en Revisión IRAC de Respuesta de acuerdo a los Criterios de la IMWG (Prueba de Log-Rank Estratificada) (Población ITT)
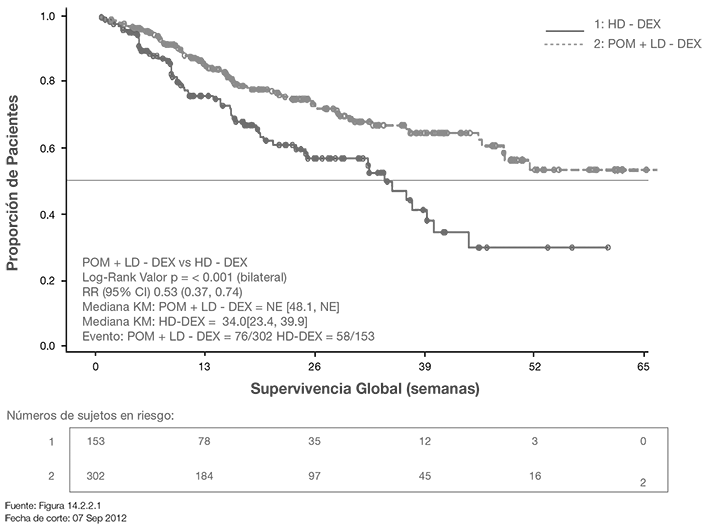
La Supervivencia Global fue uno de los puntos finales secundarios del estudio. Un total de 226 (74.8%) de los sujetos del grupo Pom + LD dex y 95 (62.1%) sujetos del grupo HD-dex estaban vivos a la fecha de corte (07 Sep 2012). La mediana del tiempo OS a partir de los estimados de Kaplan-Meier no se alcanzó para Pom + LD-dex, pero se espera sea cuando menos 48 semanas, el límite inferior del IC 95%. La mediana del tiempo OS para el grupo HD- dex fue 34 semanas (IC 95%: 23.4, 39.9). La tasa libre de eventos a 1 año fue 52.6% (± 5.72%) para el grupo Pom + LD-dex y 28.4% (± 7.51%) para el grupo HD-dex. La diferencia en OS entre los dos grupos de tratamiento fue estadísticamente significativas (p < 0.001).
Los resultados para la eficacia y población evaluable son consistentes con los observados en la población ITT.
La Curva de Kaplan-Meier para OS para la población ITT se presenta en la Figura 2.
Figura 2. Curva de Kaplan-Meier de Supervivencia Global (Población ITT)
Se llevaron a cabo dos estudios aleatorizados adicionales (MM-002 y IFM-2009-02) para evaluar la eficacia y seguridad de pomalidomida.
El Estudio 1 (MM-002) fue abierto, aleatorizado, multicéntrico, multinacional, de dos fases en pacientes con mieloma múltiple en recaída y refractario que habían sido refractarios a su última terapia para mieloma y habían recibido lenalidomida y bortezomib. En la primera fase del Estudio 1, se determinó que la dosis máxima tolerada (MTD) de pomalidomida, cápsulas, era 4 mg/día, administrados en los días 21/28 hasta progresión de la enfermedad. En la segunda fase del Estudio 1, se evaluó la seguridad y eficacia de pomalidomida 4 mg, 21/28 días hasta progresión de la enfermedad sola y en combinación con dexametasona (40 mg/día administrada a los días 1, 8, 15 y 22 de cada ciclo de 28 días). Los pacientes del grupo de pomalidomida sola podían agregar dexametasona en dosis baja hasta progresión de la enfermedad.
El Estudio 2 (IFM-2009-02) fue abierto, aleatorizado, multicéntrico, de pomalidomida en combinación con dosis bajas de dexametasona en pacientes con mieloma múltiple en recaída y refractario que tenían enfermedad progresiva y habían recibido tanto lenalidomida como bortezomib. En este estudio, los pacientes fueron aleatorizados a pomalidomida 4 mg/día, 21/28 días hasta progresión de la enfermedad (Grupo A) o pomalidomida 4 mg/día 28/28 días hasta progresión de la enfermedad. Tanto en el Grupo A como en el Grupo B, pomalidomida se administró en combinación con dexametasona en dosis baja (40 mg administrados los días 1, 8, 15 y 22 de cada ciclo de 28 días).
La Tabla 2 resume las características basales de los pacientes y enfermedades en los dos estudios. En ambos estudios las características demográficas basales y relacionadas con la enfermedad estuvieron bien balanceadas y fueron comparables entre los grupos de estudio.
Tabla 2. Características Demográficas Basales y Relacionadas con la Enfermedad - Estudios 1 y 2
|
Estudio 1 (MM-002) |
Estudio 2 (IFM-2009-02) |
|||
|
pomalidomida/ dexametasona 21/28 días (N = 113) |
pomalidomida sola1 21/28 días (N = 108) |
pomalidomida/dexametasona 21/28 días (N = 43) |
pomalidomida/dexametasona 28/28 días (N = 41) |
|
|
Características de los Pacientes |
||||
|
Edad (años) Mediana (Mín., Máx.) |
64 (34, 88) |
61 (37, 88) |
60 (45, 81) |
60 (42, 83) |
|
Distribución de la Edad n (%) ≤ 75 > 75 |
99 (87.6) 14 (12.4) |
95 (88.0) 13 (12.0) |
N/D |
N/D |
|
Sexo n (%) Masculino Femenino |
62 (54.9) 51 (45.1) |
57 (52.8) 51 (47.2) |
30 (69.8) 13 (30.2) |
27 (65.9) 14 (34.1) |
|
Raza/Etnicidad n (%) Blanca Negra o Afroamericana Todas las demás Razas |
92 (81.4) 17 (15) 4 (3.6) |
86 (79.6) 16 (14.8) 6 (5.6) |
N/D |
N/D |
|
Desempeño ECOG n (%) Status 0-1 |
100 (88.5) |
95 (87.9) |
34 (79.1) |
33 (80.5) |
|
Características de la Enfermedad |
||||
|
Estadio de Mieloma Múltiple2 n (%) I II III |
8 (7.1) 29 (25.7) 76 (67.3) |
8 (7.4) 29 (26.9) 71 (65.7) |
4 (9.3) 1 (2.3) 32 (74.4) |
10 (25) 4 (10) 22 (55) |
|
Citogenética3.4 |
||||
|
Anomalías n (%) |
||||
|
Alto Riesgo |
30 (34.5) |
30 (41.1) |
5 (18.5) |
8 (38.1) |
|
Riesgo Estándar |
57 (65.5) |
43 (58.9) |
22 (81.5) |
13 (61.9) |
|
Faltante |
26 (23.0) |
35 (32.4) |
16 |
20 |
|
Terapias Previas |
||||
|
Número de Terapias Previas Mediana (Mín., Máx.) |
5 (2, 13) |
5 (2, 12) |
5 (1, 13) |
5 (2, 10) |
|
Refractario a lenalidomida n (%) |
87 (77.0) |
85 (78.7) |
36 (83.7) |
39 (95.1) |
|
Refractario a bortezomib n (%) |
82 (72.6) |
75 (69.4) |
34 (79.1) |
34 (82.9) |
|
Refractario a bortezomib y a lenalidomida n (%) |
69 (61.1) |
64 (59.3)) |
32 (74.4) |
32 (78.0) |
N/D = no se realizó.
1. Se permitió a los pacientes agregar dexametasona en dosis baja después de la progresión de la enfermedad.
2.Durie-Salmon.
3.Estudio 1 alto riesgo basado en 17p13 y/o 4p16/14q32 mediante hibridación in situ fluorescente (FISH).
4. Estudio 2 alto riesgo basado en deleción de 17p y/o translocación (4;14) vía hibridación in situ fluorescente (FISH).
En el Estudio 1, con base en la valoración por parte de un Comité de Adjudicación de Revisión Independiente (IRAC), la tasa de respuesta global en la población de intención de tratamiento (ITT), fue 30%. En general 34 pacientes (30%) tuvieron una respuesta a pomalidomida incluyendo 1 paciente (0.9%) con respuesta completa y 33 pacientes (29.2%) con una respuesta parcial. La tasa de respuesta global fue consistente independientemente del tipo de terapia antimieloma previa a la que habían estado expuestos estos pacientes durante su historial de tratamiento o el último tratamiento inmediato al que se volvieron refractarios antes de recibir pomalidomida. El punto final primario de eficacia en el Estudio 1 (fase 2) fue supervivencia libre de progresión (PFS).
La mediana de la duración de la respuesta para respondedores tratados con pomalidomida más dexametasona fue aproximadamente 32.1 semanas (IC 95% = 22.1 a 39.9 semanas). La mediana de la duración de la respuesta para los sujetos tratados con pomalidomida sola aún no se ha alcanzado (Tabla 4: Resultados de PFS en los Estudios 1 y 2).
El punto final primario de eficacia para el estudio 2 fue la tasa de respuesta. Con base en la valoración por parte del IRC, la tasa de respuesta global en la población de Intención de Tratamiento (ITT) fue 35% (IC 95%: 25%-46%); no se observaron diferencias significativas entre las tasas de respuesta del Grupo A y Grupo B (p = 0.943). En general, 29 pacientes (35%) tuvieron una respuesta a pomalidomida, incluyendo 3 pacientes (4%) con una respuesta completa y 26 pacientes (31%) con una respuesta parcial.
La PFS fue de 25.1 semanas en ambos grupos de tratamiento. La mediana de la duración de la respuesta fue 45.7 semanas (IC 95%: 15.1 a 54.7 semanas) en el Grupo A, en comparación con 31.6 semanas en el Grupo B, una diferencia equivalente a aproximadamente 3 meses.
Tabla 3. Resultados de los Estudios
|
Estudio 1 (MM-002) |
Estudio 2 (IFM-2009-02) |
|||
|
pomalidomida/dexametasona 21/28 días (N = 113) |
pomalidomida sola2 21/28 días (N = 108) |
pomalidomida/dexametasona 21/28 días (Grupo A) (N = 43) |
pomalidomida/dexametasona 28/28 días (Grupo B) (N = 41) |
|
|
Respuesta |
||||
|
Tasa de Respuesta Global (ORR)1 n (%) |
34 (30.1) |
10 (9.3) |
15 (34.9) |
14 (34.1) |
|
Respuesta Completa (CR) n (%) |
1 (0.9) |
0 (0.0) |
1 (2.3) |
2 (4.9) |
|
Respuesta Parcial (PR) (RR/PR) n (%) |
33 (29.2) |
10 (9.3) |
14 (32.5) |
12 (29.2) |
|
Duración de la Respuesta (semanas) |
||||
|
Eventos n |
34 |
10 |
15 |
14 |
|
Mediana (semanas) |
32.1 |
NE |
45.7 |
31.6 |
|
PFS (semanas) |
||||
|
Eventos n (%) |
86 (76.1) |
81 (75.0) |
29 (65.1) |
32 (78.0) |
|
Mediana en semanas |
16.6 |
10.7 |
25.1 |
25.1 |
|
Razón de Riesgos [IC 95%] |
0.73 [0.54, 0.99] |
1.18 [0.71, 1.95] |
||
|
Refractario a lenalidomida |
||||
|
Tasa de Respuesta Global (ORR)1 n (%) |
22/87 (25.3) |
8/85 (9.4) |
13/36 (36.1) |
14/39 (35.9) |
|
Mediana de la Duración de Respuesta (semanas) |
30.40 |
NE |
NA |
NA |
|
Refractario a lenalidomida y bortezomib |
||||
|
Tasa de Respuesta Global (ORR)1 n (%) |
19/69 (27.5) |
6/64 (9.4) |
11/32 (34.4) |
9/32 (28.1) |
|
Mediana de la Duración de la Respuesta (semanas) |
26.8 |
NE |
NA |
NA |
NE = no establecido, NA = no aplicable.
1. Estudio 1: ORR = PR + CR conforme a los criterios EMBT, Estudio 2: ORR = PR, VGPR, CR conforme a IMWG.
2.Los resultados son previos a la adición de dexametasona.
CONTRAINDICACIONES:
• Embarazo; mujeres con potencial reproductivo, excepto cuando se cumplen todas las condiciones para prevención del embarazo.
• Hipersensibilidad a pomalidomida o a cualquiera de los excipientes.
PRECAUCIONES Y RESTRICCIONES DE USO DURANTE EL EMBARAZO Y LA LACTANCIA:
Embarazo: Se determinó que pomalidomida es teratogénica en estudios de toxicidad en el desarrollo embrio-fetal en ratas y conejos. Pomalidomida atravesó la placenta y se detectó en la sangre fetal después de la administración a conejas grávidas.
No se puede descartar un efecto teratogénico de pomalidomida en humanos. Por lo tanto, se requiere el siguiente plan de manejo de riesgos en exposición fetal.
• No se debe usar pomalidomida en mujeres embarazadas o que puedan quedar embarazadas mientras reciben el fármaco.
• Las mujeres con potencial reproductivo deben utilizar un medio anticonceptivo eficaz durante 28 días antes de la terapia, durante la terapia con pomalidomida y las interrupciones de dosis, además 28 días después de la descontinuación de la terapia con pomalidomida o abstenerse continuamente de relaciones heterosexuales reproductivas. Un régimen anticonceptivo eficaz se considera una combinación de dos métodos anticonceptivos: un método altamente efectivo (el dispositivo intrauterino (DIU), los métodos hormonales, ligadura de trompas o vasectomía de la pareja) y un método efectivo adicional (condón sintético masculino, diafragma o tapón cervical).
• Debido a que existe un mayor riesgo de VTE (eventos tromboembólicos venosos) en pacientes que reciben píldoras anticonceptivas orales combinadas, los médicos deben discutir los riesgos/beneficios de los métodos anticonceptivos con sus pacientes.
• Las mujeres con potencial reproductivo deben someterse a pruebas regulares de embarazo durante el tratamiento con pomalidomida. En caso de embarazo durante el tratamiento, descontinuar el fármaco inmediatamente. Bajo estas condiciones, referir al paciente a un ginecoobstetra experimentado en toxicidad reproductiva para evaluación y asesoría adicional.
• Es obligatorio que las mujeres con potencial reproductivo reciban asesoría para que estén informadas sobre los riesgos de pomalidomida. Pomalidomida está contraindicada en mujeres con potencial reproductivo a menos que se cumplan todos los términos de la asesoría.
• Los pacientes de sexo masculino que reciben pomalidomida deben utilizar métodos anticonceptivos eficaces durante la terapia con pomalidomida (incluyendo interrupciones de dosis) y cuando menos durante 28 días después de la descontinuación de la terapia con pomalidomida si su pareja tiene potencial reproductivo. Los pacientes masculinos no deben donar semen o esperma durante la terapia (incluso durante las interrupciones de dosis) y durante un mínimo de 4 semanas después de la descontinuación del tratamiento con pomalidomida.
• Todos los pacientes (hombres y mujeres con o sin potencial reproductivo) deben aceptar abstenerse de donar sangre mientras reciben pomalidomida (inclusive durante las interrupciones de dosis) y cuando menos durante 4 semanas después de la última dosis de pomalidomida.
Fertilidad: En un estudio de fertilidad y desarrollo embrionario temprano en ratas, se observó una disminución en el número promedio de embriones viables y un incremento en la pérdida postimplantación a todos los niveles de dosis en hembras tratadas. Se deben discutir minuciosamente con los pacientes las opciones y/o alternativas precautorias proactivas para planificación familiar.
Uso durante la lactancia: Se desconoce si pomalidomida es excretada en la leche humana. Pomalidomida puede ser detectada en la leche de ratas lactantes después de administrarlo a la madre. Debido al potencial de reacciones adversas en niños lactantes, se debería tomar una decisión si se suspende la lactancia o si se suspende el medicamento, tomando en cuenta la importancia del fármaco para la madre.
REACCIONES SECUNDARIAS Y ADVERSAS:
Datos de Estudios Clínicos:
Estudio Clínico de Reacciones Adversas al Fármaco (ADRs) a Pomalidomida en Mieloma Múltiple en Recaída y Refractario (MM-003): A continuación se presentan las reacciones adversas al fármaco (ADRs) observadas en pacientes tratados con pomalidomida/dexametasona por clase de sistema orgánico y frecuencia para todas las ADRs, ADRs grado 3/4 y ADRs graves. Las ADRs en esta sección fueron valoradas y seleccionadas por Celgene como al menos posiblemente relacionadas con pomalidomida y se presentan de conformidad con la guía del Grupo de Trabajo CIOMS III y V, con las categorías de frecuencia definidas como: muy frecuentes (≥ 1/10), frecuentes (≥ 1/100 a < 1/10); y poco frecuentes (≥ 1/1,000 a < 1/100). Las consideraciones para determinar una ADR incluyeron: factibilidad biológica/farmacológica para una relación fármaco-evento, morbilidades conocidas de población objetivo y la enfermedad que se está tratando, reacciones adversas sospechosas con fármacos de esta clase y peso de la evidencia (por ejemplo, reexposición positiva, eliminación de exposición positiva, tiempo para el inicio, ausencia de factores de confusión). Además, se aplicó el juicio médico para determinar excepciones para inclusión y exclusión, según fue necesario.
Tabla 4. Reacciones Adversas con Pomalidomida (población de seguridad)
|
Clase de Sistema Orgánico/Término Preferido |
Todas las ADRs/Frecuencia (%) |
ADRs 3-4/Frecuencia (%) |
SADRs/Frecuencia (%) |
|
Trastornos de la sangre y sistema linfático |
Muy Frecuente Anemia–45.7 Neutropenia–45.3 Trombocitopenia–27.0 Leucopenia–12.3 Frecuente Neutropenia febril–6.7 |
Muy Frecuente Anemia–27.0 Neutropenia–41.7 Trombocitopenia–20.7 Frecuente Leucopenia–8.7 Neutropenia febril–6.7 |
Frecuente Neutropenia febril–4.0 Neutropenia–2.0 Anemia–2.0 Trombocitopenia–1.7 |
|
Trastornos generales y condiciones del sitio de administración |
Muy Frecuente Fatiga–28.3 Fiebre–21.0 Edema periférico–13.0 |
Frecuente Fiebre–3.0 Fatiga–4.7 Edema periférico–1.3 |
Frecuente Fiebre–5.7 Poco frecuente Fatiga–0.7 Edema periférico–0.3 |
|
Infecciones e Infestaciones# |
Muy Frecuente Neumonía–10.7 Frecuente Infecciones del tracto respiratorio superior–9.3 Bronquitis–8.0 Nasofaringitis–6.3 Infección del tracto respiratorio–5.7 Bronconeumonía–3.0 Sepsis neutropénica–1.0 |
Frecuente Neumonía–9.0 Infecciones del tracto respiratorio superior–1.0 Infección del tracto respiratorio–1.0 Bronconeumonía–1.7 Sepsis neutropénica–1.0 Poco frecuente Bronquitis–0.3 |
Frecuente Neumonía–9.3 Infección del tracto respiratorio–1.0 Bronconeumonía–1.7 Sepsis neutropénica–1.0 Poco frecuente Infecciones del tracto respiratorio superior–0.7 Bronquitis–0.3 |
|
Trastornos gastrointestinales |
Muy Frecuente Estreñimiento–19.3 Diarrea–18.3 Náuseas–11.7 Frecuente Vómito–7.7 |
Frecuente Estreñimiento–1.7 Vómito–1.3 Diarrea–1.0 Poco frecuente Náuseas–0.7 |
Frecuente Vómito–1.0 Poco frecuente Diarrea–0.7 Estreñimiento–0.3 Náuseas–0.3 |
|
Trastornos musculoesqueléticos y del tejido conectivo |
Muy Frecuente Dolor óseo–14.7 Espasmos musculares–10.0 |
Frecuente Dolor óseo–6.3 Poco frecuente Espasmos musculares–0.3 |
Frecuente Dolor óseo–2.7 |
|
Trastornos respiratorios, torácicos y mediastínicos |
Muy Frecuente Disnea–16.7 Tos–15.0 Frecuente Embolismo Pulmonar–1.0 |
Frecuente Disnea–4.7 Poco frecuente Embolismo Pulmonar–0.7 Tos–0.3 |
Frecuente Embolismo Pulmonar–1.0 Disnea–1.3 Poco frecuente Tos–0.3 |
|
Trastornos del sistema nervioso |
Frecuente Mareo–9.0 Temblores–5.0 Neuropatía sensorial periférica–5.0 Nivel deprimido de la conciencia–1.3 |
Frecuente Nivel deprimido de la conciencia–1.0 Poco frecuente Mareo–0.7 Temblores–0.7 Neuropatía sensorial periférica–0.3 |
Poco frecuente Nivel deprimido de la conciencia–0.7 |
|
Trastornos del metabolismo y nutrición |
Muy Frecuente Apetito disminuido–10.0 Frecuente Hipercalemia–2.7 Hiponatremia–2.3 |
Frecuente Hipercalemia–1.7 Hiponatremia–1.0 Poco frecuente Apetito disminuido–0.7 |
Frecuente Hiponatremia–1.0 |
|
Trastornos de la piel y tejido subcutáneo |
Frecuente Prurito–7.0 Exantema–6.7 |
Frecuente Exantema–1.0 |
|
|
Trastornos psiquiátricos |
Frecuente Estado de confusión–3.7 |
Frecuente Estado de confusión–2.3 |
Frecuente Estado de confusión–1.0 |
|
Valores de laboratorio |
Frecuente Recuento disminuido de neutrófilos–4.7 Recuento disminuido de leucocitos–2.7 Recuento plaquetario disminuido–3.3 Incremento de alanino aminotransferasa–1.7 |
Frecuente Recuento disminuido de neutrófilos–4.0 Recuento disminuido de leucocitos–2.7 Recuento plaquetario disminuido–2.7 Incremento de alanino aminotransferasa 1.3 |
Poco frecuente Recuento plaquetario disminuido–0.3 |
|
Trastornos renales y urinarios |
Frecuente Insuficiencia renal–4.0 Retención urinaria–1.3 |
Frecuente Insuficiencia renal–3.0 Poco frecuente Retención urinaria–0.3 |
Frecuente Insuficiencia renal–2.7 Retención urinaria–1.0 |
|
Trastornos del oído y laberinto |
Frecuente Vértigo–3.0 |
Frecuente Vértigo–1.0 |
Poco frecuente Vértigo–0.3 |
|
Trastornos del sistema reproductivo mamario |
Frecuente Dolor pélvico–1.7 |
Frecuente Dolor pélvico–1.3 |
|
|
Trastornos vasculares |
Frecuente Trombosis venosa profunda–1.3 |
Poco frecuente Trombosis venosa profunda–0.3 |
Poco frecuente Trombosis venosa profunda–0.3 |
|
Trastornos hepatobiliares |
Poco frecuente Hiperbilirrubinemia–0.9 |
Poco frecuente Hiperbilirrubinemia–0.9 |
Poco frecuente Hiperbilirrubinemia–0.9 |
# Todos los terminos preferidos bajo el SOC de Infecciones e Infestaciones (incluyendo infecciones bacterianas, virales y fúngicas), excepto por las infecciones raras de interés de Salud Pública que deberán considerarse listadas.
Datos Postcomercialización: Las siguientes reacciones adversas al fármaco se han identificado a partir de la experiencia postcomercialización a nivel mundial con pomalidomida.
Debido a que estas reacciones son reportadas voluntariamente de una población de tamaño incierto, no siempre es posible estimar confiablemente su frecuencia o establecer una relación causal con la exposición al fármaco.
Trastornos de la sangre y del sistema linfático: Pancitopenia.
Trastornos gastrointestinales: Hemorragia gastrointestinal.
Trastornos hepatobiliares: Hepatitis, aumento de las pruebas de función hepática.
Trastornos del sistema inmune: Reacciones alérgicas (por ejemplo, angioedema, urticaria).
Infecciones e infestaciones: Reactivación viral (como el virus de la hepatitis B y herpes zoster).
Neoplasias benignas, malignas y no especificadas (incluyendo quistes y pólipos): Síndrome de lisis tumoral, carcinoma de células basales y carcinoma de células escamosas de la piel.
Trastornos respiratorios, torácicos y mediastínicos: Enfermedad pulmonar intersticial (ILD), neumonitis.
Trastornos cutáneos y subcutáneos: Síndrome de Stevens-Johnson (SJS), necrólisis epidérmica tóxica, reacción farmacológica con eosinofilia y síntomas sistémicos (DRESS).
PRECAUCIONES Y RELACIÓN CON EFECTOS DE CARCINOGÉNESIS, MUTAGÉNESIS, TERATOGÉNESIS Y SOBRE LA FERTILIDAD:
Genotoxicidad/Carcinogenicidad: La pomalidomida no fue mutagénica en ensayos de mutaciones bacterianas y de mamíferos y no indujo aberraciones cromosómicas en los linfocitos de sangre periférica humana o formación de micronúcleos en eritrocitos policromáticos en médula ósea de ratas administrados hasta 2000 mg/kg/día. No se han realizado estudios de Carcinogenicidad.
Fertilidad y Desarrollo Embrionario Inicial: En un estudio de fertilidad y desarrollo embrionario temprano en ratas, pomalidomida se administró a machos y hembras en dosis de 25, 250 y 1000 mg/kg/día. Las ratas macho se dosificaron empezando 28 días antes de la cohabitación y continuando hasta el día previo a la necropsia; las ratas hembra fueron dosificadas empezando 14 días antes de la cohabitación, durante el periodo de apareo y hasta el Día de Gestación 7. El examen uterino en el Día 13 de Gestación mostró una disminución en la media del número de embriones viables y un incremento en la pérdida postimplantación a todos los niveles de dosis. Por lo tanto, el NOAEL para estos efectos observados fue < 25 mg/kg/día (Exposición 99 veces mayor a la dosis más baja relativa probada a una dosis de 4 mg). Cuando los machos tratados en este estudio fueron apareados con hembras no tratadas, todos los parámetros uterinos fueron comparables con los controles. Con base en estos resultados, los efectos observados fueron atribuidos al tratamiento de hembras.
Desarrollo Embrio-Fetal: Se determinó que pomalidomida es teratogénica tanto en ratas como en conejos cuando se administra durante el periodo principal de organogénesis. En el estudio de toxicidad del desarrollo embrio-fetal en ratas se observaron malformaciones de ausencia de vejiga urinaria, ausencia de glándula tiroides y fusión y mala alineación de los elementos vertebrales lumbares y torácicos (arcos central y/o neural) en todos los niveles de dosis (25, 250 y 1000 mg/kg/día). No se observó toxicidad materna en este estudio. Por lo tanto, el NOAEL materno fue 1000 mg/kg/día y el NOAEL para toxicidad en el desarrollo fue < 25 mg/kg/día (AUC24h = 34340 ng• h/mL el día de Gestación 17; la exposición 85 veces más alta a la dosis relativa probada a la dosis clínica de 4 mg). En conejos, pomalidomida en rangos de dosis de 10 a 250 mg/kg produjo malformaciones en el desarrollo embrio-fetal. Se observaron más anomalías cardiacas a todas las dosis con incrementos significativos a 250 mg/kg/día. A 100 y 250 mg/kg/día, hubo ligeros incrementos en la pérdida postimplantación y una ligera disminución de los pesos corporales fetales. A los 250 mg/kg/día, las malformaciones fetales incluían anomalías de extremidades (delanteras y/o traseras flexionadas y/o rotadas, desprendidos o dígitos ausentes) y malformaciones esqueléticas asociadas (metacarpo no osificado, falanges y metacarpos mal alineados, dígito ausente, falange no osificada y corta, tibia no osificada o flexionada); dilatación moderada del ventrículo lateral del cerebro; colocación anormal de la arteria subclavia derecha; lóbulo intermediario ausente en los pulmones; implantación baja de los riñones; morfología hepática alterada; pelvis no osificada u osificada incompletamente; un incremento promedio para costillas torácicas supernumerarias y un promedio reducido de tarsos osificados. Ligera reducción en el aumento de peso corporal materno, reducción significativa en triglicéridos y disminución significativa en pesos absolutos y relativos del bazo se observaron a 100 y 250 mg/kg/día. El NOAEL materno fue 10 mg/kg/día y el NOAEL de desarrollo fue < 10 mg/kg/día (AUC24h = 418 ng• h/ml el Día 19 de Gestación; exposición de 1 vez a la dosis más baja probada con respecto a una dosis clínica de 4 mg).
INTERACCIONES MEDICAMENTOSAS Y DE OTRO GÉNERO: Potencial de que pomalidomida afecte otros fármacos
Pomalidomida no inhibe CYP1A2, CYP2A6, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, CYP2E1 o CYP3A4/5 in vitro. Además, pomalidomida no induce CYP1A2, CYP2B6, CYP2C9, CYP2C19 o CYP3A4/5 in vitro.
Pomalidomida no es un inhibidor de la glucoproteína P (P-gp) y tiene poco o ningún efecto inhibitorio sobre la proteína resistente al cáncer de mama (BCRP, por sus siglas en inglés), la Proteína transportadora de Aniones Orgánicos (OATP)1B1, OATP1B3, Transportadores de Aniones Orgánicos OAT1 y OAT3 y Transportador de Cationes Orgánicos OCT2 con base en estudios in vitro.
No se anticipa que pomalidomida cause interacciones medicamentosas farmacocinéticas clínicamente relevantes debido a la inhibición o inducción de enzimas o inhibición de transportadores cuando se coadministra con sustratos de estas enzimas o transportadores. El potencial de interacciones medicamentosas, incluyendo el potencial impacto de pomalidomida sobre la exposición a anticonceptivos orales, no se ha evaluado clínicamente.
Potencial de que Otros Fármacos Afecten Pomalidomida: Pomalidomida es metabolizada parcialmente por CYP1A2 y CYP3A4/5. También es un sustrato para la glucoproteína P. La coadministración de pomalidomida con el fuerte inhibidor de CYP3A4/5 y P-gp, ketoconazol o el fuerte inductor CYP3A4/5 carbamazepina, no tuvo efecto clínicamente relevante sobre la exposición a pomalidomida.
La coadministración del fuerte inhibidor CYP1A2 fluvoxamina, con pomalidomida en presencia de ketoconazol, incrementó la exposición media a pomalidomida en un 107% con un intervalo de confianza al 90% [91% a 124%] en comparación con pomalidomida más ketoconazol. En un estudio secundario que evalúa la contribución de un inhibidor de la CYP1A2 a los cambios metabólicos, la co-administración de la fluvoxamina sola en combinación con pomalidomida incrementó la exposición media a pomalidomida en 125% con un intervalo de confianza del 90% [98% a 157%] en comparación con pomalidomida sola. Reduzca en un 50% la dosis de pomalidomida si son co-administrados inhibidores fuertes de la CYP1A2 con la pomalidomida.
Dexametasona: La coadministración de 4 mg de pomalidomida con 20 mg a 40 mg de dexametasona (un inductor débil a moderado de varias enzimas CYP incluyendo CYP3A) a pacientes mieloma múltiple no tuvo efecto sobre la farmacocinética de pomalidomida en comparación con pomalidomida administrada sola. La dexametasona es un inductor débil a moderado de enzimas y se desconoce su efecto sobre la warfarina. Se recomienda monitorear estrechamente la concentración de warfarina durante el tratamiento.
ALTERACIONES EN LOS RESULTADOS DE PRUEBAS DE LABORATORIO: El paciente puede presentar alteraciones de las pruebas de funcionamiento hepático y biometría hemática.
PRECAUCIONES GENERALES:
Advertencia sobre Embarazo: Pomalidomida es un análogo de la talidomida. Se sabe que la talidomida es un teratógeno humano que causa defectos de nacimiento graves potencialmente mortales. Se encontró que la pomalidomida es teratogénica tanto en ratas como en conejos cuando se administra durante el periodo de organogénesis principal. Si se toma pomalidomida durante el embarazo, no se puede descartar un efecto teratogénico de pomalidomida en humanos.
Eventos Tromboembólicos: Los pacientes que reciben pomalidomida han desarrollado eventos tromboembólicos venosos (VTE), reportados como eventos adversos graves. Se recomienda terapia anticoagulación (a menos que esté contraindicada) (como aspirina, warfarina, heparina o clopidogrel). Se debe tomar cuidadosamente una decisión para realizar medidas profilácticas después de una valoración de los factores de riesgo subyacentes de un paciente individual.
Reacciones Alérgicas y Reacciones Serias en la Piel: Se ha reportado angioedema y reacciones dermatológicas severas, incluyendo síndrome de Stevens-Johnson (SJS), necrólisis epidérmica tóxica (TEN) y reacción a fármacos con eosinofilia y síntomas sistémicos (DRESS, por sus siglas en inglés). Se puede presentar DRESS con una reacción cutánea (tal como exantema o dermatitis exfoliativa), eosinofilia, fiebre y/o linfadenopatía con complicaciones sistémicas como hepatitis, nefritis, neumonitis, miocarditis y/o pericarditis. Estos eventos pueden ser fatales.
Los pacientes con antecedentes de reacciones alérgicas serias asociadas con talidomida o lenalidomida fueron excluidos de los estudios clínicos, porque pueden tener mayor riesgo de hipersensibilidad y no deben recibir pomalidomida. Se debe considerar la interrupción o descontinuación del tratamiento con pomalidomida en casos de exantema cutáneo Grado 2-3. Se debe descontinuar pomalidomida en caso de angioedema, exantema Grado 4, exantema exfoliativo o buloso, o si se sospecha SJS, TEN o DRESS, y no se debe reiniciar después de la descontinuación por estas reacciones.
Mareos y Confusión: Se han reportado casos de mareo y confusión. Indicar a los pacientes que tengan precaución en situaciones en las que los mareos o la confusión pueden ser un problema.
Malignidades secundarias: Se han reportado segundas malignidades primarias en pacientes que reciben pomalidomida. No se ha esclarecido la significancia clínica de estas observaciones. Los médicos deben evaluar cuidadosamente a los pacientes antes y durante el tratamiento usando detección de cáncer estándar para segundas malignidades primarias e instituir tratamiento según lo indicado.
Trastornos hepáticos: Se han observado niveles marcadamente elevados de alanina aminotransferasa y bilirrubina en pacientes tratados con pomalidomida. También ha habido casos de hepatitis que dieron lugar a la suspensión del pomalidomida. Se recomienda el control regular de la función hepática.
Infección: Raramente se ha reportado la reactivación de hepatitis B en pacientes que reciben pomalidomida en combinación con dexametasona quienes previamente han sido infectados con el virus de hepatitis B (HBV, por sus siglas en inglés). Algunos de estos casos ha progresado a falla hepática aguda, resultando en la descontinuación de pomalidomida.
Se debe tener precaución cuando la pomalidomida en combinación con dexametasona es usada en pacientes infectados previamente con HBV. Estos pacientes deben ser monitoreados cercanamente por si presentan signos y síntomas de una infección activa de HBV durante la terapia.
No compartir, desecho adecuado de las cápsulas sin usar: Se debe instruir a los pacientes que nunca proporcionen este medicamento a otra persona y que depositen las cápsulas no utilizadas en los contenedores de residuos de envases.
Donación de sangre o esperma: Todos los pacientes (hombres y mujeres con o sin potencial reproductivo) deben aceptar abstenerse de donar sangre o esperma mientras reciben pomalidomida (incluyendo interrupciones de dosis) y durante un mínimo de 4 semanas después de la última dosis de pomalidomida.
Síndrome de Lisis Tumoral: Puede ocurrir síndrome de lisis tumoral (SLT) en pacientes tratados con pomalidomida. Los pacientes en riesgo de SLT son aquellos con alta carga tumoral antes del tratamiento. Estos pacientes deben ser monitoreados estrechamente y se deben tomar las precauciones apropiadas.
Mieloma Múltiple en Recaída/Refractario:
Hematopoyesis: La reacción adversa hematológica reportada con mayor frecuencia fue neutropenia grado 3/4 en sujetos con mieloma múltiple en recaída/refractario, seguida por anemia y trombocitopenia.
Se debe monitorear a los pacientes para prevenir toxicidades hematológicas, especialmente neutropenia. Se recomienda monitorear la biometría hemática semanalmente durante las primeras 8 semanas y mensualmente a partir de entonces. Es posible que se requiera una modificación de la dosis. Los pacientes pueden requerir el uso de productos hematológicos y/o factores de crecimiento.
EFECTOS SOBRE LA CAPACIDAD PARA CONDUCIR VEHÍCULOS Y OPERAR MAQUINARIA: No se han realizado estudios sobre los efectos de pomalidomida sobre la capacidad para conducir u operar maquinaria. Se han reportado casos de confusión, fatiga, nivel de depresión de la conciencia y mareos con el uso de pomalidomida. Por consiguiente, se recomienda precaución al conducir u operar maquinaria en personas que reciben pomalidomida.
PRECAUCIONES ESPECIALES DE DESECHO Y OTRO MANEJO: No abrir o triturar las cápsulas. Si el polvo de pomalidomida entra en contacto con la piel, lavar inmediata y minuciosamente la piel con agua y jabón. Si la pomalidomida entra en contacto con las membranas mucosas, lavar con agua corriente en forma minuciosa.
DOSIS Y VÍA DE ADMINISTRACIÓN:
Administración: Pomalidomida se debe tomar por vía oral aproximadamente a la misma hora cada día. Las cápsulas no se deben abrir, romper o masticar. Las cápsulas de pomalidomida se deben ingerir enteras, de preferencia con agua, con o sin alimentos.
Adultos:
Mieloma Múltiple:
Dosis recomendada: La dosis inicial recomendada de pomalidomida es 4 mg/día por vía oral los Días 1-21 de ciclos repetidos de 28 días (21/28 días) hasta progresión de la enfermedad. La dosis recomendada de dexametasona es 40 mg/día los Días 1, 8, 15 y 22 de cada ciclo de tratamiento de 28 días.
La dosis se continúa o se modifica con base en los hallazgos clínicos y de laboratorio.
Modificación o Interrupción de la Dosis: Las instrucciones para interrupciones y reducciones de la dosis de pomalidomida se presentan en la Tabla 5 (toxicidades hematológicas).
Tabla 5. Instrucciones para Modificación de la Dosis de Pomalidomida por Toxicidades Hematológicas
|
Toxicidad |
Modificación de Dosis |
|
Neutropenia • ANC < 500/μl o Neutropenia febril (fiebre ≥ 38.5°C y ANC < 1,000/μL) • Retorno de ANC a ≥ 500/μL |
Interrumpir el tratamiento con pomalidomida, seguir semanalmente la Biometría Hemática. Agregar G-CSF (a discreción del médico tratante) Reanudar pomalidomida a 3 mg al día |
|
• Para cada caída subsecuente < 500/μL • Retorno a ≥ 500/μL |
Interrumpir el tratamiento con pomalidomida Reanudar pomalidomida a 1 mg menos que la dosis previa |
|
Trombocitopenia • Plaquetas < 25,000/μL • Retorno de plaquetas a > 50,000/μL |
Interrumpir el tratamiento con pomalidomida, llevar seguimiento semanal de Biometría Hemática Reanudar tratamiento con pomalidomida a 3 mg al día |
|
• Para cada caída subsecuente a < 25,000/μL • Retorno a ≥ 50,000/μL |
Interrumpir el tratamiento con pomalidomida Reanudar pomalidomida a 1 mg menos que la dosis previa |
Para iniciar un nuevo ciclo de pomalidomida, el recuento de neutrófilos debe ser ≥ 500/μL, el recuento plaquetario debe ser ≥ 50,000/μL.
Para otras toxicidades grado 3/4 que se consideren relacionadas con pomalidomida, suspender el tratamiento y reiniciar el tratamiento con 1 mg menos que la dosis previa cuando la toxicidad se haya resuelto a ≤ grado 2 a discreción del médico.
Si se presentan toxicidades después reducciones de dosis a 1 mg, se debe descontinuar el fármaco.
Ajustes de la dosis por la co-administración de los inhibidores de la CYP1A2: Reduzca la dosis de pomalidomida en un 50% si ésta se co-administra con inhibidores fuertes de la CYP1A2 (por ejemplo, fluvoxamina, ciprofloxacino).
Descontinuación de pomalidomida: Se debe considerar la interrupción o descontinuación de pomalidomida por rash cutáneo grado 2-3. Pomalidomida debe ser descontinuada por angioedema, rash grado 4, rash exfoliativo o buloso, o si se sospecha de Síndrome de Stevens-Johnson (SJS), necrólisis epidérmica tóxica (TEN) o reacción farmacológica con eosinofilia y síntomas sistémicos (DRESS) y no se debe reanudar después de la descontinuación por estas reacciones.
Pacientes pediátricos: No existen datos disponibles sobre administración de pomalidomida para sujetos pediátricos o adolescentes (< 18 años de edad).
Pacientes geriátricos: No se requiere ajuste de dosis para pomalidomida.
Para pacientes mayores de 75 años de edad, la dosis inicial de dexametasona es 20 mg/día los Días 1, 8, 15 y 22 de cada ciclo de tratamiento de 28 días.
Pacientes con Función Renal Deteriorada: No se requieren ajustes en la dosis de pomalidomida para los pacientes con deterioro renal. En días de hemodiálisis, la pomalidomida se debe tomar después de la hemodiálisis.
Pacientes con Función Hepática Deteriorada: Los pacientes con bilirrubina sérica > 2.0 mg/dL fueron excluidos de los estudios de eficacia. No se requieren ajustes de dosis de pomalidomida para pacientes con deterioro hepático, definidos por el criterio de Child-Pugh.
MANIFESTACIONES Y MANEJO DE LA SOBREDOSIFICACIÓN O INGESTA ACCIDENTAL: Se han estudiado dosis de pomalidomida tan altas como 50 mg como dosis única en voluntarios sanos y 10 mg como dosis múltiples una vez al día en pacientes con mieloma múltiple sin reportar eventos adversos graves relacionados con sobredosis. La pomalidomida fue removida por la hemodiálisis.
PRESENTACIONES: Caja con 21 cápsulas de 1 mg, 2 mg, 3 mg ó 4 mg.
RECOMENDACIONES SOBRE ALMACENAMIENTO: Consérvese de 20ºC a 25°C (68º F-77ºF); variaciones permitidas de 15-30ºC (59 ºF-86ºF).
Consérvese la caja bien cerrada.
LEYENDAS DE PROTECCIÓN:
Vía de administración: oral. Dosis: La que el médico señale. Su venta requiere receta médica. Léase instructivo anexo. Precaución: Potenciales defectos humanos de nacimiento. No se deje al alcance de los niños. Este medicamento deberá ser administrado únicamente por médicos especialistas con experiencia. No se use en el embarazo y lactancia, ni en menores de 18 años.
Reporte las sospechas adversas a los correos: farmacovigilancia@cofepris.gob.mx y a
dsmexico@celgene.com
Toda sospecha de exposición del feto a pomalidomida debe ser informada inmediatamente a Celgene Logistics SARL, al correo electrónico: dsmexico@celgene.com
Hecho en Suiza por:
Celgene International Sàrl
Route de Perreux 1
2017 Boudry, Suiza
o
Hecho en Reino Unido por:
Penn Pharmaceutical Services Limited
Units 23-24, Tafarnaubach Industrial Estate
Tafarnaubach, Tredegar
Gwent, NP22 3AA, Reino Unido
Distribuido y representante legal en México:
CELGENE LOGISTICS SARL
CPA Logistics Center Tlalnepantla
Edificio 1, Bodega 7, Almacén 12
Km 12.5 Vía Gustavo Baz,
Col. San Pedro Barrientos,
Tlalnepantla de Baz
C.P. 54010, México
Reg. Núm. 128M2016, SSA IV



